


Abstract
Compared with traditional algorithms for long metagenomic sequence classification, characterizing microorganisms’ taxonomic and functional abundance based on tens of millions of very short reads are much more challenging. We describe an efficient composition and phylogeny-based algorithm [Metagenome Composition Vector (MetaCV)] to classify very short metagenomic reads (75–100 bp) into specific taxonomic and functional groups. We applied MetaCV to the Meta-HIT data (371-Gb 75-bp reads of 109 human gut metagenomes), and this single-read-based, instead of assembly-based, classification has a high resolution to characterize the composition and structure of human gut microbiota, especially for low abundance species. Most strikingly, it only took MetaCV 10 days to do all the computation work on a server with five 24-core nodes. To our knowledge, MetaCV, benefited from the strategy of composition comparison, is the first algorithm that can classify millions of very short reads within affordable time.
INTRODUCTION
Recent advances in next-generation sequencing (NGS) technologies have opened a new era in the field of metagenomics (1–5), by providing much higher throughput and lower cost to sequence DNA directly taken from environmental samples. The application of NGS technologies on studies of microorganisms in human gut (6–9), oral cavity and skin, has greatly revealed the relationship of microorganisms to human disease, like adiposity, high blood pressure and dental cavity. NGS technologies also benefit the studies of bacterial communities in soil (1,2), deep ocean (3,4) and even ancient specimens (5). The fast expansion of NGS-based metagenomics calls for new bioinformatic algorithms which can handle the vast amount of sequence data more efficiently and effectively. Yet, the biggest concern for bioinformaticians is not only on the amount of the sequencing data but also on their read length. As the most popular NGS platforms, Illumina and Roche/454 output sequences as short as 75–400 bp, which bring more challenges and difficulties to environmental biologists.
There are currently two types of approaches to deal with metagenomic sequences. One is alignment based, like Megan (10), which compares short reads against coding sequences in public databases of coding genes using BlastX and then assigns them to their latest common ancestors (LCA) of targeted organisms. BlastX-based comparisons are commonly adopted to obtain fairly similar alignment between query sequences and reference proteins. Although many efforts have been made to improve the alignment speed (11,12), this process is still computationally expensive when working on tens of millions of very short reads. A possible solution is to first assemble the short reads into long contigs, and all subsequent analyses are based on these assembled contigs. Yet, almost all currently available assembly algorithms are designed for single genomes and require an even coverage distribution across chromosome. Applying them to assemble mass sequences from multiple species can result in heterozygous and chimeric contigs contributed by closely related species/strains and repetitive elements. Most importantly, assembly-based approaches may fail to assemble a certain amount of data, especially for those low abundance species.
The other type of approaches, i.e. Phymm (13), TETRA (14) and PhyloPythia (15) and CD-hit (16), uses information of oligonucleotide/oligopeptide composition for microorganism identification, because different organisms tend to have different composition patterns (17), e.g. GC content, codon usage or recognition sites of restriction endonuclease, which could be used to recognize taxonomic source of sequenced environmental reads. Composition-based strategy greatly facilitates the fast classification of microbial communities, compared with the BlastX-based algorithms. However, most of these algorithms (14,15) aim to work on sequence >1 kb, which require a pre-assembly of short reads into contigs and thus meet the same problem from misassembly. Furthermore, due to the complexity of microbial communities and limited number of sequenced genomes, distant related organisms are less likely to be detected by nucleotide-based algorithms comparing with those by BlastX.
In this study, we present a new composition-based approach, Metagenome Composition Vector (MetaCV), which directly works on raw short sequencing reads. Unlike any other composition-based methods, MetaCV first translates a nucleotide sequence into six-frame peptides, and these translated six-frame peptides are further decomposed to k-strings (oligopeptides of fixed length K), which are weighted and selected for further taxonomical classification based on their frequency in a pre-built reference protein database. Benefiting from this new composition-based strategy, MetaCV performs nearly as good as BlastX (even better at the Genus level), but significantly reduces the computing time (∼300 × faster) on a huge amount of metagenomic sequences. More importantly, besides focusing on taxonomic classification, MetaCV can also functionally annotate those unassembled short reads, to address the question ‘what they are doing’ after investigating ‘who they are’, and allows researchers to study the metabolic activities in microbial communities.
MATERIALS AND METHODS
The nature of this algorithm is to classify short reads to their most likely taxonomic group and gene function class by comparing with known proteins in the aspect of k-string similarity. As shown in Supplementary Figure S1, we employ a five-step strategy to process the comparison:
(1) Representing coding sequences by composition vectors of oligopeptide
MetaCV adopts frequencies of amino acid strings as features for classification as they are more conserved among far related species. Given a protein sequence with length L, we count the number of appearances of strings of a fixed length K in the sequence, and there are totally 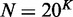 possible types of such strings. The collection of such frequencies is also known as ‘term frequency’ (TF) in the field of text mining. We also adopt ‘inverse document frequency’ (IDF) to weight different types of strings by different signal strength for inferring composition similarities between gene pairs. Given a k-string
possible types of such strings. The collection of such frequencies is also known as ‘term frequency’ (TF) in the field of text mining. We also adopt ‘inverse document frequency’ (IDF) to weight different types of strings by different signal strength for inferring composition similarities between gene pairs. Given a k-string 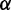 and an organism tree
and an organism tree 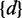 of the protein set, IDF of the string is defined as
of the protein set, IDF of the string is defined as 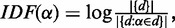 while
while 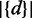 represents the total number of nodes in the organism tree and
represents the total number of nodes in the organism tree and 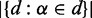 stands for the number of nodes including organisms containing this string and internal nodes on the paths to their LCA.
stands for the number of nodes including organisms containing this string and internal nodes on the paths to their LCA.
