


Abstract
Direct modulation of gene expression by targeting oncogenic transcription factors is a new area of research for cancer treatment. ERG, an ETS-family transcription factor, is commonly over-expressed or translocated in leukaemia and prostate carcinoma. In this work, we selected the di-(thiophene-phenyl-amidine) compound DB1255 as an ERG/DNA binding inhibitor using a screening test of synthetic inhibitors of the ERG/DNA interaction followed by electrophoretic mobility shift assays (EMSA) validation. Spectrometry, footprint and biosensor-surface plasmon resonance analyses of the DB1255/DNA interaction evidenced sequence selectivity and groove binding as dimer. Additional EMSA evidenced the precise DNA-binding sequence required for optimal DB1255/DNA binding and thus for an efficient ERG/DNA complex inhibition. We further highlighted the structure activity relationships from comparison with derivatives. In cellulo luciferase assay confirmed this modulation both with the constructed optimal sequences and the Osteopontin promoter known to be regulated by ERG and which ERG-binding site was protected from DNaseI digestion on binding of DB1255. These data showed for the first time the ERG/DNA complex modulation, both in vitro and in cells, by a heterocyclic diamidine that specifically targets a portion of the ERG DNA recognition site.
INTRODUCTION
Development of therapeutic strategies for inhibiting transcription is of major interest for modulating gene expression associated with various diseases. Transcription factors are key regulators of gene expression, and their deregulation, direct or indirect, is often associated with oncogenesis, cancer development, invasiveness and metastasis. However, in spite of their important cancer generation/progression roles, transcription factors have not been extensively evaluated as targets for cancer treatment strategies (1,2). As transcription factors are considered as “undruggable” targets because of difficulty to directly modulate protein/DNA binding, most drug development strategies act at the protein–protein interaction or protein degradation levels. An example is the treatment of acute promyelocytic leukaemia expressing the fusion protein ProMyelocytic Leukemia–Retinoic Acid Receptor alpha using retinoid acid derivatives that target the DNA binding activity of the RAR moiety (3). Alternatively, other approaches were recently developed to target protein–protein interactions using structurally specific competitive drugs such as nutlin-3 that binds to MDM2 and avoids p53 degradation resulting from p53/MDM2 complex formation in numerous cancers (4). Another approach was developed to target transcription factor activities using compounds that block protein–DNA interactions such as S3I-201 inhibiting Stat3/DNA binding (5), the isoquinolone alkaloid compound berberine interfering with TATA binding protein (6) or synthetic polyamides, specifically designed for transcription factor/DNA modulation through their sequence-selective binding to the minor groove of the DNA helix (7). Such targeted transcription factor/DNA complexes include NF-κB, EVI1 and ETS-1, leading to a decrease in the expression of controlled genes (8–10). Non-specific DNA targeting is a major limitation to the development of transcription factor modulators as illustrated by echinomycin that targets both HIF-1 and Myc/Max transcription factors binding DNA (11). To bypass this drawback, identifying new DNA-binding compounds and evaluating them for DNA-binding selectivity using molecular studies are essential to obtain more effective DNA sequence–specific compounds.
With this aim, we focussed on the synthesis and DNA-binding activities of heterocyclic diamidines for directly targeting the DNA minor groove in a sequence-selective manner. Previous work highlighted the ability of the phenyl-furan-benzimidazole diamidine DB293 to inhibit Pit-1 and Brn-3 transcription factor/DNA complex (12). Because the used TranSignal protein/DNA array also evidenced a much smaller effect on transcription factor interactions to the ETS-binding site (EBS) (12), we then focussed on the modulation of transcription factors that interact with EBS. The minimal EBS core is the consensus 5′-GGA(A/T)-3′ known to be recognized by the ETS family of transcription factors through their highly conserved winged helix-turn-helix DNA-binding domain (ETS-domain) (13,14). The ETS family is divided in 12 subgroups based on structural homologies, among which ERG (ETS-related gene) is of particular interest for its oncogenic function. ERG, together with FLI1 and FEV, belongs to the ERG subgroup (15) on four recently defined subclasses based on their preferred ETS DNA–binding sequences (16). The ETS proteins have regulatory functions in embryonic development and physiological processes including proliferation, apoptosis, vasculogenesis, differentiation and haematopoiesis (17). However, aberrant expression could be associated with cancer diseases. In the case of ERG, fusion of the androgen-regulated gene TMPRSS2 to ERG sequences induces an over-expression of ERG associated with ∼50% of prostate cancers with poor prognosis in >90% of TMPRSS2-ERG-positive prostate cancers (18,19). Other ETS fusion proteins (TMPRSS2-ETV1, TMPRSS2-ETV4, TMPRSS2-FLI1) are also detected in 5–10% of prostate cancers (18,20). Moreover, over-expression of ERG is observed in acute megakaryoblastic, myeloblastic and lymphoblastic leukaemia, associated with poor prognosis and frequent relapses (21–23). Fusion proteins (FUS/TLS-ERG and ELF4-ERG) resulting from translocations have also been associated with those leukaemia, resulting in aberrant expression of ERG transcription factor (24,25). Furthermore, EWS-FLI1 and EWS-ERG fusion proteins are commonly observed in Ewing sarcoma (26,27). Despite their frequent implication in cancer disease, those ETS transcription factors are poorly studied in terms of inhibition and are currently not used in targeted therapy. A few studies investigated the inhibition of EWS-FLI1 in Ewing sarcoma by using, for instance, the mimetic peptide ESAP1 that directly binds EWS-FLI1 and alters its oncogenic function, or YK-4-279 that blocks EWS-FLI1 interaction with the RNA helicase A, leading to the decrease in EWS-FLI1 transcriptional activity. Moreover, YK-4-279 also targets ERG and ETV1 transcription activities reducing motility and invasion of cancer cells without targeting ETS/DNA binding (28–30). Alternatively, the DNA minor groove alkylating agent trabectedin (ET743) inhibits EWS-FLI1/DNA binding and modulates a known EWS-FLI1 downstream target in Ewing sarcoma cell lines (31). Using a high-throughput screening strategy, the same group evidenced similar in vitro and in vivo results with mithramycin (32). Alternatively, another targeting approach was based on the epigenetic modulation of ERG-positive tumors by histone deacetylase inhibitors (33,34).
Our approach was to investigate ERG/DNA binding inhibition using heterocyclic diamidine compounds that target the minor groove of DNA. After selecting DB1255 as the most active compound, its DNA binding selectivity and that of ERG were studied to highlight their respective optimal binding site. The efficiency of DB1255 in ERG/DNA binding inhibition was identified at the molecular and cellular levels. The results of this research provide new directions in anticancer drug design.
MATERIAL AND METHODS
Chemicals and plasmids
All DB compounds (Table 1) were prepared using methodologies previously reported (35,36) and prepared as 5 or 10 mM solutions in DMSO. CT-DNA was purchased from Sigma Aldrich (France). Oligonucleotides EBS-WT (5′-GATCTCGAGCAGGAAGTTCGA), EBS-Em (5′-GATCTCGAGCACCAAGTTCGA), EBS-Dm (5′-GATCTCGAGCAGGAAGTGGGA), EBS-m (5′-GATCTCGAGCAAGAAGTTCGA) and all EBS-mutated oligonucleotides were purchased from Eurogentec (Belgium). The ERG-sensitive reporter plasmid Pye-LUC, containing four copies of the artificial Py-enhancer-element (Pye), has been described in Carrere et al. (37). pSG5, pSG5-ERG, pSG5-FLI1, Pye-Luc, murine Osteopontin (OPN) promoter reporter vectors OPN−136/+77-Luc and OPN−136/+77mutant1-Luc were obtained as previously described (38,39). pSG5-ETS-1 and -ETS-2 were generous gifts from Dr Marc Aumercier (40), whereas pRSV-ERM, PEA-3 and ETV1 were kind gifts from Dr Jean-Luc Baert (41). pGL3-EBS-WT-1R, pGL3-EBS-WT-6R, pGL3-EBS-Em-6R and pGL3-EBS-Dm-6R reporter plasmids were obtained on ligation between NheI and XhoI restriction sites of an oligonucleotide with single EBS-WT or 6-fold tandem repeat of EBS-WT, EBS-Em or EBS-Dm elements, respectively, positioned 5′ upstream to the minimal SV40 promoter.
Table 1.
