


Abstract
Transcription is initiated when RNA polymerase recognizes the duplex promoter DNA in the closed complex. Due to its transient nature, the closed complex has not been well characterized. How the initial promoter recognition occurs may offer important clues to regulation of transcription initiation. In this article, we have carried out single-base pair substitution experiments on two Escherichia coli promoters belonging to two different classes, the −35 and the extended −10, under conditions which stabilize the closed complex. Single-base pair substitution experiments indicate modest base-specific effects on the stability of the closed complex of both promoters. Mutations of base pairs in the −10 region affect the closed complexes of two promoters differently, suggesting different modes of interaction of the RNA polymerase and the promoter in the two closed complexes. Two residues on σ70 which have been suggested to play important role in promoter recognition, Q437 and R436, were mutated and found to have different effects on the closed-complex stability. DNA circular dichroism (CD) and FRET suggest that the promoter DNA in the closed complex is distorted. Modeling suggests two different orientations of the recognition helix of the RNA polymerase in the closed complex. We propose that the RNA polymerase recognizes the sequence dependent conformation of the promoter DNA in the closed complex.
INTRODUCTION
The first step in the transcription process is the formation of the closed complex in which the promoter DNA is recognized in the double-stranded form (1). The recognition of the promoter DNA in the duplex state is followed by a series of steps, leading to the separation of strands, the open-complex formation and the initiation of phosphodiester bond formation (2). Several base pairs have been found to be important for the overall transcription initiation process (3). However, it is often not clear at which step(s) these base pairs exert their effect. One important goal is to identify the kinetic steps and determine the role of each base pair in each step (2) leading to a structural understanding of the open-complex formation process. Roles of some of the bases, such as −11A have been identified (4). The closed complex has proven to be difficult to study at room temperature or above, due to its transient nature. In a study of λ-PR by deHaseth and co-workers (5), a quadruple mutant of the σ70-holoenzyme was used to stabilize the closed complex and partial single-base pair substitution experiments were performed to identify base pairs important for the closed-complex formation. They observed weak base-specific effects on the stability of the closed complex.
If such weak base-specific effects are the rule in the closed-complex recognition, then a more quantitative technique to study the contribution of each base pair to the binding energy may yield more accurate results. Fluorescence anisotropy has been widely used to obtain quantitative equilibrium binding data (6). It has been established that at 4°C RNA polymerase forms a stable closed complex (7). We have previously used fluorescence anisotropy at 4°C to obtain quantitative binding isotherms of the RNA polymerase and the duplex promoter DNA (8). Using the same methodology, in this article, we have studied the effect of single-base pair substitution on closed complexes of two different classes of promoters. Both the promoters exhibit weak base substitution effects in the −10 region, but the role of individual base pairs are different in two promoters. We thus conclude that the mode of recognition of duplex DNA in individual promoters are different, but involves indirect readout.
MATERIALS AND METHODS
Materials
RNA polymerase holo-enzyme and core-enzyme were purchased from Epicenter. Sephadex G-50, Ni-NTA sepharose and pre-packed FPLC column MonoQ were from GE Healthcare. Netropsin dihydrochloride, BSA and DTT were from Sigma Chemical Company (St Louis, MO, USA). MES was from JT Baker. Oregon green isothiocyanate 488, succinimidyl ester of 5(6)-Alexa Fluor 488 carboxylic acid and 5(6)-Alexa Fluor 594 carboxylic acid were from Molecular Probes, Invitrogen. DNA grade acetonitrile, anhydrous acetonitrile, dichloromethane, trichloroacetic acid/dichloromethane, tetrazole/acetonitrile, 1-methylimidazole/tetrahydrofuran, acetic anhydride/pyridine/tetrahydrofuran, 0.02 M iodine/water/pyridine/tetrahydrofuran, dAbz, T, dGiBu and dCbz phosphoramidite and all starting columns with immobilized nucleosides were purchased from Applied Biosystem. 5′TFA-amino C6 modifier was from Biosearch Technologies. Some 5′-C6-amino-linked oligonucleotides were synthesized in-house by a 3400 DNA synthesizer (Applied Biosystems) and some others were purchased from Trilink BioTechnologies.