(2) Building of a k-string composition database for reference genes
MetaCV calculates TF–IDF values for all possible strings as components to form a composition vector for each gene. A reference database contains a collection of all gene vectors and a correlation matrix for each pair of genes. The correlation 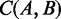 between any two sequences A and B is calculated as the cosine similarity of the two representative vectors in the N-dimensional composition space as described in (18). MetaCV calculates correlations for any pair of proteins with at least three 6-mers shared by them, and the final correlation matrix includes 3 × 109 values, which used 12 Gb space by using strategy of compressed sparse row. In this analysis, which contains 5 million proteins from 1691 organisms, a memory of 26 Gb (almost identical to the sum of db files) is required for downstream read binning.
between any two sequences A and B is calculated as the cosine similarity of the two representative vectors in the N-dimensional composition space as described in (18). MetaCV calculates correlations for any pair of proteins with at least three 6-mers shared by them, and the final correlation matrix includes 3 × 109 values, which used 12 Gb space by using strategy of compressed sparse row. In this analysis, which contains 5 million proteins from 1691 organisms, a memory of 26 Gb (almost identical to the sum of db files) is required for downstream read binning.
(3) Comparing query reads with reference genes
Query nucleotide sequences are translated into coding sequences by considering six-frame translations. Here MetaCV adopts an open reading frame (ORF) searching strategy similar to OrfPredictor (19), which is designed for expressed sequence tags (ESTs). Each frame is evaluated individually to give complete/partial ORF candidates in cases of having/lacking of start/stop codons. These candidates are then compared with all proteins in the database, and only the one who has the optimal correlation score, is considered as the correct translation and kept for further analysis. Users could also use a third party gene finders, i.e. MetaGene (20) to translate short reads into amino acid sequences as an alternative input for MetaCV. For paired-end reads which are resulted from DNA libraries with short insert size, both two ends of a given read pair are evaluated together, and the strand information is also taken into account.
(4) Tree construction for query read and targeted genes to obtain taxonomic and functional information of the read
Denote matched genes of a query read by 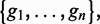 which are sorted descendingly according to their correlation values against the query read represented by
which are sorted descendingly according to their correlation values against the query read represented by  The first m genes
The first m genes  are selected by maximizing
are selected by maximizing 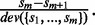 The method UPGMA (21) is then applied on the sequence set including selected sequences
The method UPGMA (21) is then applied on the sequence set including selected sequences 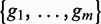 and the query read to build a phylogenetic tree. The aim of this step is to collect genes, denoted by
and the query read to build a phylogenetic tree. The aim of this step is to collect genes, denoted by 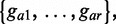 in the sister-group of the query read on the tree, to infer taxonomic and functional information of the query.
in the sister-group of the query read on the tree, to infer taxonomic and functional information of the query.
The taxonomic position of the query is defined as the LCA of organisms who contains the gene set  in NCBI’s taxonomic tree (22), while the functional categories of the query is defined as the KEGG class (23) who have the most votes from annotation of the gene set
in NCBI’s taxonomic tree (22), while the functional categories of the query is defined as the KEGG class (23) who have the most votes from annotation of the gene set 
(5) Post-analysis and comparison of taxonomic and functional enrichment of different samples
MetaCV outputs category enrichment at various taxonomic levels from genus to phylum and also for each level of KEGG functions. It can also compare multiple samples and provide differentially enriched taxonomic or functional categories. The output and comparison can be displayed as tables and figures in an integrated R package.
Complete prokaryotic proteomes as references
We have included all prokaryotic complete genomes that had been publicly available by April 2012 at the National Center for Biotechnology Information (NCBI) (22). Altogether 1691 organisms from 1023 prokaryotic species distributed in 578 genera, 230 families, 113 orders, 53 classes and 32 phyla are included in our complete genome collection. When a genome consists of more than one chromosome, we would collect all the translated sequences.