Structure of the chemical compounds. The general sheme of the compounds is presented on the top of the table. The external A1 and A2 and the central B1 and B2 cycles are as described in the following table
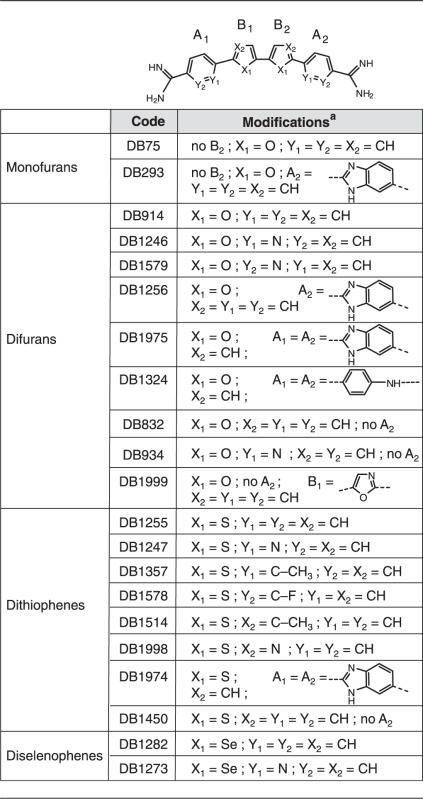 |
aAll compounds are hydrochloride salts except DB1256, DB832, and DB934, which are acetates.
Electrophoretic mobility shift assay
All double-strand oligonucleotides were 5′-radiolabelled as described (12). The binding experiments were performed as previously detailed (12), with some modifications. Briefly, 5 µg of HT29 nuclear extracts (Nuclear extraction kit, Panomics, Fremont, CA, USA) or 2 µl of ERG or other ETS proteins expressing reticulocyte lysates (TNT®-coupled reticulocyte lysate system, Promega, Madison, WI, USA) were incubated for 20 min in binding buffer at 4°C with the radiolabelled DNAs and DB compounds at specified concentrations or 50-fold of the same EBS oligonucleotide (specific) or EBS-m (non-specific) as unlabelled competitors. Free DNA was separated from protein–DNA complexes on a 6% non-denaturating polyacrylamide gel under electrophoresis for 3 h 30 min at 300 V in 0.5× TBE buffer. Gel revelations were performed using a Molecular Dynamics STORM 860 and analysed with the ImageQuant-3.3 software.
DNase I footprint assay
pGL3-EBS-WT-1R, pGL3-EBS-WT-6R, pGL3-EBS-Em-6R and pGL3-EBS-Dm-6R plasmids were digested by ClaI and BglII restriction enzymes and radiolabelled with [α-32P]dGTP at 3′-termini as reported (42). The OPN promoter DNA sequence was obtained by HindIII and MluI restriction enzymes digestion of the OPN−136/+77-Luc vector. The generated DNA fragment was 3′-end radiolabelled using [α-32P]dCTP as specified previously. DNA sequences were deduced from guanine positions cleaved on DMS and piperidine treatment (G-track).
ELISA-derived protein/DNA binding inhibition assay
Streptavidin-coated 96-well plates (ThermoFicher) were blocked with 200 µl of blocking solution Tris Buffer Saline (TBS: 10 mM Tris–HCl, pH 8, 150 mM NaCl, 0.5% Tween and 5% BSA) for 1 h at room temperature and 5.7 nM of biotinylated EBS-containing oligonucleotide was immobilized per well for 1 h at room temperature. After two washes for 5 min with 200 µl of washing solution (TBS-Tween 0.5%) at room temperature, ERG proteins expressed from reticulocyte lysate system (Promega, Madison, WI, USA) (2 µl of lysate previously diluted to one-fifth in sterile water) were incubated for 30 min at 4°C with/without 5 µM of the indicated DB compounds in binding buffer (10 mM Tris–HCl, pH 7.5, 1 mM EDTA, 1 mM DTT, 75 mM NaCl, 6% glycerol, 10 µg BSA). After three washes, the ERG/DNA complex was incubated with ERG antibody (sc-354, Santa Cruz Biotechnology) in TBS-Tween 0.5%–0.5% BSA solution 1 h at room temperature. After three washes, ERG/DNA complexes were detected by the incubation of the rabbit horseradish peroxidase-conjugated secondary antibody (Santa Cruz Biotechnology) in TBS-Tween 0.5%–BSA 0.5% solution for 1 h at room temperature. After two washes of 5 min with 200 µl of washing solution and one with TBS alone, 100 µl of TMB (Promega, Madison, WI, USA) was added and incubated for 15 min at room temperature. The reaction was stopped with 100 µl of 0.5 M H2SO4, and the absorbance was measured at 450 nm on the microplate spectrometer (Molecular Devices Versamax). All collected data was analysed using SoftMax Pro software.
Spectrometric analysis of DNA binding
DNA melting temperature, UV/Visible spectroscopy and circular dichroism experiments were performed as previously reported (43), with the following modifications. For melting temperature studies (ΔTm), the indicated ratios are expressed as drug/base pair. For UV spectroscopy, DB1255 (20 µM) was incubated with increasing concentrations of CT-DNA (0.1–200 µM) or EBS-WT oligonucleotide (0.1–50 µM). For circular dichroism spectrometry, 200 µM (bp) of CT-DNA or EBS-WT oligonucleotide was incubated with increasing concentrations of DB1255 from 1 to 50 µM and 0.01 to 10 µM, respectively. All three experiments were performed in BPE buffer (6 mM Na2HPO4, 2 mM Na2H2PO4, 1 mM EDTA, pH 7).
Surface plasmon resonance analysis of the DNA binding
Surface plasmon resonance (SPR) measurements were performed with a four-channel Biacore T200 optical biosensor system (Biacore, GE Healthcare, Inc.). 5′-biotin labelled hairpin oligonucleotides EBS-WT 5′-GCGTCGAACTTCCTGCTtctcAGCAGGAAGTTCGACGC-3′ (the EBS-minimal sequence is underlined, loop in lower case and stem in caps letters) or EBS-Dm 5′-GCGTCCCACTTCCTGCTtctcAGCAGGAAGTGGGACGC-3′ (mutated bases are in bold and italic letters) were immobilized onto streptavidin-coated sensor chips (Biacore SA) as previously described (44,45), with the difference that the compounds were injected in a solution containing 10% DMSO to avoid drug precipitation in the flow cells at the highest drug concentrations.
Cell culture
Human cervical HeLa, colon HT29 and prostatic VCaP carcinoma cells were grown at 37°C under 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM, Gibco) supplemented with 10% foetal bovine serum, penicillin (1000 U/ml) and streptomycin (10 μg/ml).
Transient transfections
Using HT29 cell line, 15 × 104 cells/well were cultured overnight in 1 ml of supplemented medium in 12-well plate. Transfection was performed by adding a mixture of 3 µl of lipofectamine 2000 (Invitrogen), 1.5 µg of reporter plasmid (pGL3-EBS-WT-6R, pGL3-EBS-Em-6R, pGL3-EBS-Dm-6R or pGL3), 0.5 µg of pmaxGFP as a normalising plasmid qsp 100 µl of OptiMEM medium (Gibco, Invitrogen) previously incubated for 20 min at room temperature. After 6 h, the medium was changed by complete medium supplemented, or not, with DB1255 at the indicated concentrations, and cells were cultured for 24 h at 37°C. The transfected cells were collected, rinsed with PBS before lysis for 20 min at 4°C by lysis passive buffer (Promega, Madison, Wisconsin, USA). After a 15-min centrifugation at 13 000g, supernatants were collected. Luciferase activity was quantified using Luciferase assay system (Promega, Madison, Wisconsin, USA) according to manual recommendations, whereas the fluorescence of GFP protein was detected using a 96-well fluorimeter (Mithras, Berthold) at λexcitation = 485 nm and λemission = 535 nm. For exogenous ERG expression in HeLa cells, 2 × 105 cells/well were plated in 12-well plates 5 h before transfection using polyethylenimine (Eurogentec) according to the manufacturer’s instructions. DNA mixture included 500 ng of the indicated firefly luciferase reporter gene with or without 200 ng of the expression vectors pSG5-ERG and 5 ng of a control plasmid tk-luciferase (tk-Renilla, Promega). After 6 h, cells were challenged with 2.5 µM of DB1255 for 20 h. Cells were harvested and luciferase activities were detected using the Dual-Glo Luciferase Assay System (Promega). Luciferase activity was measured on a luminometer (Berthold Biolumat centro LB960). Firefly luciferase values were normalized to those of the control renilla luciferase.