Labeling of oligonucleotides
After purification by MONO-Q, oligonucleotides were labeled in 100 µl solution containing 1 M sodium carbonate/bicarbonate buffer (pH 9.0): DMF: water = 5:2:3. Reaction was carried out for 20 h at 25°C with a dye to oligonucleotide ratio of 20:1. After incubation, the reaction mixture was loaded onto a Sephadex G-50 (GE Healthcare) column and eluted and dialyzed against H2O.
Synthesis and purification of oligonucleotides
The complementary oligonucleotides were annealed in 10 mM Tris–HCl buffer, pH 7.4 containing 100 mM NaCl and 1 mM EDTA by placing the tube containing the mixed oligonucleotides in water at 95°C for 10 min and cooling at room temperature without any disturbance. The duplex oligonucleotides were then purified by MONO-Q column in FPLC using 0–1 M KCl gradient in 1× PBS (5 mM potassium phosphate buffer, pH 7.4 containing 137 mM NaCl and 2.7 mM KCl) and dialyzed against H2O.
Fluorescence anisotropy
Fluorescence anisotropy measurements were performed at 0°C and 4°C by using Quantamaster 6 (PTI) T-geometry fluorometer. The titrations were carried out in 50 mM MES buffer, pH 6.4 containing 0.2 M NaCl, 10 mM MgCl2, 100 μg/ml BSA, 1 mM DTT, 1 mM EDTA and 10% glycerol. The temperature was maintained by circulating water bath and water vapor was purged from the chamber by a steady stream of dry nitrogen gas. Fluorescence anisotropy was measured with excitation at 490 nm and emission at 526 nm using bandwidths of 5 nm. The DNA concentrations were 1 nM for the galP1-promoter and 2 nM for the PR-promoter. When higher DNA concentrations are used, it is explicitly stated in the figure legend. After mixing with RNA polymerase, it was incubated for 3 min before fluorescence anisotropy measurements. Each point was measured five to six times and an average anisotropy value was obtained and fitted to a single-site binding equation. Some selective binding isotherms are shown in Supplementary Figure S3.
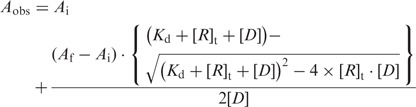 |
where Aobs is the observed anisotropy, Af is the anisotropy of the protein–DNA complex, Kd is the apparent dissociation constant, Ai is the anisotropy of the free template, [R]t is the total RNA polymerase concentration and [D] is the total template concentration.
Correction of the observed dissociation constant for end-binding
For an oligonucleotide duplex, D, we assume that there is only two binding modes. These are promoter binding and end binding. If RNA polymerase is represented by R the following multiple equilibrium would represent the RNA polymerase binding to the oligonucleotide. We also assume that under the conditions studied, the DNA is largely singly ligated (that is one ligand is bound). This is reasonable as in most of the titrations the RNA polymerase concentrations are not very high compared to the lowest dissociation constants.
| (1) |
| (2) |
| (3) |
where K represents the promoter binding constant, Ke represents the end binding constants and Equations (2) and (3) represent binding to two ends of the oligonucleotide.
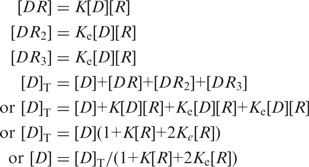 |
(4) |
Fractional saturation Y may be written as
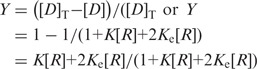 |
(5) |
The observed binding isotherm was fitted to a single-site binding equation to extract an apparent binding constant Kapp.