Metagenomic reads simulation
Dataset 1
To evaluate the performance of MetaCV on new species detection, we used inGAP (24) to simulate single-end short reads from each read donor genome. Among 578 known genera in RefSeq, 154 of them contain at least two species groups. For each of these 154 genera, only one strain from one species was selected as a read donor genome, while all the strains from the sister-species were used as references. For each donor genome, 1000 reads were generated for each different read length (as 100 bp, 200 bp, 400 bp, 600 bp, 800 bp and 1 kb, respectively). This procedure was repeated five times to estimate the variance of evaluation, and finally 4.6 million reads were generated. All the four methods, MetaCV, Phymm (13)3.2, BlastX2.2.24 and RAPSearch2 (11) 2.04 were applied to classify these reads against the same reference data set, in which Phymm utilized the nucleotide genomic sequences while MetaCV, BlastX and RAPSearch2 used the protein sequences.
Dataset 2
We also tested the sensitivity and specificity of MetaCV by using ‘benchmarked’ simulation datasets. Among the currently available 1691 prokaryotic genomes in the NCBI datasets, 428 of them were released after January 2011 and we used them as query genomes. The remaining 1263 genomes released before 2011 were regarded as references. Similar to the first dataset simulation procedure, by randomly selecting 1000 reads for each length variances from each genome and after a five-time repeat, a total of 12.8 million reads were simulated, among which 2.1 million reads were of 100 bp. Because this test tripled the number of simulation reads and doubled that of reference genomes comparing with Dataset 1, only MetaCV was applied to all simulated reads of different lengths, both Phymm and BlastX were restrictively applied to the classification of those 100-bp reads by considering the enormous time they consumed.RAPSearch2 was also applied to the 100-bp simulation data.
All the simulated data and lists of both reads-donor and reference organisms are available online at http://metacv.sourceforge.net/.
Real NGS short reads from human gut samples
Qin et al. (6) studied the structure of bacterial communities from 124 human gut samples by using paired-end Illuminia-sequencing technology. In their article, short reads were assembled into contigs, on which BlastX search against NR database (22) were applied, to obtain relative enrichment of non-divergent prokaryotes comparing with those sequences available genomes. Here we applied MetaCV on the same sequence data to predict the enrichment of microorganisms, then compared the most redundant organisms shared by cohorts of healthy people with the ‘core set’ provided by Qin et al. since assembled contigs are more likely to be from relatively enriched species.
RESULTS
We compared the performance between MetaCV and BlastX, which is time consuming but is still considered as the most accurate algorithm to perform homology search. The downstream taxonomic classification of the short reads based on the results of BlastX is proceeded by Megan (10). Phymm (13), a method adopted an Interpolated Markov model and is capable of classifying reads as short as 100 bp, and RAPSearch2, a fast alignment tool, were also applied on the same data for comparison. MetaCV adopted a score of 20 as a minimal value to filter the classification results for 100 bp reads (see ‘Discussion’ section for longer reads). A cutoff e-value of 1 e − 1 was applied to both BlastX and RAPSearch2 for all datasets. As Phymm did not claim a confidence threshold for its binning scores, no cutoff value was set.
Species-mask comparison of MetaCV with other approaches on Dataset 1
We first evaluated the performance of the four methods on detecting new species. Till now, there are 1691 complete prokaryotic genomes available in NCBI. By selecting 154 out of 578 genera in which two or more species are sequenced, we simulated short reads from one species of a genus with various lengths (see ‘Materials and Methods’ section for details), while the sequences from the sister species were used to build reference profiles, to perform a species-mask comparison. As expected, due to higher conservation of genes in the amino acid level than in the nucleotide level within and among species, MetaCV could classify 100-bp reads within their donor genera with sensitivity/specificity as 41%/80%, which are very comparable to those based on BlastX (41%/87%) and RAPSearch2 (41%/86%), and are considerably higher than those of Phymm (24%/26%). Among all the 154 genera we employed, MetaCV outperformed Phymm in 131 of them on sensitivity and all genera on specificity as shown in Figure 1 and Supplementary Table S1. Interestingly, the average sensitivity of MetaCV on the genus level performs better than that of BlastX of up to 6%, the longer the simulated read length is, the higher sensitivity MetaCV reaches (Figure 2). This is because Megan (10), the methods used here to process BlastX results, tends to assign reads to the higher taxonomic levels than MetaCV does (Supplementary Figure S2). That also explains why the effectivity of BlastX-based binning outperforms that of MetaCV on higher level comparison (Figure 2.). In general, MetaCV detects novel species on short reads as effectively as BlastX does while it operates much more efficiently when comparing computational speed as shown in Figure 3A.
Figure 1.

Performance comparison on the species-mask testing of MetaCV, Phymm, BlastX and RAPSearch2 on the classification of 100-bp simulated reads from 154 genera. Sensitivities (top) and specificities (bottom) of MetaCV, Phymm, BlastX and RAPSearch2on each genus are displayed vertically in one dot, represented in blue, red green and orange, respectively. The 154 genera are sorted according to BlastX sensitivities (green dots) in descending.
Figure 2.

Sensitivity and specificity comparisons of MetaCV (blue), Phymm (red), BlastX (green) and RAPSearch2 (orange) on different read lengths. Sub-figures from left to right, represent sensitivities (top) and specificities (bottom) of the methods from genus to phylum level.
Figure 3.