Western blotting
Transfected cells were washed once with PBS and incubated in ice-cold lysis buffer (50 mM Tris–HCl, 10 mM EDTA, 1% SDS, pH 8) for 2 h at 4°C. After a 20-min centrifugation at 4°C and 13 000g, the protein extracts were collected, quantified (Bradford) and equal amounts of proteins was denatured in loading buffer (100 mM Tris–HCl, pH 6.8, 200 mM DTT, 4% SDS, 0.2% bromophenol blue and 20% glycerol) 3 min at 90°C and loaded on SDS-PAGE. After gel electrophoresis, the proteins were transferred onto a nitrocellulose membrane (GE Healthcare, France), which was blocked for 1 h at room temperature with TBS blocking solution (Tris–HCl 10 mM pH 8, NaCl 150 mM, Tween 0.5%) containing 5% of BSA before immunodetection using polyclonal anti-ERG antibody (sc-353, Santa Cruz, Biotechnology) and revelation by ECL detection system (GE Healthcare) using an HRP-conjugated secondary antibody (Santa Cruz, Biotechnology). Anti-β-actin monoclonal antibody (Sigma Aldrich) was used as an internal control.
RESULTS
Screening heterocyclic diamidines for inhibition of ERG/DNA binding
Because previous researches highlighted some inhibitory effects of DB293 on the ability of proteins to complex with EBS sequence present on TranSignal protein/DNA array, we first investigated a series of transcription factors belonging to the ETS family (ETS-1, ETS-2, ERG, FLI1, ERM, PEA-3 and ETV1) for binding to this EBS-containing sequence. Only ERG, and to a much lesser extent FLI1, shows impressive band shift (data not shown) and was therefore selected for inhibition with the lead compound, DB293. The band shift was inhibited in a similar dose–response manner using both ERG-expressing reticulocyte lysate and HT29 nuclear extracts with full inhibition using 25 µM of DB293 (Supplementary Figure S1A). In a DNaseI footprinting assay (Supplementary Figure S1B), binding of DB293 at a site (grey box) located on the 3′-side of the minimal EBS (underlined in black) was observed, as exemplified by a strong negative differential cleavage from 1.25 µM. The minimal EBS site was not sufficient for ERG/DNA binding, which is known to require specific 5′ and 3′ adjacent bases and the 3′ binding site of DB293 is in agreement with interference of ERG/DNA binding.
We then challenged a new series of heterocyclic diamidines of varied structure for ERG/DNA binding inhibition using an ELISA-derived Protein/DNA binding inhibition assay (which we call here EPDBi). This method can screen and quantify in one step using 96-well plates the inhibitory effect of a large series of compounds for protein/DNA complex formation. A new structural sub-type of diamidine compounds (Table 1) gave promising inhibitory results (Figure 1). Structure/activity analyses showed that di-thiophene derivatives were more active than their di-furan or di-selenophene counterparts. Among di-thiophenes, the di-thiophene-phenyl-amidine DB1255 was a particularly efficient inhibitor with more than 90% of complex inhibited using 5 µM of DB1255. At the same concentration, DB293 only induced a 20% inhibition. Such relative inhibition efficiency was confirmed using EMSA (Figure 2). Results for ERG/DNA binding inhibition using EMSA were consistent with those resulting from EDPBi assay where the more efficient competitors present two thiophenes as central rings rather than furans or selenophenes. In the di-thiophene series, the substitution of the di-phenyl by di-pyridine (DB1247) or di-benzimidazole (DB1974) heterocycles decreases the inhibition efficiency by only 40–60%. In conclusion, the DB1255 is selected as the best inhibitor of the ERG/DNA binding. The protein/DNA complex obtained with HT-29 nuclear extracts was also inhibited on addition of increasing concentrations of DB1255 (Figure S2).
Figure 1.

EPDBi for screening the ERG/EBS-WT binding inhibitors. EPDBi was performed on streptavidin-coated plates in which biotinylated EBS-WT oligonucleotides were fixed. ERG proteins were added together with the indicated DB compounds at 5 µM. Control corresponds to 100% of ERG/DNA complex. EBS-WT DNA-binding specificity was validated by addition of 50-fold excess of EBS-WT (50× S) or non-specific oligonucleotide (50× NS) in the binding buffer. Results are means ± s.e.m. from two experiments both performed in triplicate.
Figure 2.

Validation of the ERG/EBS complex inhibition by the evaluated diamidine compounds. EMSA was performed with the EBS-WT radiolabelled oligonucleotide incubated with ERG protein expressed in reticulocyte lysate systems or empty lysate made with the equivalent empty vector, in the presence of thiophene (A), selenophene or furan (B–D) derivatives. Bound ERG/EBS-WT complexes (b, indicated by a black arrow) were competed with increasing concentration of DB compounds (µM) and separated to free EBS-WT oligonucleotide (f, indicated by black arrow) by electrophoresis migration. DNA binding specificity was determined by addition of 50-fold excess of the specific unlabelled EBS-WT (50× S) or a non-specific unlabelled oligonucleotide EBSm (50× NS).
DNA-binding properties of DB1255 and derivatives
DB1255 was selected for DNA binding studies using UV–visible spectrometry, DNA melting temperature studies and circular dichroism analysis of the mode of binding. First, interaction of DB1255 with DNA was analysed using spectroscopic analyses in the presence of EBS oligonucleotide (EBS-WT) or CT-DNA (Figure 3 and Supplementary Figure S3). The variation of the DNA melting temperature indicates strong binding of DB1255 to the EBS-containing oligonucleotide, even at low drug/DNA ratio (Figure 3A). UV/visible absorbance spectroscopy evidenced hypochromic effects at low concentration (0–4 µM) of EBS-WT oligonucleotide (high drug/DNA ratios) that is probably due to stacking of the compound outside of the double helix. The spectra then evidenced bathochromic and hyperchromic effects from 4 to 50 µM with an isosbestic point at 460 nm that argues for a stronger, single binding mode at the lower drug/DNA ratios (Figure 3B). Circular dichroism spectrometry was then performed to get an insight in the orientation of the compound relatively to the DNA helix (groove binding or intercalation) on addition of increasing concentrations of DB1255. In the presence of DB1255 (Figure 3C), the biphasic intrinsic CD signal is modified with the appearance of a strong bisignate CD around 420 nm that argues for a groove binding interaction of DB1255 and is consistent with dimer binding to the EBS-containing oligonucleotide (46). Similar conclusions could be obtained on binding to CT-DNA (Figure S3).
Figure 3.
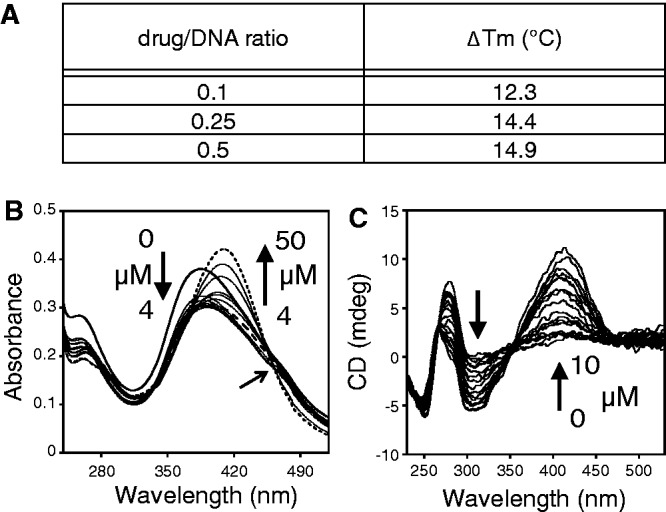
DNA-binding characteristics of the selected compound DB1255. (A) Temperature melting assay of the EBS-WT oligonucleotide in presence of the DB1255. ΔTm values were obtained from the equation: ΔTm = (Tm(drug/DNA) – Tm(DNA)). (B) UV-spectroscopy properties of the complex DB1255/DNA was observed using a fixed concentration of DB1255 and increasing concentrations of EBS-WT oligonucleotide. (C) Circular dichroïsm properties of EBS-WT oligonucleotide in presence of increasing concentrations of DB1255. Major changes in spectra (B and C) were indicated by black arrows.
The precise binding localization of DB1255 or derivatives relative to the EBS site was determined using DNase I footprinting assays as described previously for the DB293 (Figure 4). For this purpose, we used the ClaI-BglII DNA fragment containing the 6-fold repeated EBS site from pGL3-EBS-WT-6R reporter vector. The radiolabelled DNA was incubated with increasing concentrations of the various selected compounds before DNase I mild digestion. The densitometric analyses were performed for each gel. A representative gel is presented in Figure 4 using the active compounds DB1255 and DB1998 for comparison with the inactive DB1974 or DB1975. The portion of the gel that is covered by DB1255 and DB1998 encompasses the 5′-gAAGTTc site. The other compounds that appeared to be less active from EPDBi (Figure 1) were less selective or bound with lower affinity to the same portion of the DNA (Supplementary Figure S4). Such binding only partially covers (underlined) the EBS minimal binding site 5′-GGAA, but also spans the important EBS binding sequence, 5′-GTT, located 3′ after it. In conclusion, active compounds determined by EPDBi showed an excellent DNA-binding recognition and affinity for the EBS binding site.