| (6) |
| (7) |
| (8) |
| (9) |
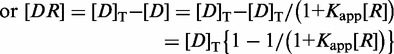 |
From these equations, one can obtain a fractional saturation (Y),
| (10) |
By comparing Equations (5) and (10)
We may conclude that
Fluorescence resonance energy transfer
5′-C6 amino-link oligonucleotides were purchased from Trilink BioTechnologies and reverse phase HPLC purified before use. The oligonucleotides were reacted with a succinimidyl ester of 5(6)-Alexa Fluor 488 carboxylic acid (Alexa 488) or 5(6)-Alexa Fluor 594 carboxylic acid (Alexa 594). The coupling reaction was performed by mixing the DNA strand with a 10-fold excess of the dye molecule in 200 mM sodium carbonate buffer (pH 9.0) and leaving the mixture standing in the dark overnight at room temperature. After the unreacted dye had been removed by gel filtration, the labeled strand was purified by FPLC on a MonoQ column using 0–1 M KCl gradient in 1× PBS. After FPLC, salt was removed by gel filtration. Concentrations of the labeled DNA strands and the labeling ratio (dye/DNA) were determined spectrophotometrically using extinction coefficients of 71 000 M/cm at 495 nm for Alexa 488 and 73 000 M/cm at 590 nm for Alexa 594. For DNA concentration measurement, the dye absorbance at 260 nm was corrected using the following formula: Areal = Aobs − (Aλmax × CF). Aλmax is the peak absorbance value of the coupled dye and CF is the correction factor (0.27 for Alexa 488 and 0.40 for Alexa 594). In this manner, we were able to confirm that each DNA strand contained only one dye molecule. Double-stranded DNA was made by mixing the appropriate complementary strands in an equimolar ratio (for the doubly dye-labeled DNA), heating to 95°C H2O for 10 min and cooling slowly to room temperature over a period of 12–14 h.
Steady-state fluorescence spectra were recorded at 4°C by using Quantamaster 6 (PTI) T-geometry fluorimeter. The bindings were carried out in 50 mM MES buffer, pH 6.4 containing 0.2 M NaCl, 10 mM MgCl2, 100 μg/ml BSA, 1 mM DTT, 1 mM EDTA and 10% glycerol. The fluorescence spectra of oligonucleotides and the oligonucleotide complexes were taken at oligonucleotide concentration of 4 nM and RNA polymerase concentration of 40 nM, respectively. The buffer-only spectrum was subtracted from the oligonucleotide spectra and the protein-only spectrum was subtracted from the complex spectra. Fluorescence excitation spectra were recorded from 480 to 610 nm at the emission maximum set to 620 nm with slits set at 5 nm for both excitation and emission.
Circular dichroism spectroscopy
CD measurements were done on a JASCO (Tokyo, Japan) J720 spectropolarimeter using a 1-cm pathlength quartz cuvette. The scan speed was 120 nm/min. Five scans were signal averaged to increase the signal to noise ratio. The CD spectra of oligonucleotides and the oligonucleotide complexes were taken at oligonucleotide concentration of 200 nM and RNA polymerase concentration of 400 and 600 nM, respectively. The buffer-only spectrum was subtracted from the oligonucleotide spectra and the protein-only spectrum was subtracted from the complex spectra. These experiments were carried out in 50 mM MES buffer, pH 6.4 containing 0.2 M NaCl, 10 mM MgCl2, 100 μg/ml BSA, 1 mM DTT, 1 mM EDTA and 10% glycerol at 4°C.
Site-directed mutagenesis, cloning and purification of the σ70 mutants
The mutant proteins were purified following the protocol of Jin and co-workers (9) with certain modifications. Site-directed mutagenesis were done at Arginine 436 and Glutamine 437 of E. coli σ70 to generate the alanine mutants using the His-tagged wild-type plasmid by PCR-based site-directed mutagenesis kit (Agilent Technologies). Both the clones were done in pET28a. Sequencing of the whole gene showed that the plasmids contain the desired mutations only.
The plasmids encoding the mutant proteins were transformed in BL21 (DE3) competent cells. The cells were grown overnight in 100 ml LB medium with 2% glucose and 100 μg/ml kanamycin at 37°C. The next day, the entire 100-ml culture was diluted 1:10 in fresh LB medium (1000 ml) with 2% glucose and 100 μg/ml kanamycin and was grown at 37°C until A600 ∼ 0.6. IPTG was added to a final concentration of 1 mM. The culture was grown at 30°C for an additional 4 h and then harvested.