(A) Computation time of MetaCV (blue), Phymm (red), BlastX (green) and RAPSearch2 (orange) on different lengths of reads, compared with a reference database of 5 × 106 genes. For 100-bp reads, MetaCV could process one million reads with ∼5 h on a single thread. (B) Accuracy comparison of MetaCV between taxonomic classification and functional assignment on simulated reads.
However, out of the 154 genera, 20 have sensitivity values <10% by MetaCV as shown in Figure 1, and the scores were not significantly improved even when the length of simulated reads was extended from 100 to 1 kb (Figure 2 and Supplementary Table S2). Among the 20 genera, we noticed that 86% of the 100-bp reads simulated from Escherichia were classified to Shigella or into a higher level (Enterobacteriaceae family). The controversy on the phylogeny definition of Shigella and Escherichia has been existing for a long time (25) and it is debated Shigella should be redefined as an species group within Escherichia. If so, the sensitivity of MetaCV would be saved (to 95%) on the evaluation of family-prediction level.
Another interesting case came from genus Anabaena. On the first submission of this work, NCBI taxonomy showed that it had two species, Anabaena variabilis (strain ATCC 29413) and Nostoc azollae (strain 0708) and MetaCV classified 71% of the simulated reads from Anabaena variabilis into an organism from its neighbour genus, Nostoc punctiforme (strain PCC 73102). A further BlastP comparison was made to observe the protein-level divergence among the three organisms. As shown in Supplementary Figure S3, there are only 41% proteins on Anabaena variabilis have matches on its sister-species, Nostoc azollae, with high identity (>50%). In contrast, 76% of proteins in Anabaena variabilis matched to Nostoc punctiforme, which indicates that Anabaena variabilis is phylogenetically closer to Nostoc punctiforme. A similar observation was also made based on the characteristics of cellular fatty acids (26). Interestingly but not unexpectedly, NCBI taxonomy updated the phylogenetic relationship among them in 2012 (Supplementary Figure S3), where Nostoc azollae was put under a new genus Trichormus, thus consequently avoided this controversy but still could not answer the challenge of MetaCV result.
We further performed an all-against-all BlastP comparison for the proteins in 154 genera, to figure out the correlation between BlastP-based similarity and MetaCV accuracy rate. As shown in Supplementary Figure S4, the higher ratio of homologues shared within genus, the higher accuracy rate reached by MetaCV. To monitor the correlation between the composition scores and BlastX identities, we inspected them on 100-bp short reads from four genera by considering various homologue identities of BlastX from 60% to 90%, as shown in Supplementary Figure S5. Most of the reads with composition scores > 20 (the cutoff value) were correctly classified. Likewise, falsely classified reads usually have composition scores lower than the cutoff. We noticed that there were still a few reads with high composition scores and BlastX identities, but were falsely classified by both methods. For example, a 100-bp read was simulated from a RNA-directed DNA polymerase of Pectobacterium atrosepticum SCRI1043. However, it was classified to Erwinia tasmaniensis Et1/99. This is because the donor gene has a 91% similar homologue in Erwinia but no homologue in its sister-species.
Moreover, we observed that a small proportion of reads were assigned to a higher taxonomic level. This is due to the reason that the donor genes are highly conserved among many species and the reads were thus assigned to their last common ancestors. For example, we found that a read simulated from an acyltransferase of Pyrobaculum aerophilum (strain IM2) was classified into the Archaea domain, because this gene was highly conserved in many archaeal genera, i.e. Thermoproteus, Vulcanisaeta, Sulfolobus and Metallosphaera.
Performance evaluation on simulated ‘randomized’ metagenome datasets (Dataset 2)
The real metagenomic samples might have relatively simple community structures, reflected by DNA sampling from, i.e. acid mine drainage biofilms (27). However, in most cases, environmental samples may include thousands of organisms from a wide spectrum of taxonomy. To build simulated datasets including random organisms to test these methods further, we chose the 428 genomes released after than January 2011 to simulate short reads, while the rest 1263 genomes published earlier to build reference database. The ‘random’ division of organisms into queries and references by their release dates could be used both to make a randomized mixture of read donor genomes and to inspect coverage of taxonomic tree by recent sequenced organisms.
Among the 428 query genomes distributed in 219 genera, 117 organisms (27%) come from 103 genera, averagely one organism per genus, which are brand new and distantly related to the reference organisms. Therefore, the simulated reads from the 117 organisms are considered as ‘negative’ in the comparison of genus level. On average, MetaCV could correctly assign 58% of 100-bp reads to their donor genera, slightly lower than that of BlastX (61%) and RAPSearch2 (60%) and much better than Phymm (28%) as shown in Figure 4. On the comparison of family level, only 21 organisms (5%) from 17 new families are considered as ‘negative’, the rest 96 new-genus organisms are from ‘known’ families. Adding them reduces the overall sensitivity to 52% due to their divergence to any references, while the specificity remains the same.
Figure 4.