Figure 4.

Sequence selectivity of diphenyl di-thiophene diamidine compounds. (A) DNase I footprint assays showed the interaction sequences of the DB compounds. The radiolabelled DNA fragment containing the six repeats of the EBS-WT sequence was incubated with the indicated concentrations of compounds (µM) and submitting to electrophoresis migration. The track labelled “G” is as in Supplementary Figure S1. (B) Densitometric analysis derived from the gel and protected sites observed are indicated by black boxes.
Looking for DB1255 and ERG optimal binding sites
To get an insight in the precise bases that are crucial for the DNA interaction, DNA binding capacity of the selected compound DB1255 was evaluated in terms of sequence selectivity using a DNase I footprint assay on DNA fragments containing the wild-type EBS sequence or EBS mutated at one position per experiment to get information with all four nucleotides options at each position. All nucleotides from positions −1 to +7 relative to the minimal EBS site G−1G0A+1A+2 were mutated and evaluated for DB1255 binding selectivity (Figure 5A and B). Radiolabelled DNA contained the three mutated EBS surrounded with two wild-type EBS. At the external positions −1, 0 and +7, no major changes in the differential cleavages from densitometric analyses were observed whatever the nature of the nucleotide at those positions suggesting that DB1255 has no selective binding to those bases (Figure S5). By contrast, mutations in central positions from +1 to +5 affected (decreased or abolished) DB1255 binding. Globally, DNase I footprinting data demonstrate the importance of the A+1, A+2, T+4 and T+5 bases for DB1255 selective binding to DNA. Mutations at position +3 and +6, however, affected the compound binding to a lesser extent (Figure S5). Values from the densitometric analyses were used to deduce the predicted logo of the frequencies of DNA recognition with 0.8 µM of DB1255 using each single base pair mutation (Figure 5C).
Figure 5.

Sequence selectivity of ERG and DB1255 DNA binding. (A) Sequence selectivity of the DB1255/DNA interaction. DNase I footprint assays were performed with a DNA fragment containing three EBS sequences mutated at the indicated positions (positions −1 to +7, DNase I footprint gels and densitometric analyses for the position −1 and 0 and position +3 to +7 are presented in Figure S5A). For each mutation incubation with DB1255 at the indicated concentrations (µM) revealed protected sites of DB1255 interaction. The track labelled “G” is as in Figure S1. (B) Densitometric analyses derived from the gels and EBS-WT (WT) sequence or EBS-mutated sequence are indicated by black boxes or a grey boxes, respectively. (C) The logos were drawn using enoLOGOS (47), where the size of the letter is directly proportional to the binding affinity at 0.8 µM of the compound resulting for densitometric analysis (data shown in Figure S5B). (D) Sequence selectivity of the ERG binding to the EBS-WT sequence. ERG binding sequence selectivity are observed by EMSA using radiolabelled EBS oligonucleotide WT (EBS WT) or mutated (EBS mXN) at the indicated position (position X of base N) incubated with ERG protein expressed in reticulocyte lysate system or empty lysate. EBS sequences with mutated positions from −5 to +7 were evaluated but only the EMSA with mutated positions +3 to +7 are shown here (EMSAs of mutated position −5 to +2 are shown in Figure S6A). (E) Representation of the percentage of ERG/DNA complex derived from the gel quantification as means ± s.e.m. relatively to EBS-WT. (F) The logos were drawn as above (full data presented in Figure S6B).
In parallel, the DNA binding selectivity of the ERG transcription factor was evaluated in EMSA using 5′-end labelled oligonucleotides containing the EBS-WT sequence or mutants at each single position from −5 to +7 as indicated in Figure 5D and Supplementary Figure S6. Globally, the mutations affecting the EBS minimal site abolish the ERG binding as expected. In addition, the same mutations at positions −2 (A→C) or −4 (G→A) and to a lesser extent +5 (T→G) increases ERG/DNA binding intensity. For the surrounding positions −3, +3 and +4 the relative percentage of ERG/DNA binding decreased relatively to EBS-WT suggesting their relevance for the ERG/DNA binding. By contrast, changing bases at the most external positions −5, +6 and +7 did not significantly modify ERG/DNA binding propensity. From those data, the predicted logo for ERG/DNA binding selectivity is presented in Figure 5F. For each mutated sequence that could form ERG/DNA complexes (mutations 3A, all mutated points at positions +5 to +7), we evaluated the propensity of DB1255 at a fixed concentration (1 µM) to inhibit ERG/DNA binding. A strong decrease of the inhibition potency is observed as expected for all three mutations (A, C and G) at +5 position (Figure 6A). This result is in agreement with the consensus DNA binding site for DB1255 showing that the presence of a T+5 is essential for the binding (Figure 5) as well as for the ERG/DNA complex inhibition (Figure 6A). In conclusion, comparison of DB1255 and ERG consensus binding sites exemplifies the necessity of the presence of a common 5′-AA(G/N)T-3′ site that encompasses the minimal EBS for optimal DB1255 interaction and for best ERG/DNA complex inhibition.
Figure 6.

Specificity of ERG/DNA and DB/DNA binding on mutated on EBS sequences or on DB binding sequences. (A) ERG/DNA binding inhibition depending on mutated EBS sequences. The capacity of a fixed concentration of DB1255 (1 µM) to inhibit ERG/DNA binding was evaluated by EMSAs on mutated EBS sequences when a binding was possible. Binding complexes were quantified and expressed as following. ERG/DNA binding inhibition by DB1255 with mutated EBS were measured as a percentage of the inhibition obtained with EBS-WT. ERG/DNA binding inhibition by DB1255 obtained with EBS-WT sequence was scaled up to 1 and the percentage of inhibition with mutated EBS relative to EBS-WT were plotted as fold-reduction of 1255 inhibition capacity. (B) Densitometric analysis of DB/DNA interaction derived from a DNase I footprint (gels not shown). DNase I footprint assays were performed with DNA fragments containing six repeats of EBS-WT, EBS-Em (mutated on the EBS element) or EBS-Dm (mutated on the compound site interaction) and compounds at the indicated concentrations. (C) SPR Sensorgrams for DB1255 on the EBS-WT (left panel) or EBS-Dm (right panel) hairpin oligonucleotides. (D) Fitting analysis of SPR sensorgrams. RU values from the steady-state region of SPR sensorgrams are plotted against free DB1255 concentration (M). The corresponding K1 and K2 values are calculated from one or two site(s) fitting models for EBS-Dm (Red) and EBS-WT (Blue) sequences. (E) ERG/EBS-WT and ERG/EBS-Dm binding modulation by DB1255. Quantification of the percentage of ERG/DNA complex formation was performed from EMSA gels (not shown).