The cell pellet was resuspended in 40 ml buffer B containing 20 mM Tris–HCl buffer, pH 8.0, 500 mM NaCl, 20 mM imidazole, 1 mM PMSF, 1 mM β-Me, 10% glycerol and 0.1% Triton X and then disrupted by sonication. The cell lysate was centrifuged for 40 min at 23 000 g at 4°C. The supernatant was allowed to bind for 1 h with 1 ml Ni-sepharose pre-equilibrated with buffer B. It was loaded onto a column and then washed subsequently with 20 ml buffer B and 10 ml buffer B containing 80 mM imidazole. Nickel bound proteins were eluted with 80–300 mM imidazole gradient in buffer B. The fractions containing mutant σ70 were initially dialyzed against 50 mM Tris–HCl buffer, pH 8.0 containing 50 mM NaCl, 10% glycerol, 0.1 mM EDTA, 1 mM β-Me and 0.1% Triton X. It was finally dialyzed against buffer containing 50 mM Tris–HCl buffer, pH 8.0 containing 50 mM NaCl, 50% glycerol, 0.1 mM EDTA, 1 mM DTT and then stored at −70°C. Both R436A-σ70 and Q437A-σ70 proteins were purified according to the above protocol.
RNA polymerase holo-enzyme containing mutants σ70 were then reconstituted by mixing core RNA polymerase and the mutant σ70 in ratio 1:1.3 in protein storage buffer (50 mM Tris–HCl buffer, pH 8.0 containing 50 mM NaCl, 50% glycerol, 0.1 mM EDTA, 1 mM DTT). After mixing, it was incubated at 25°C for 30 min and finally stored at −20°C.
Modeling
The model of σ70 subunit of E. coli RNA polymerase was docked to the λ-PR and gal promoter models with expert interface of HADDOCK 2.1. HADDOCK (10,11) is an information-driven flexible docking tool for modeling of biomolecular complexes. The starting structure for docking was the homology-modeled structure of E. coli σ70 RNA polymerase. The homology model was built for residues 359–613 using RNA polymerase holoenzyme of Thermus thermophilus (PDB id: 1IW7) (12) as template, using Insight II (Accelrys Inc., San Diego, CA, USA). The λ-PR and gal promoters were modeled using 3D-DART (13). The bases from −40 to −1 were modeled for λ-PR and −30 to −1 for gal promoters. For introduction of bending in the DNA models, twist, roll and slide base pair step parameters for the TATA bases (corresponding to TATG in gal and GATA in λ-PR promoter) and the base −26 were changed within conformational space allowed for these bases (14,15).
The residues 589, 433, 434 and 437 of the sigma subunit of the RNA polymerase were defined as active residues for docking with λ-PR promoter. The bases −12, −10, −11 from −10 region and −35 from −35 region were defined as active residues for λ-PR promoter. Distance restraints were provided between bases −11, −12 and −35, and residues 433, 437 and 589, respectively. For docking with gal promoter, residues 433, 434 and 458 of RNA polymerase were considered as active residues whereas bases −10, −11 and −14 were considered active residues for the gal promoter. Both the DNA molecules were considered as fully flexible. Docking was performed in solvated mode with water as solvent. The docking protocol consists of three stages, a rigid-body energy minimization, a semi-flexible refinement in torsion angle space and a final refinement in explicit solvent. In expert interface, during the rigid body energy minimization, 1000 structures were calculated and the best 200 solutions, based on the intermolecular energy, were used for the semi-flexible simulated annealing followed by an explicit water refinement.
RESULTS AND DISCUSSION
Transient molecular associations, like the closed complex, are difficult to analyze due to their short-lived nature. One of the best ways to study a transient species is to stabilize it under certain conditions. It has been observed that at 4°C, the closed complex does not proceed to the open complex and hence it can be studied under equilibrium conditions (16). One-time tested way to explore the important interactions in protein–nucleic acid complexes is through systematic base and amino acid substitutions (17). Single-base pair substitution effects are most effectively surveyed if the binding assays are done at equilibrium and quantitatively. Fluorescence anisotropy is an established technique to study protein–DNA interactions at equilibrium. This assay requires an end-labeled oligonucleotide duplex and hence interference from RNA polymerase end-binding can be significant under certain conditions (18). In previous studies, conditions were reported in which the end-binding is substantially reduced, making the quantitative assay of promoter binding in the closed complex possible (8). Thus, we have chosen fluorescence anisotropy assay at 4°C to obtain binding isotherms of RNA polymerase to promoter DNA in the closed-complex state.