Evaluation of classification sensitivities (top) and specificities (bottom) of the four methods on ‘randomized’ metagenomic data by simulating reads from 428 genomes released after January 2011, compared with 1263 genomes published earlier. Ratio values of MetaCV, Phymm, BlastX and RAPSearch2 are coloured by blue, red, green and orange, respectively. Five groups from left to right are the results of these methods on different taxonomic level (genus to phylum, respectively). Only MetaCV is applied to classify simulated reads >100 bp.
Behaviour of MetaCV to deal with human DNA contaminations and prokaryotic non-coding sequences
It is worthy to note that some environmental samples are likely possible to be contaminated by eukaryotic DNA, especially for host-associated bacterial communities. In practice, an initial step to process potentially contaminated metagenomic data is to align them to known eukaryotic genomes and then to filter all the mapped reads. However, there still is a possibility that some host sequences pass the filtering step and are used to determine their ‘bacterial original’. To test the robustness of MetaCV, we simulated 1 million 100–1000 bp random DNA segments from chromosome 1 of the human genome and applied MetaCV to do taxonomic classification. As shown in Supplementary Figure S6, 95% of 100-bp human sequences were not assigned to any bacterial taxon when using a minimal identity score of 20 as a cutoff. The cutoff value reduces along with the increasing of read length at the same false positive rate of 5% (Supplementary Figure S6). These cutoff values were applied all through this work.
As suggested by the anonymous reviewer, there is a possibility that non-coding sequences of prokaryotes could share similarities with reference sequences in the view of composition. To test this hypothesis, we inspected the classification results of the 66 817 non-coding reads (100 bp, on average 8.7% per genome) from the first simulated dataset (see ‘Materials and Methods’ section). Among them, 82.8% obtained a composition-identity score <20 (the cutoff value), compared with reference proteins. In this sense, not all metagenomic DNA sequences could be utilized for the estimation of taxonomic abundance by MetaCV: the more non-coding regions an organism has, the lower ratio of its short reads could pass the filter. On the other hand, we found 2572 of non-coding reads (0.3%) shown composition identity as high as 100 to reference proteins by using MetaCV, and they were further approved by using BlastX. This indicates these reads might come from real coding regions in the donor genome and were recognized as ‘non-coding’ due to incomplete annotation. As expected, MetaCV classified these reads with sensitivity as 86.0% and specificity as 94.7% on genus level (96.2% and 97.7% on family level, respectively).
A case study on 109 human gut metagenomic data
To further evaluate the performance of MetaCV on real datasets, we applied it to classify and annotate 371-Gb NGS reads from Meta-HIT (6). In Qin et al.’s study (6), short reads were first assembled into contigs, and then BlastX searches against NR databases (22) were applied to obtain the relative enrichment of identified microorganisms. Here we applied MetaCV on the same sequence data to predict the enrichment of microorganisms at a single-read resolution. To avoid bias of classification accuracy on different read lengths, 109 of 124 human faecal samples were selected which adopted Illumina 2 × 75-bp paired-end sequencing technology. Among 4952 million reads in the raw data, 1530 million of them had composition identity scores >20 and were kept for further statistical analyses. As shown in Supplementary Figure S7, Bacteroidetes, as the most abundant phylum reported by Qin et al. (6), occupied 54% of the total reads from all 109 samples assigned by MetaCV, followed by Firmicutes (34%) and Proteobacteria (5%). It should be noted that MetaCV successfully identified an archaeal genus, Methanobrevibacter, which was recently reported as a human gut-associated archaeon and played an important role in the metabolism of hydrogen (28). As shown in Figure 5B, healthy cohorts contain much less Methanobrevibacter, compared to the samples of ulcerative colitis or Crohn’s disease patients, except for sample MH0011. Among the 271 490 reads contributed to a draft assembly (6) of Methanobrevibacter (∼1.6 Mb), 74% of them were further confirmed as Methanobrevibacter by MetaCV. These reads could be evenly distributed along the contigs (Figure 5C), and we also identified a number of intraspecific polymorphic sites on them (Figure 5D), which may help investigate the relative enrichment of Methanobrevibacter strains at a deeper taxonomic level.
Figure 5.

Display of taxonomic classification of MetaCV on 109 Meta-HIT samples. (A) Box plots of read coverage for the top 44 most abundant genera on average of 109 samples. Whiskers show the lowest and highest value among samples, while the boxes denote the first, median (line) and the third quartiles. Total number of reads in each sample is normalized to one million. (B) Relative abundance of selected prokaryotes among 109 samples, in which 70 samples are from healthy human (marked as ‘Healthy’), 27 samples are from ulcerative colitis patients (as ‘UC’) and 12 from Crohn’s disease patients (as ‘CD’). (C) The alignment of MetaCV-classified reads to the 1.6-Mb scaffolds of Methanobrevibacter. (D) Mapping details of a 3-kb region in (C) reveal nucleotide polymorphisms in Methanobrevibacter contigs.