To evaluate the inhibition of ERG/DNA binding in cells using luciferase assays, we selected from previous analyses the ERG/DNA binding mutated sequences (EBS-Em) and the DB1255/DNA binding mutated sites (EBS-Dm) as being 5′-GGAA → 5′CCAA and 5′-GGAAGTTCGA → 5′-GGAAGTGGGA, respectively (mutated bases are underlined). The efficiency for binding of DB1255 (Figure 6B) and other derivatives (data not shown) to EBS-Em but not EBS-Dm sites was confirmed from comparison with the EBS-WT sites using DNaseI footprinting assays on 6-fold repeated sequences generated from the pGL3-EBS-6R, pGL3-EBS-Em-6R and pGL3-EBS-Dm-6R reporter vectors as described in the Materials and Methods section. As expected, inactive compounds such as DB1974 and DB1975 did not present any footprint on the three DNA fragments (data not shown), whereas the active compounds DB1255 and DB1998, and to a lesser extent DB1357 and DB1578 (Figure 6B and data not shown), displayed huge footprints characterized by negative differential cleavages for EBS-WT and EBS-Em but not for EBS-Dm sequences. To get more quantitative analysis of the sequence binding and selectivity of DB1255 on EBS-WT, SPR experiments were conducted with DNA hairpin duplexes containing EBS-WT or EBS-Dm sequences (Figure 6C). Because of the poor solubility of DB1255, the experiments were performed in a buffer containing 10% DMSO during the whole experimental process. The impact of DMSO in this context was evaluated using the DB1998 derivative, which is more water soluble. The equilibrium and kinetic constants obtained in the presence of 10% DMSO (Figure S7) and in low concentrations of DMSO (data not shown) were roughly the same and any correction is less than experimental error. The sensorgrams for DB1998 (Figure S7A) showed fast kinetics of association and dissociation, whereas that for DB1255 presented slower association/dissociation rates, suggesting better binding kinetics for DB1255 over DB1998. The steady state RU values were plotted against Cf (free compound concentration, in M) and fitted to a single or two sites model (Figures 6D and S7B) and showed that both DB1255 and DB1998 present similar equilibrium constants for EBS-WT but different ones for EBS-Dm sequence. Both compounds have a single strong binding site for the WT sequence (K1), with a second site that is 100–200 times weaker (K2). The binding of DB1255 to the EBS-Dm sequence is much weaker than that to EBS-WT, suggesting that such mutations strongly affect the DNA binding propensity of DB1255 as also observed earlier using DNase I footprinting assays (Figure 6B). By contrast, DB1998 binds strongly to EBS-Dm sequence, and K1 is only reduced by a factor of 5 compared with EBS-WT, suggesting lower binding selectivity (Figure S7B). Therefore DB1255 appears to be the most interesting compounds in this series and was therefore retained for further cellular experiments. As part of our sequence validation, we finally showed that the ERG protein binds to EBS-WT and EBS-Dm, but not EBS-Em oligonucleotide (data not shown), which confirms the above data showing that the two guanines at positions −1 and 0 are essential for the ERG binding but not the bases at positions +5 and +6. In competition studies, ERG binding to DNA is abolished by increasing concentrations of DB1255 for the EBS-WT sequence but not for the EBS-Dm sequences (Figure 6E). Thus, those three sequences could be used for luciferase assays.
Modulation of the ERG/EBS complex inhibition in cellulo
This experiment was performed in HT29 cell line, which expresses the ERG transcription factor as visualized by a Western blot (Supplementary Figure S8A). The cell uptake and nuclear localization of DB1255 was evidenced using fluorescence microscopy based on DB1255 intrinsic fluorescence properties in comparison with the cytoplasmic localization of MitoFluor Red (a mitochondrial tracer) (Supplementary Figure S8B). Such nuclear localization is in agreement with genomic DNA as a target for DB1255. Therefore, the luciferase assays were performed in cells transfected with pGL3-EBS-WT-6R, pGL3-EBS-Em-6R and pGL3-EBS-Dm-6R for 6 h before the addition of DB1255 from 1 to 5 µM during 24 h, conditions for which DB1255 is not cytotoxic (Figure S8C). After normalization relatively to GFP expression and empty vectors, we evidenced relative ERG-dependent transactivation using EBS-WT and EBS-Dm but not EBS-Em element, highlighting the crucial role of the GG bases of the EBS site that are mutated in EBS-Em sequences (Figure 7A). In the presence of increasing concentrations of DB1255, the ERG-mediated transactivation at EBS-WT sites decreased in a dose-dependent manner, consistent with the inhibition of ERG/DNA binding observed in vitro in EMSA (Figure 6E). No inhibitory effect is obtained using EBS-Dm-containing reporter vector in agreement with previous EMSA analyses (Figure 6E).
Figure 7.
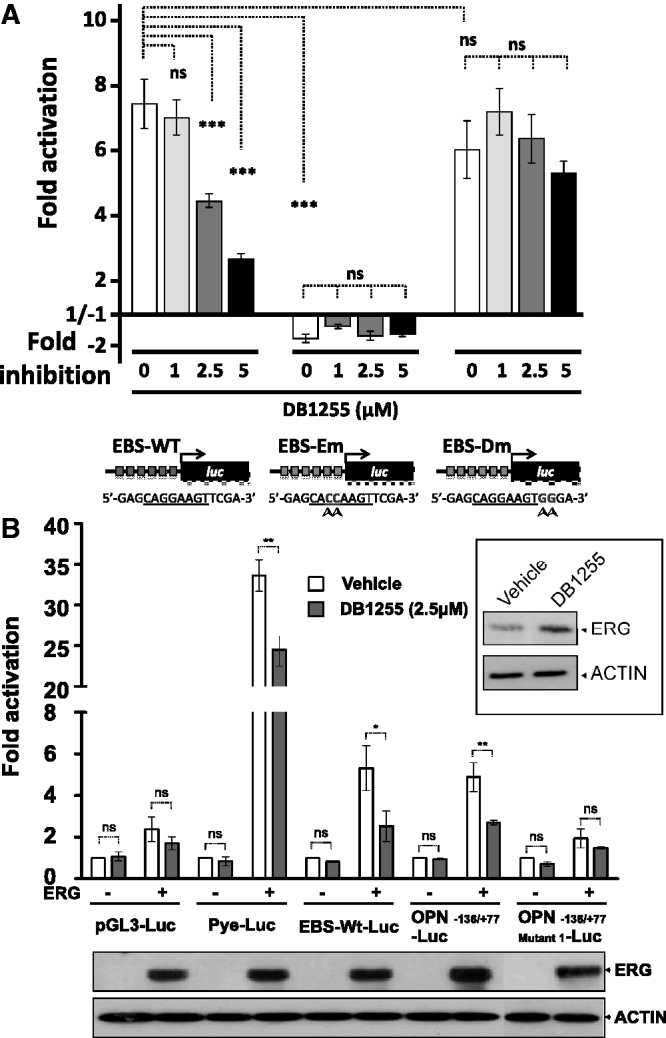
In cellulo inhibition of the ERG/DNA complex by DB1255. (A) Modulation of the ERG/DNA transcriptional activity on a 6-fold repeated EBS-containing reporter vector. HT29 cells transfected with luciferase vector with or without 6 repeat of the EBS-WT, EBS-Em or EBS-Dm elements (point mutations indicated with arrow heads) located upstream the cDNA of the luciferase (luc) gene, as indicated, were incubated with increasing concentrations of DB1255 (24 h). The fold-activation values correspond to the normalized luciferase activity in comparison with cells transfected with empty vector. Results are means ± s.e.m. from three independent experiments performed in triplicate. Statistical analyses were performed using analysis of variance test. (B) In cellulo inhibition of the transcriptional activity of ERG-target gene promoter by the DB1255 compound on the OPN promoter. Transfections were performed in HeLa cells using ERG-expressing vector or empty control vector. For transcriptional modulation, different reporter vectors were used: pGL3-Luc as empty reporter, Pye-Luc as the reference of polyomavirus enhancer, EBS-WT-Luc similar to pGL3-EBS-WT-6R, OPN−136/+77-Luc as the OPN promoter and OPN−136/+77 Mutant1-Luc containing the OPN promoter mutated on EBS. After transfection, HeLa cells were treated or not with DB1255 (2.5 µM, 24 h). ERG and β-actin expressions were detected by Western blotting (bottom and embedded panels). Results are means ± s.e.m. from at least two experiments. Statistical significance was determined using one-way analysis of variance, followed by Bonferroni’s multiple comparison post test. *P < 0.05, **P < 0.01, ***P < 0.001, ns, non-specific (P > 0.05).
To confirm this modulation we finally evaluated the competitive activity of DB1255 on promoters that have been described to be regulated by ERG protein such as the OPN promoter (39). The DNA recognition of DB1255 at the EBS site was addressed using DNaseI footprinting assays (Figure S9). The luciferase assay was performed in HeLa cells as described previously (39). Because HeLa cells do not express ERG protein, those cells were co-transfected with the ERG-expressing vector or empty vector as control (Figure 7B, bottom panel). The pGL3-EBS-WT-6R (EBS-WT-Luc) previously evaluated in HT-29 cells and the reference polyomavirus enhancer (Pye) were first evaluated from comparison with the control to validate the model in HeLa cell line. An inhibition of the ERG-mediated transactivation by DB1255 was obtained for both EBS-WT and Pye models. Such inhibition could not be attributed to a decrease of the transcription of ERG-expressing vector itself as visualized in the embedded Western blot panel from comparison with actin expression. Moreover, DB1255 also inhibits ERG-mediated transactivation from the OPN promoter (OPN−136/+77-Luc) in a manner that is dependent on the presence of the ERG-binding site because no significant effect is obtained using the sequence mutated at the EBS at position −119 to −115 (5′-GGAGGAAG mutated to 5′-GGTAAAAG in OPN−136/+77 Mutant1-Luc) (Figure 7B). Both luciferase assays bring out the inhibitory effect of DB1255 on ERG/DNA binding in cells.