Base–σ interactions in the λ-PR promoter in the closed complex
Since many of the previous studies on the closed complex were performed on λ-PR promoter, we chose this promoter to study the −35 class of promoters (Figure 1A). Figure 1B shows the binding isotherm of RNA polymerase and wild-type λ-PR promoter (contained in an oligonucleotide duplex: named PR) at 4°C using fluorescence anisotropy. As a control, a binding isotherm of oligo dA.dT (dAT-20) and RNA polymerase is also shown (Figure 1C). The dissociation constants derived from promoter binding isotherms are considered as apparent dissociation constants (Kdapp) as the binding isotherm is a result of both promoter (stronger) and end (weaker) binding. The dAT-20 binding isotherm is substantially weaker than the promoter binding and likely results from the end binding of RNA polymerase. The dAT-20:RNA polymerase Kd was used to correct the effect of competing end binding reactions in the promoter binding isotherms to obtain the actual dissociation constant (Kdact). The details of the correction procedure is given in ‘Materials and Methods’ section. Figure 1D shows the ΔΔG values of single-base substitution effects in the −10 region for the wild-type λ-PR promoter. The base substitution strategy used was to substitute non-complementary purines for pyrimidines and vice versa, e.g. G to T, A to C, etc. The most striking feature of single-base pair substitution effects was that it is weak, if any, for all the base pairs studied. Only significant effects in the −10 region in the wild-type template are seen for −13 (weakening) and −12 (strengthening upon G to T substitution) positions. Strengthening of binding of similar magnitude upon G to T substitution at −12 position was also observed previously (5). To our knowledge, the effect of −13-bp substitution on the closed complex has not been observed before in the λ-PR promoter. deHaseth and co-workers (5) also observed that T7C substitution led to ∼2.4-fold weakening of the binding constant in the G12T mutant background. Thus, equilibrium binding was also studied in the T7C-substituted template in the G12T background. We also observed ∼2-fold weakening of RNA polymerase binding under closed-complex conditions. Almost identical results obtained with two different techniques strongly suggest that there is only weak base specificity in the closed complex of λ-PR.
Figure 1.

Effect of base pair substitution in λ-PR promoter. (A) The sequence of λ-PR promoter. The red-colored bases are the −35 element and the blue-colored bases are the −10 elements. (B) Binding isotherm of E. coli RNA polymerase with wild-type λ-PR promoter using fluorescence anisotropy at 4°C. (C) Binding isotherm of dAT-20 oligomer duplex under identical conditions. (D) ΔΔG values of single-base pair mutants of λ-PR promoter.