A list of 44 genera, which includes at least 400 reads per million sequences on average of all samples, is shown in Figure 5A and Supplementary Table S4. Of the 44 genera, 31 were found to present in at least 50 gut specimens (Supplementary Table S4 and Supplementary Figure S8). Compared with the list of 27 genera reported by Qin et al. based on short read assembly, 13 of 27 genera have complete genomes available in the reference database of MetaCV, and all of them are top abundant in the MetaCV classifications, which implies that more abundant organisms tend to have a higher possibility to be assembled. MetaCV further investigated the genera with relatively lower abundance but present in a majority of cohorts. Most of them were considered as gastrointestinal bacteria by the Human Microbiome Project (HMP) (29) (Supplementary Table S4). MetaCV also identified a few gut-associated genera not listed in HMP (Figure 5B and Supplementary Table S4): Ethanoligenens, reported to have a possible correlation with Irritable Bowel Syndrome in rats by a recent study based on microarray analysis (30); Burkholderia, of which a type of bacteriophage is found to be enriched among viral community of human faeces (31); Spirochaeta, which may have a phylogenetically close relative in pig gut microbiota (32); and Fibrobacter, a type of polysaccharide-utilizing bacteria commonly existing in gut of rumen (33).
In the studies of metagenomics, instead of only analysing abundance of species, it may be also important to detect the existence of specific genes or pathways, especially of the genes easily to be transferred horizontally. Furthermore, as most enriched functional categories are usually for basic metabolic processes (Supplementary Figure S9), it makes the studies on low abundant pathways necessary. Abundance of functional categories assigned by MetaCV shows less fluctuation compared to the taxonomic classifications (Supplementary Figures S10 and S11). This was further confirmed by using the simulation data, on which the accuracy of function prediction outperforms that of taxonomic classification on different read lengths, as shown in Figure 3B. A possible reason is that the definition systems of gene functions, like KEGG (23) and eggnog (34), etc. are based on homology comparison, and provide a relatively ‘fair’ standard than that of the definition of taxonomic groups which depends on the collections of phenotypes and metabolism features.
DISCUSSION
Although limited prokaryotes have whole genome sequence available at present and most of them are human disease related, more and more organisms are being sequenced and assembled in laboratories worldwide. Bergey’s Manual Trust initialized a large-scale genome sequencing project, which aims to cover most phyla of prokaryotes. Department of Energy of the USA supported a project of ‘A Genomic Encyclopedia of Bacteria and Archaea’ for the same purpose. Classifications of taxon-unknown sequences based on homology search will largely benefit from the sharp increase of whole prokaryotic genomes in the next few years. At present, divergent organisms, which lack closely related references, are hard to be correctly classified by supervised (or reference-guided) methods including MetaCV. In an evaluation on genus-masking comparison (Supplementary Figure S12), none of the methods involved have acceptable sensitivities to trace short reads back to their donor families, although the specificities of MetaCV and BlastX are >60% on average. This problem can be partially overcome by incorporating with unsupervised methods, like TETRA (14) etc., which can efficiently discover new genera or even far related organisms. The integration of two or more types of algorithms calls for further study.
Another way to detect very divergent organisms, when sharing enough oligopeptides between query reads and reference genes are rare possible, is to allow mutations in the process of composition comparison, i.e. to avoid the requirement of exact matching of k-strings. It is well known that the replacement of some residues by other ones with the same physical properties, basic or acidic, hydrophobic or hydrophilic, might not affect the folding of proteins and are tolerated by natural selection during evolution. This has been also reflected more or less in scoring matrices adopted by BlastP alignment. Naturally, simplifying the alphabet of 20 amino acids in MetaCV will be a candidate solution.
The only parameter in this method is the length of oligopeptides K, of which the selection has been discussed to highlight the phylogenetic signals within compositions for phylogenetic study of prokaryote relationships (18,35). In this study, we tested the performance of MetaCV under different K-values. Taking calculating time into account, K = 6 runs 20 times faster than K = 5 (Supplementary Figure S13), and faster further than K = 4. For accuracy of classification, K = 6 has sensitivities almost identical to K = 5 but has much higher specificities (Supplementary Figure S14). Therefore, K is set as 6 by default in MetaCV and is also adopted throughout this work. K-values > 6 are not considered, because the number of oligopeptide types is over 1 × 109, which makes the composition database too large to load in many workstations.
In general, MetaCV not only performs taxonomic classification at various levels from genus to phylum but also provides functional annotation for short reads without any assembly. In addition, it can compare multiple samples and report differentially enriched taxonomic or functional categories. MetaCV is implemented in C++ and is integrated with R scripts to generate figures and tables. Comparing with BlastX, MetaCV speeds up computing >300-fold faster, which mainly benefits from its searching strategy of compositions instead of doing alignment. Computation time of MetaCV is linearly correlated with length of reads as shown in Figure 3A. It takes MetaCV 10 days to compare 5 billion reads (2 × 75-bp paired-end sequencing, 371 Gb in total) of 109 human faecal samples against 1691 prokaryotic organisms on a mini server with five 24-core nodes, averagely 0.2 million reads per hour per core. Its classification results could be visualized by Megan (10), which provides user-friendly graphical interface for checking community structure of environmental organisms. The source code of MetaCV and the corresponding database can be freely accessed at http://metacv.sourceforge.net/
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Tables 1–4 and Supplementary Figures 1–14.