DISCUSSION
Transcriptional modulation of oncogenic transcription factors is a new and promising area to develop targeted therapies, particularly in cancer. Despite the difficulty to target this class of proteins, many groups have investigated their inhibition by using several approaches. One method under development consists of targeting the protein–DNA binding interaction using small DNA-binding compounds. Such compounds specifically interact with the same DNA site and consequently decrease the transcription factor/DNA complexes leading to the diminution of gene transcription. By using this competition approach of transcription factor inhibition, we first showed the possibility of this inhibition concept using the heterocyclic diamidine DB293, which targets Pit-1 and Brn-3 (12). In that project, we showed that if compound binding to a sequence (ATGA) common to that bound by a transcription factor was important, it does not mean that all transcription factors containing that sequence would be affected. Indeed, the IRF-1 transcription factor consensus interaction site also contains an ATGA sequence, but that complex was not affected by the presence of DB293. This result shows that the mode of binding of each transcription factor needs to be taken into account in inhibition design. Based on previous data using TranSignal Protein/DNA arrays (12), we focussed here on the ERG transcription factor and showed that the inhibition of ERG binding to the DNA is obtained through the recognition of a sequence that partially spans the minimal ERG-binding site. It is known that the binding of the ETS-domain to DNA requires direct recognition of conserved arginine residues with the core 5′-GGA(A/T)-3′ in the major groove as well as contacts with the phosphate backbone within the minor groove at bases flanking the minimal 5′-GGA(A/T)-3′ core (48). Thus, targeting one portion of the EBS consensus may be sufficient to impede ERG/DNA binding. This is what we observed here using DB1255 and related derivatives. In terms of structure/activity relationships, changing di-thiophene rings in DB1255 for di-furans or di-selenophenes abolishes sequence-selectivity and the ERG/DNA binding inhibitory effect.
Among the di-thiophene series in which the di-phenyl rings of DB1255 were changed for di-benzimidazole or in which di-methyl-thiophene or di-thiazole substituted the di-thiophen central rings, DB1255 remains the most active. Such modifications may increase the stearic hindrance relative to DB1255, thus leading to the loss of DNA recognition. Sulphur atoms are less electronegative than oxygen atoms and develop weaker hydrogen bonding but present better stacking properties than furan groups. However, the size of the sulphur atoms is larger than that of oxygen (but smaller than selenium atoms) and this can increase the C-S-C bond angle of the thiophene rings of DB1255 in comparison with the C-O-C angle in furan rings (DB914). The selenium atoms may induce a much larger angle that is no longer compatible with proper DNA binding (DB1282). The optimal recognition seems to require a balance between thiophene properties and the curvature of this type of molecule for matching the minor groove of the double helix of DNA. The different capacity of inhibition was confirmed by an EMSA assay (Figure 2) where the more active compounds detected in the EDPBi assay decrease the ERG/DNA complex in contrast with the inactive compounds. As observed for DB293, the inhibition potency of active compounds strictly correlates with their ability to partially cover the EBS minimal 5′GGA(A/T) and to span the 5′-GTT sequence located 3′ to the minimal EBS sequence (Figure 4). The active di-thiophene heterocyclic diamidine compounds inhibit ERG/DNA complex formation through DNA interaction on one portion of the EBS, leading to the destabilization of ERG/DNA binding. As DB1255 is clearly the best ERG/DNA complex inhibitor, we focused on that compound for fine analysis of sequence and cellular validation. As usual for this class of compound, DB1255 is a (minor) groove DNA binder as evidenced using both non-specific CT-DNA and a sequence-specific EBS-containing oligonucleotide (Figures 3 and Supplementary Figure S3). The strong bisignate CD spectra observed for this compound suggest a groove interaction as a dimer similar to DB293 (46,49). The SPR results suggest that the difference in inhibition effects of DB1255 relative to other compounds such as DB1998 is due to several factors. DB1255 is more specific than DB1998 and should therefore have more DB1255 molecules focused at specific sites and not lost in non-specific binding. DB1255 also dissociates from the specific site more slowly than DB1998 and this may help in modulation of protein–DNA complexes and function.
About the sequence-selective DNA recognition by DB1255, we brought out the importance of the nucleotides 5′-(g)TT following the core 5′-GGAA that showed an AT-rich sequence interaction separated by a G base as observed for other compounds (49). In parallel, studies on ERG selective binding showed, as expected, the necessity of the core 5′-GGAA-3′ for optimal ERG/DNA complex formation. These results also showed the importance of adjacent bases for binding, particularly the 5′-GT dinucleotide following the core 5′-GGAA (DNaseI footprinting and SPR, Figure 6).
Interestingly, such consensus binding site strictly correlates with that deduced earlier by Wei et al. (16) from ChIP-sequence analyses. This reinforces the therapeutic interest in our compound with the objective to target in vivo the strong and selective ERG/DNA binding sites as defined using ChIP-sequencing. At the cellular level, the transient transfection assays confirmed the gradual decrease by DB1255 of ERG/DNA binding in the cellular model using both a synthetic promoter model and the OPN gene promoter (Figure 7).
As a transcriptional factor, ERG interacts with the DNA to positively or negatively regulate target gene expression depending on the cell context. Thus, our selected compound may positively or negatively interfere with the transcription of ERG-controlled genes, but in a way that may induce a change in the phenotype of ERG-dependent cancers. However, DB1255 interacts with DNA on one portion of the defined ERG-binding site, suggesting that it may not modulate all ERG-regulated genes but a portion of those that include the proper DB1255 binding site as defined here (Figure 5C). Thus, further experiments will be required to characterize the implication of the ERG/DNA binding, modulated by the selected compound regarding changes in gene expression. Genome-wide analyses (ChIP-seq and transcriptome analysis) in physiological conditions, disease conditions and in presence of the DNA ligand would be of major interest and could be compared with cells invalidated for ERG expression using RNA interference. One part of the question is provided by recent data of the invalidation of ERG expression in the TMPRSS2-ERG positive VCaP prostate cancer cell line evidencing the upregulation of genes associated with differentiated luminal prostate epithelial cells (50). In HUVEC endothelial cells, similar transcriptome profiling evidenced the cytosolic histone-deacetylase-6 gene (HDAC6) as a direct ERG transcriptional target implicated in cell migration (51). The vascular endothelial adhesion molecule VE-cadherin was also described as a target for ERG, associated with angiogenesis (52). Those genes and approaches would be of major interest to validate the effect of ERG/DNA binding inhibitory compounds such as DB1255.
In conclusion, we selected a di-(phenyl-thiophene-amidine) compound as a DNA-binding modulator that specifically targets the ERG/DNA complex. To our knowledge, this is the first in cellulo modulation of the ERG/DNA binding by a small molecule targeting the protein/DNA interaction. As a strong inhibitor, DB1255 thus offers a therapeutic window for targeting ERG oncogenecity both in leukaemia disease and in prostate cancer.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Figures 1–9.
FUNDING
The Fonds Européen de Développement Régional (FEDER, European Community) together with the Région Nord/Pas-de-Calais (to M.-H.D.-C. and M.D.-C.); the Ligue Nationale Contre le Cancer (Comité du Pas-de-Calais, Septentrion), the Association Laurette Fugain and the Association pour la Recherche sur le Cancer (to M.-H.D.-C.); NIH NIAID [064200 to W.D.W. and D.W.B.]; the CHRU de Lille and the Région Nord/Pas-de-Calais for a PhD fellowship (to R.N.); the Institut National du Cancer (INCa) for post-doctoral fellowships (to X.D. and S.F.); the Institut pour la Recherche sur le Cancer de Lille (IRCL) for technical expertise (to S.D.). Funding for open access charge: IRCL.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We thank the CHRU de Lille and the Région Nord/Pas-de-Calais for a PhD fellowship (to R.N.); the Institut National du Cancer (INCa) for post-doctoral fellowships (to X.D. and S.F.), and the Institut pour la Recherche sur le Cancer de Lille (IRCL) for technical expertise (to S.D.). The IMPRT-IFR114 is acknowledged for giving access to the Storm 860 equipment.