Base pairs that stabilize the closed complex at galP1 are different from λ-PR
Promoters in E. coli has several elements that influence their activity. They are −10 region, −35 region, UP element, the TGx sequence, the discriminator base and the spacer between the −10 and −35 regions (19). In one major class of promoter, the −35 element plays a crucial role (−35 class). In another major class of promoters −10 and TGx sequence at −14 plays the crucial role (extended −10) (20). The strength of −35 class promoters are strongly dependent on the nucleotide sequence present in the −35 region whereas the activity of extended −10 promoters does not depend on nucleotide sequences in the −35 region; instead they are strongly dependent on the nature of the nucleotide present at the −14 position. The λ-PR promoter belongs to the −35 class. Hence we have attempted to identify the base pairs that are recognized in the closed complex of a promoter belonging to the extended −10 class. We chose the galP1 promoter of E. coli as it is one of the most intensively studied promoters of this class (21). The galP1 promoter however, partially overlaps with another promoter, the galP2 (Figure 2A). The galP2 transcription start site is 5 bp upstream of the start site of the galP1 (22). Initially, four mutations at −14, −13, −12 and −11 of the galP2 with respect to the P2 transcription start site (+1) was chosen as the template to reduce competition from RNA polymerase binding at galP2. Single-base pair substitution experiments with this template (called P2−) indicate that like the λ-PR, base substitutions effects are also weak when compared to base substitution effects in lambda operator sites; however, unlike the λ-PR, 3 bp (−14, −10 and −8) contribute significant-specific binding energy to the closed complex (Figure 2B). Interestingly no contribution is made by −12 and −7 bp, ones that contribute significant binding energy to the λ-PR promoter (−7 in G12T background). Among these 3 bp, the −10 bp appears to contribute the most base-specific binding energy, followed by the −8. Contribution of the −14 bp appears to be the weakest. The galP1 triple mutant (−14, −10 and −8) in the P2− background also binds weakly when compared to the wild-type galP1. Similar results were obtained in another mutant background which contained mutations in both galP2 and another weak promoter galP3, present in the oligonucleotide sequence used (see later). In a previous article, Burr et al. (21) have concluded that the removal of the TG elements results in significant lowering of the isomerization constant, kf but not the KB. This is fully consistent with our result that the −14 bp contributes very little specific binding energy to the closed complex formation. Interestingly, C to A substitution in the −13 position results in tightening of binding, suggesting that the presence of a purine in this position contributes some specific binding energy to the closed complex. Previous bioinformatic analysis suggested the importance of purines in the −13 position (23). The effect of substitution in the −13 position is opposite in the λ-PR promoter, indicating significant difference in the structure of the two closed complexes. Despite the difference in the contribution of the individual base pairs in the −10 region to the specific binding energy, the contribution of base-specific binding energy to the overall binding energy is similar in the closed complex in a different class of promoter as well. We have also performed binding experiments for selected mutants at 0°C, as extended −10 promoters form open complexes at lower temperatures than the −35 class promoters (16). Binding isotherms determined at 0°C gave almost identical results as those at 4°C (Figure 2C).
Figure 2.

Effect of single-base pair substitution on galP1 promoter. (A) Sequence of galP1 promoter. The red-colored base is the −14 and the blue-colored bases are the −10 elements. (B) ΔΔG values with respect to galP1+P2−P3+ promoter, plotted for all the single-base pair mutants tested at 4°C. The sequence at the top shows the P2− mutations shown in green. (C) ΔΔG values of selected mutants at 0°C with respect to galP1+P2−P3+ promoter. (D) ΔΔG values of single-base pair mutants in the galP2=P3− background at 4°C. All the experimental conditions are described in ‘Materials and Methods’ section. The sequence above shows the P2= and P3− mutant gal promoter with P2= mutations in magenta color and P3− mutations are in orange color.
Since the galP2 also belongs to the extended −10 class, it may be argued that the four mutations introduced in the galP2− template may not be sufficient to significantly weaken polymerase binding to the galP2 at the closed-complex level as two important bases (the −10 and the −8) that contribute significant specific binding energy in the galP1 are left out (8). In addition, the oligonucleotide sequence used here for binding experiments also contains a cryptic promoter, the galP3 (24). We thus created another template which contains −14. −10 and −8 mutation with respect to the galP2 as well as the galP3 (termed P2=P3−). Sequences of all the oligonucleotides are given in the Supplementary Tables S1–S11. Selective mutations with respect to the galP1 in this template (P2=P3−) indicated same base pairs (particularly −10 and −8 with respect to galP1) of galP1 contributing to the base-specific binding energy in the closed complex (Figure 2D). This suggests that the results obtained in experiments described above are not significantly impacted by the presence of galP2 and galP3 promoters. Since extended −10 class promoters elements are confined within the −1 to −20 bp, and the specific binding energy is contributed by −14, −10 and −8 bp, we reasoned that a shorter DNA fragment from +1 to −20 may bind well to the RNA polymerase. A 21-bp duplex oligonucleotide (P1–21) containing the sequence of +1 to −20 binds to RNA polymerase with high affinity (Kdact = 4.3 ± 1.6 nM) containing the galP2− mutations (−14 through −11). When the three mutations (−14, −10 and −8) with respect to the galP1 was introduced in this template, it bound with ∼10-fold lower affinity, indicating that this smaller oligonucleotide faithfully reproduces the binding characteristics of the larger oligonucleotides (Supplementary Figure S1). Thus, it is very likely that in the galP1, only 3 bp contribute any significant base-specific binding energy to the closed complex. The differential sensitivity of the λ-PR and the galP1 to base substitutions are strongly suggestive of two different modes of interaction of the RNA polymerase with two different classes of promoters.