FUNDING
The National Natural Science Foundation of China [31100094 to J.Q., 31100952 to F.Z.]; open fund of State Key Laboratory of Freshwater Ecology and Biotechnology [2012FB16]. Funding for open access charge: National Natural Science Foundation of China [31100094 to J.Q., 31100952 to F.Z.].
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Profs Bailin Hao (Fudan University), Jingchu Luo (Peking University) and Liping Zhao (Shanghai Jiaotong University) for discussion and comments. This research is supported by the biological supercomputing server of Computing Center of Beijing Institutes of Life Science.
REFERENCES
- 1.Leininger S, Urich T, Schloter M, Schwark L, Qi J, Nicol GW, Prosser JI, Schuster SC, Schleper C. Archaea predominate among ammonia-oxidizing prokaryotes in soils. Nature. 2006;442:806–809. doi: 10.1038/nature04983. [DOI] [PubMed] [Google Scholar]
- 2.Tringe SG, von Mering C, Kobayashi A, Salamov AA, Chen K, Chang HW, Podar M, Short JM, Mathur EJ, Detter JC, et al. Comparative metagenomics of microbial communities. Science. 2005;308:554–557. doi: 10.1126/science.1107851. [DOI] [PubMed] [Google Scholar]
- 3.DeLong EF. Microbial community genomics in the ocean. Nat. Rev. Microbiol. 2005;3:459–469. doi: 10.1038/nrmicro1158. [DOI] [PubMed] [Google Scholar]
- 4.Venter JC, Remington K, Heidelberg JF, Halpern AL, Rusch D, Eisen JA, Wu D, Paulsen I, Nelson KE, Nelson W, et al. Environmental genome shotgun sequencing of the Sargasso Sea. Science. 2004;304:66–74. doi: 10.1126/science.1093857. [DOI] [PubMed] [Google Scholar]
- 5.Poinar HN, Schwarz C, Qi J, Shapiro B, Macphee RD, Buigues B, Tikhonov A, Huson DH, Tomsho LP, Auch A, et al. Metagenomics to paleogenomics: large-scale sequencing of mammoth DNA. Science. 2006;311:392–394. doi: 10.1126/science.1123360. [DOI] [PubMed] [Google Scholar]
- 6.Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe BA, Affourtit JP, et al. A core gut microbiome in obese and lean twins. Nature. 2009;457:480–484. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gill SR, Pop M, Deboy RT, Eckburg PB, Turnbaugh PJ, Samuel BS, Gordon JI, Relman DA, Fraser-Liggett CM, Nelson KE. Metagenomic analysis of the human distal gut microbiome. Science. 2006;312:1355–1359. doi: 10.1126/science.1124234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kurokawa K, Itoh T, Kuwahara T, Oshima K, Toh H, Toyoda A, Takami H, Morita H, Sharma VK, Srivastava TP, et al. Comparative metagenomics revealed commonly enriched gene sets in human gut microbiomes. DNA Res. 2007;14:169–181. doi: 10.1093/dnares/dsm018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huson DH, Auch AF, Qi J, Schuster SC. MEGAN analysis of metagenomic data. Genome Res. 2007;17:377–386. doi: 10.1101/gr.5969107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhao Y, Tang H, Ye Y. RAPSearch2: a fast and memory-efficient protein similarity search tool for next-generation sequencing data. Bioinformatics. 28:125–126. doi: 10.1093/bioinformatics/btr595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kent WJ. BLAT–the BLAST-like alignment tool. Genome Res. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brady A, Salzberg SL. Phymm and PhymmBL: metagenomic phylogenetic classification with interpolated Markov models. Nat. Methods. 2009;6:673–676. doi: 10.1038/nmeth.1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Teeling H, Waldmann J, Lombardot T, Bauer M, Glockner FO. TETRA: a web-service and a stand-alone program for the analysis and comparison of tetranucleotide usage patterns in DNA sequences. BMC Bioinformatics. 2004;5:163. doi: 10.1186/1471-2105-5-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McHardy AC, Martin HG, Tsirigos A, Hugenholtz P, Rigoutsos I. Accurate phylogenetic classification of variable-length DNA fragments. Nat. Methods. 2007;4:63–72. doi: 10.1038/nmeth976. [DOI] [PubMed] [Google Scholar]
- 16.Li W, Wooley JC, Godzik A. Probing metagenomics by rapid cluster analysis of very large datasets. PLoS One. 2008;3:e3375. doi: 10.1371/journal.pone.0003375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Karlin S, Burge C. Dinucleotide relative abundance extremes: a genomic signature. Trends Genet. 1995;11:283–290. doi: 10.1016/s0168-9525(00)89076-9. [DOI] [PubMed] [Google Scholar]
- 18.Qi J, Wang B, Hao BI. Whole proteome prokaryote phylogeny without sequence alignment: a K-string composition approach. J. Mol. Evol. 2004;58:1–11. doi: 10.1007/s00239-003-2493-7. [DOI] [PubMed] [Google Scholar]