REFERENCES
- 1.Darnell JE., Jr Transcription factors as targets for cancer therapy. Nat. Rev. Cancer. 2002;2:740–749. doi: 10.1038/nrc906. [DOI] [PubMed] [Google Scholar]
- 2.Koehler AN. A complex task? Direct modulation of transcription factors with small molecules. Curr. Opin. Chem. Biol. 2010;14:331–340. doi: 10.1016/j.cbpa.2010.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huang ME, Ye YC, Chen SR, Chai JR, Lu JX, Zhoa L, Gu LG, Wang ZY. Use of all-trans retinoic acid in the treatment of acute promyelocitic leukaemia. Blood. 1988;72:567–572. [PubMed] [Google Scholar]
- 4.Vassiley LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammalott U, Lukacs C, Klein C, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;203:844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 5.Siddiquee K, Zang S, Guida WC, Blaskovich MA, Greedy B, Lawrence HR, Yip ML, Jove R, McLaughlin MM, Lawrence NJ, et al. Selective chemical probe inhibitor of stat3, identified through structure-based virtual screening, induces antitumor activity. Proc. Natl Acad. Sci. USA. 2007;104:7391–7396. doi: 10.1073/pnas.0609757104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang Y, Kheir MM, Chai Y, Hu J, Xing D, Lei F, Du L. Comprehensive study in the inhibitory effect of the berberine on gene transcription, including TATA box. PLoS One. 2011;6:e23495. doi: 10.1371/journal.pone.0023495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Doss RM, Marques MA, Foister S, Chenoweth DM, Dervan PB. Programmable oligomers for the minor groove DNA recognition. J. Am. Chem. Soc. 2006;128:9074–9079. doi: 10.1021/ja0621795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Raskatov JA, Meier JL, Puckett JW, Yang F, Ramakrishnan P, Dervan PB. Modulation of the NF-κB-dependent gene transcription using programmable DNA minor groove binders. Proc. Natl Acad. Sci. USA. 2012;109:1023–1028. doi: 10.1073/pnas.1118506109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang Y, Sicot G, Cui X, Vogel M, Wuertzer CA, Lezon-Geyda K, Wheeler J, Harki DA, Muzikar KA, et al. Targeting a DNA binding motif of the EVI1 protein by a pyrrole-imidazole polyamide. Biochemistry. 2011;50:10431–10441. doi: 10.1021/bi200962u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dickinson LA, Gulizia RJ, Trauger JW, Baird EE, Mosier DE, Gottesfeld JM, Dervan PB. Inhibition of RNA polymerase II transcription in human cells by synthetic DNA-binding ligands. Proc. Natl Acad. Sci. USA. 1998;95:12890–12895. doi: 10.1073/pnas.95.22.12890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vlaminck B, Toffoli S, Ghislain B, Demazy C, Raes M, Michiels C. Dual effect of echinomycin on hypoxia-inducible factor-1 activity under normoxic and hypoxic conditions. FEBS J. 2007;274:5533–5542. doi: 10.1111/j.1742-4658.2007.06072.x. [DOI] [PubMed] [Google Scholar]
- 12.Peixoto P, Liu Y, Depauw S, Hildebrand MP, Boykin DW, Bailly C, Wilson WD, David-Cordonnier MH. Direct inhibition of the DNA-binding activity of POU transcription factors Pit-1 and Brn-3 by a selective binding of a phenyl-furn-benzimidazole dication. Nucleic Acids Res. 2008;10:3341–3353. doi: 10.1093/nar/gkn208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Karim FD, Urness LD, Thummel CS, Klemsz MJ, McKercher SR, Celada A, Van Beveren C, Maki RA, Gunther CV, Nye JA. The ETS-domain: a new DNA-binding motif that recognizes a purine-rich core DNA sequence. Genes Dev. 1990;4:1451–1453. doi: 10.1101/gad.4.9.1451. [DOI] [PubMed] [Google Scholar]
- 14.Nye JA, Petersen JM, Gunther CV, Jonsen MD, Graves BJ. Interaction of murine ets-1 with GGA-binding sites establishes the ETS domain as a new DNA-binding motif. Genes Dev. 1992;6:975–990. doi: 10.1101/gad.6.6.975. [DOI] [PubMed] [Google Scholar]
- 15.Hollenhorst PC, Shah AA, Hopkins C, Graves BJ. Genome-wide analyses reveal properties of redundant and specific promoter occupancy within the ETS gene family. Genes Dev. 2007;21:1882–1894. doi: 10.1101/gad.1561707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wei GH, Badis G, Berger MF, Kivioja T, Palin K, Enge M, Bonke M, Jolma A, Varjosalo M, Gehrke AR, et al. Genome-wide analysis of ETS-family DNA –binding in vitro and in vivo. EMBO J. 2010;29:2147–2160. doi: 10.1038/emboj.2010.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oikawa T, Yamada T. Molecular biology of the Ets family of transcription factors. Gene. 2003;303:11–34. doi: 10.1016/s0378-1119(02)01156-3. [DOI] [PubMed] [Google Scholar]
- 18.Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, Sun XW, Varambally S, Cao X, Tchinda J, Kuefer R, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310:644–648. doi: 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- 19.Demichelis F, Fall K, Perner S, Andren O, Schmidt F, Setlur SR, Hoshida Y, Mosquera JM, Pawitan Y, Lee C, et al. TMPRSS2:ERG gene fusion associated with lethal prostate cancer watchful waiting cohort. Oncogene. 2007;26:4596–4599. doi: 10.1038/sj.onc.1210237. [DOI] [PubMed] [Google Scholar]
- 20.Rajput AB, Miller MA, De Luca A, Boyd N, Leung S, Hurtado-Coll A, Fazli L, Jones EC, Palmer JB, Gleave ME, et al. Frequency of the TMPRSS2:ERG gene fusion is increased in moderate to poorly differentiated prostate cancers. J. Clin. Pathol. 2007;60:1238–1243. doi: 10.1136/jcp.2006.043810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Salek-Ardakani S, Smooha G, de Boer J, Sebire NJ, Morrow M, Rainis L, Lee S, Williams O, Izraeli S, Brady HJ. ERG is a megakaryocytic oncogene. Cancer Res. 2009;69:4665–4673. doi: 10.1158/0008-5472.CAN-09-0075. [DOI] [PubMed] [Google Scholar]
- 22.Baldus C, Burmeister T, Martus P, Schwartz S, Gökbuget N, Bloomfield CD, Hoelzer D, Thiel E, Hofmann WK. High expression of the ETS transcription factor ERG predicts adverse outcome in acute T-lymphoblastic leukemia in adults. J. Clin. Oncol. 2006;24:4714–4720. doi: 10.1200/JCO.2006.06.1580. [DOI] [PubMed] [Google Scholar]
- 23.Metzeler KH, Dufour A, Benthaus T, Hummel M, Sauerland MC, Heinecke A, Berdel WE, Büchner T, Wörmann B, Mansmann U, et al. ERG expression is an independent prognostic factor and allows refined risk stratification in cytogenetically normal acute myeloid leukemia: a comprehensive analysis of ERG, MN1, and BAALC transcript levels using oligonucleotide microarrays. J. Clin. Oncol. 2009;27:5031–5038. doi: 10.1200/JCO.2008.20.5328. [DOI] [PubMed] [Google Scholar]
- 24.Moore SD, Offor O, Ferry JA, Amrein PC, Morton CC, Dal Cin P. ELF4 is fused to ERG in a case of acute myeloid leukemia with a t(X;21)(q25-26;q22) Leuk. Res. 2006;30:1037–1042. doi: 10.1016/j.leukres.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 25.Kim J, Park TS, Song J, Lee KA, Hong DJ, Min YH, Cheong JW, Choi JR. Detection of FUS-ERG chimeric transcript in two cases of acute myeloid leukemia with t(16;21)(p11.2;q22) with unusual characteristics. Cancer Genet. Cytogenet. 2009;194:111–118. doi: 10.1016/j.cancergencyto.2009.06.010. [DOI] [PubMed] [Google Scholar]
- 26.Sorensen PHB, Lessnick SL, Lopez-Terrada D, Liu XF, Triche TJ, Denny CT. A second Ewing’s sarcoma translocation, t(21;22), fuses the EWS gene to another ETS-family transcription factor, ERG. Nat. Genet. 1994;6:146–151. doi: 10.1038/ng0294-146. [DOI] [PubMed] [Google Scholar]
- 27.Delattre O, Zucman J, Melot T, Garau XS, Zucker JM, Lenoir GM, Ambros PF, Sheer D, Turc-Carel C, Triche TJ, et al. The Ewing family of tumors—a subgroup of small-round-cell tumors defined by specific chimeric transcripts. N. Engl. J. Med. 1994;331:294–299. doi: 10.1056/NEJM199408043310503. [DOI] [PubMed] [Google Scholar]
- 28.Erkizan HV, Scher LJ, Gamble E, Barber-Rotenberg JS, Sajwan KP, Üren A, Toretsky JA. Novel peptide binds EWS-FLI1 and reduces the oncogenic potential in Ewing tumors. Cell Cycle. 2011;10:3397–3408. doi: 10.4161/cc.10.19.17734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Erkizan HV, Kong Y, Merchant M, Schlottman S, Barber-Rotenberg JS, Yuan L, Abaan OD, Chou TH, Dakshanamurthy S, Brown ML, et al. A small molecule blocking oncogenic protein EWS-FLI1 interaction with RNA helicase A inhibits growth of Ewing’s sarcoma. Nat. Med. 2009;15:750–756. doi: 10.1038/nm.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rahim S, Beauchamp EM, Kong Y, Brown ML, Torestky JA, Üren A. YK-4-279 inhibits ERG and ETV1 mediated prostate cancer cell invasion. PLoS One. 2011;6:e19343. doi: 10.1371/journal.pone.0019343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grohar PJ, Griffin LB, Yeung C, Chen Q-R, Pommier Y, Khanna C, Khan J, Helman LJ. Ecteinascidin 743 interferes with the activity of EWS-FLI1 in Ewing sarcoma cells. Neoplasia. 2011;13:145–153. doi: 10.1593/neo.101202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grohar PJ, Woldemichael GM, Griffin LB, Mendoza A, Chen QR, Yeung C, Currier DG, Davis S, Khanna C, Khan J, et al. Identification of an inhibitor of the EWS-FLI1 oncogenic transcription factor by high-troughput screening. J. Natl Cancer Inst. 2011;103:962–978. doi: 10.1093/jnci/djr156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Björkman M, Iljin K, Halonen P, Sara H, Kaivanto E, Nees M, Kallioniemi OP. Defining the molecular action of HDAC inhibitors and synergism with androgen deprivation in ERG-positive prostate cancer. Int. J. Cancer. 2008;123:2774–2781. doi: 10.1002/ijc.23885. [DOI] [PubMed] [Google Scholar]
- 34.Fortson WS, Kayarthodi S, Fujimura Y, Xu H, Matthews R, Grizzle WE, Rao VN, Bhat GK, Reddy ES. Histone deacetylase inhibitors, valproic acid and trichostation-A induce apoptosis and affect acetylation status of p53 in ERG-positive prostate cancers cells. Int. J. Oncol. 2011;39:111–119. doi: 10.3892/ijo.2011.1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ismail MA, Boykin DW, Stephens CE. An efficient synthesis of 2,5′-diarylbichalcophenes. Tetrahedron Lett. 2006;47:795–797. [Google Scholar]
- 36.Ismail MA, El Bialy SA, Brun R, Wenzler T, Nanjunda R, Wilson WD, Boykin DW. Dicationic phenyl-2, 2′-bichalcophenes and analogues as antiprotozoal agents. Bioorg. Med. Chem. 2011;19:978–984. doi: 10.1016/j.bmc.2010.11.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carrere S, Verger A, Flourens A, Stehelin D, Duterque-Coquillaud M. Erg proteins, transcription factors of the Ets family, form homo, heterodimers and ternary complexes via two distinct domains. Oncogene. 1998;16:3261–3268. doi: 10.1038/sj.onc.1201868. [DOI] [PubMed] [Google Scholar]
- 38.Duterque-Coquillaud M, Niel C, Plaza S, Stehelin D. New human erg isoforms generated by alternative splicing are transcriptional activators. Oncogene. 1993;8:1865–1873. [PubMed] [Google Scholar]
- 39.Flajollet S, Tian TV, Flourens A, Tomavo N, Villers A, Bonnelye E, Aubert S, Leroy X, Duterque-Coquillaud M. Abnormal expression of the ERG transcription factor in prostate cancer cells activates osteopontin. Mol. Cancer Res. 2011;9:914–924. doi: 10.1158/1541-7786.MCR-10-0537. [DOI] [PubMed] [Google Scholar]
- 40.Baillat D, Leprivier G, Régnier D, Vintonenko N, Bègue A, Stehelin D, Aumercier M. Stromelysin-1 expression is activated in vivo by Ets-1 through palindromic head-to-head Ets binding sites present in the promoter. Oncogene. 2006;25:5764–5776. doi: 10.1038/sj.onc.1209583. [DOI] [PubMed] [Google Scholar]
- 41.Baert JL, Monte D, Verreman K, Degerny C, Coutte L, De Launoit Y. The E3 ubiquitin ligase complex component COP1 regulates PEA3 group member stability and transcriptional activity. Oncogene. 2010;29:1810–1820. doi: 10.1038/onc.2009.471. [DOI] [PubMed] [Google Scholar]
- 42.Genès C, Lenglet G, Depauw S, Nhili R, Prado S, David-Cordonnier MH, Michel S, Tillequin F, Porée FH. Synthesis and biological evaluation of N-substituted benzo[c]phenanthrolines and benzo[c]phenanthrolinones as antiproliferative agents. Eur. J. Med. Chem. 2011;46:2117–2131. doi: 10.1016/j.ejmech.2011.02.065. [DOI] [PubMed] [Google Scholar]
- 43.Racane L, Tralic-Kulenivic V, Kraljevic Pavelic S, Ratkaj I, Peixoto P, Nhili R, Depauw S, Hildebrand M-P, David-Cordonnier M-H, Pavelic K, et al. Novel diamidino-substituted derivatives of phenyl benzothiazolyl and dibenzothiazolyl furans and thiophenes: synthesis, antiproliferative and DNA binding properties. J. Med. Chem. 2010;53:2418–2432. doi: 10.1021/jm901441b. [DOI] [PubMed] [Google Scholar]
- 44.Liu Y, Kumar A, Depauw S, Nhili R, David-Cordonnier MH, Lee MP, Ismail MA, Farahat AA, Say M, Chackal-Catoen S, et al. Water-mediated binding of agents that target the DNA minor groove. J. Am. Chem. Soc. 2011;133: 10171–10183. doi: 10.1021/ja202006u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rahimian M, Kumar A, Say M, Bakunoy SA, Boykin DW, Tidwell RR, Wilson WD. Minor groove binding compounds that jump a GC base pair and bind to adjacent AT base pair sites. Biochemistry. 2009;48:1573–1583. doi: 10.1021/bi801944g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Eriksson M, Norden B. Linear and circular dichroism of drug-nucleic acids complexes. In: Chaires JB, Waring MJ, editors. Drug-nucleic Acids Interactions. Vol. 340. 1 edn. 2001. pp. 68–98. [DOI] [PubMed] [Google Scholar]
- 47.Workman CT, Yin Y, Corcoran DL, Ideker T, Stormo GD, Benos PV. enoLOGOS: a versatile web tool for energy normalized sequence logos. Nucleic Acids Res. 2005;33:W389–W392. doi: 10.1093/nar/gki439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Szymczyna BR, Arrowsmith CH. DNA binding specificity studies of four ETS proteins support an indirect read-out mechanism of protein-DNA recognition. J. Biol. Chem. 2000;275:28363–28370. doi: 10.1074/jbc.M004294200. [DOI] [PubMed] [Google Scholar]
- 49.Bailly C, Tardy C, Wang L, Armitage B, Hopkins K, Kumar A, Schster GB, Boykin DW, Wilson WD. Recognition of ATGA sequences by the unfused aromatic dication DB 293 forming stacked dimers in the DNA minor groove. Biochemistry. 2001;40:9770–9779. doi: 10.1021/bi0108453. [DOI] [PubMed] [Google Scholar]
- 50.Tomlins SA, Laxman B, Varambally S, Cao X, Yu J, Helgeson BE, Cao Q, Prensner JR, Rubin MA, Shah RB, et al. Role of the TMPRSS2-ERG gene fusion in prostate cancer. Neoplasia. 2008;10:177–188. doi: 10.1593/neo.07822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Birdsey GM, Dryden NH, Shah AV, Hannah R, Hall MD, Haskard DO, Parsons M, Mason JC, Zvelebil M, Gottgens B, et al. The transcription factor Erg regulates expression of histone deacetylase 6 and multiple pathways involved in endothelial cell migration and angiogenesis. Blood. 2012;119:894–903. doi: 10.1182/blood-2011-04-350025. [DOI] [PubMed] [Google Scholar]
- 52.Birdsey GM, Dryden NH, Amsellem V, Gebhardt F, Sahnan K, Haskard DO, Dejana E, Mason JC, Randi AM. Transcription factor Erg regulates angiogenesis and endothelial apoptosis through VE-cadherin. Blood. 2011;111:3498–3506. doi: 10.1182/blood-2007-08-105346. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.