- 19.Min XJ, Butler G, Storms R, Tsang A. OrfPredictor: predicting protein-coding regions in EST-derived sequences. Nucleic Acids Res. 2005;33:W677–W680. doi: 10.1093/nar/gki394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Noguchi H, Park J, Takagi T. MetaGene: prokaryotic gene finding from environmental genome shotgun sequences. Nucleic Acids Res. 2006;34:5623–5630. doi: 10.1093/nar/gkl723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sokal R, Michener C. A statistical method for evaluating systematic relationships. Univ. Kansas Sc. Bull. 1958;38:1409–1438. [Google Scholar]
- 22.Sayers EW, Barrett T, Benson DA, Bolton E, Bryant SH, Canese K, Chetvernin V, Church DM, DiCuccio M, Federhen S, et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2011;39:D38–D51. doi: 10.1093/nar/gkq1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kanehisa M, Goto S, Furumichi M, Tanabe M, Hirakawa M. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 2010;38:D355–D360. doi: 10.1093/nar/gkp896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qi J, Zhao F, Buboltz A, Schuster SC. inGAP: an integrated next-generation genome analysis pipeline. Bioinformatics. 2010;26:127–129. doi: 10.1093/bioinformatics/btp615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bouvet OM, Lenormand P, Guibert V, Grimont PA. Differentiation of Shigella species from Escherichia coli by glycerol dehydrogenase activity. Res. Microbiol. 1995;146:787–790. doi: 10.1016/0923-2508(96)81074-6. [DOI] [PubMed] [Google Scholar]
- 26.Caudales R, Wells JM. Differentiation of free-living Anabaena and Nostoc cyanobacteria on the basis of fatty acid composition. Int. J. Syst. Bacteriol. 1992;42:246–251. doi: 10.1099/00207713-42-2-246. [DOI] [PubMed] [Google Scholar]
- 27.Tyson GW, Chapman J, Hugenholtz P, Allen EE, Ram RJ, Richardson PM, Solovyev VV, Rubin EM, Rokhsar DS, Banfield JF. Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature. 2004;428:37–43. doi: 10.1038/nature02340. [DOI] [PubMed] [Google Scholar]
- 28.Hansen EE, Lozupone CA, Rey FE, Wu M, Guruge JL, Narra A, Goodfellow J, Zaneveld JR, McDonald DT, Goodrich JA, et al. Pan-genome of the dominant human gut-associated archaeon, Methanobrevibacter smithii, studied in twins. Proc. Natl Acad. Sci. USA. 108(Suppl. 1):4599–4606. doi: 10.1073/pnas.1000071108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peterson J, Garges S, Giovanni M, McInnes P, Wang L, Schloss JA, Bonazzi V, McEwen JE, Wetterstrand KA, Deal C, et al. The NIH Human Microbiome Project. Genome Res. 2009;19:2317–2323. doi: 10.1101/gr.096651.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nelson TA, Holmes S, Alekseyenko AV, Shenoy M, Desantis T, Wu CH, Andersen GL, Winston J, Sonnenburg J, Pasricha PJ, et al. PhyloChip microarray analysis reveals altered gastrointestinal microbial communities in a rat model of colonic hypersensitivity. Neurogastroenterol Motil. 2011;23:169–177. doi: 10.1111/j.1365-2982.2010.01637.x. e141–e162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Breitbart M, Hewson I, Felts B, Mahaffy JM, Nulton J, Salamon P, Rohwer F. Metagenomic analyses of an uncultured viral community from human feces. J. Bacteriol. 2003;185:6220–6223. doi: 10.1128/JB.185.20.6220-6223.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leser TD, Amenuvor JZ, Jensen TK, Lindecrona RH, Boye M, Moller K. Culture-independent analysis of gut bacteria: the pig gastrointestinal tract microbiota revisited. Appl. Environ. Microbiol. 2002;68:673–690. doi: 10.1128/AEM.68.2.673-690.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Flint HJ, Bayer EA, Rincon MT, Lamed R, White BA. Polysaccharide utilization by gut bacteria: potential for new insights from genomic analysis. Nat. Rev. Microbiol. 2008;6:121–131. doi: 10.1038/nrmicro1817. [DOI] [PubMed] [Google Scholar]
- 34.Powell S, Szklarczyk D, Trachana K, Roth A, Kuhn M, Muller J, Arnold R, Rattei T, Letunic I, Doerks T, et al. eggNOG v3.0: orthologous groups covering 1133 organisms at 41 different taxonomic ranges. Nucleic Acids Res. 2012;40:D284–D289. doi: 10.1093/nar/gkr1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zuo G, Xu Z, Yu H, Hao B. Jackknife and bootstrap tests of the composition vector trees. Genomics Proteomics Bioinformatics. 8:262–267. doi: 10.1016/S1672-0229(10)60028-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.