Residues on σ70 play different roles in GalP1 and λ-PR closed complexes
The two different modes of interaction of RNA polymerase with two different classes of promoter inferred from the base substitution experiments suggest that amino acid residues on σ70 subunits may also have different roles in the two closed complexes. Two such possible residues are Q437 and R436. Darst and co-workers (25,26) have predicted that Q437 of Ecσ70 may be close to the base pair −12 of −35 class promoters and may even interact with the base pair. Gralla and co-workers (27) have used an EMSA assay to show that two mutations, R436S and Q437S results in abrogation of promoter binding at 4°C in the former case and tightening of promoter binding in the latter case. We have thus attempted to use fluorescence anisotropy assay to explore the roles of the two residues in the closed complex of the galP1 and the λ-PR promoters. Figure 3A shows the binding isotherms of wild-type and Q437Aσ70RNAP with the wild-type λ-PR promoter. The Kdact of wild-type RNAP:λ-PR promoter is 7.7 ± 0.3 nM. In contrast, Kdact derived from the binding isotherm of the Q437Aσ70-holoenzyme is 14.9 nM, which is ∼2-fold weaker than that of the wild-type RNA polymerase. In a similar way the Kdact of galP1:Q437Aσ70RNAP was derived from the binding isotherm and was found to be 0.76 nM. This value is almost identical to that of the galP1–wtRNAP complex, suggesting that Q437 does not contribute specific binding energy in the formation of the closed complex with the galP1 promoter. The roles of the residue 437 is different in the two closed complexes. A similar exercise was carried out with R436A mutation and the ΔΔG values are shown in Figure 3B. The binding defect detected in this case with λ-PR template is smaller than that of the R436S on the same promoter detected with EMSA. Gralla and co-workers (27) reported that they have detected almost complete abrogation of duplex binding upon R436S substitution. In our assays, we see significant weakening of binding upon R436A mutation, but the effect appears to be much smaller than that of Gralla and co-workers. This could be due to the serine versus alanine effect, but more likely it could reflect the difference between two assays. As was stressed above, the fluorescence anisotropy method is a true equilibrium technique, whereas the EMSA involves kinetic separation of two or more species in the gel. Under certain conditions, EMSA tends to overestimate binding defects (28). We discuss other possibilities later. In order to find out if Q437A and R436A mutant RNA polymerase is behaving like the wild-type polymerase in promoter recognition, we have carried out binding of these mutant polymerases with several mutant galP1 templates. Figure 3C shows the ΔΔG values of the mutant templates in reference to the corresponding non-mutant template. Clearly, the ΔΔG values are qualitatively similar to the wild-type polymerase, suggesting that the binding modes of the two mutant polymerases are similar to that of the wild-type polymerase. We can tentatively conclude that the RNA polymerase recognizes two different promoters differently in the closed complex.
Figure 3.

Effect of σ70 mutations on the binding in the closed complex. (A) Binding isotherms of E. coli RNA polymerase (open circles) and E. coli RNA polymerase-σ70-Q437A (solid circle) with wild-type λ-PR promoter at 4°C. (B) ΔΔG values of Q437A and R436A mutants for wild-type galP1 and λ-PR promoters with respect to the wild-type RNA polymerase at 4°C. (C) ΔΔG values of Q437A and R436A mutant RNA polymerases for different galP1 mutant promoters from the corresponding wild-type template at 4°C.
Promoter DNA is distorted in the closed complex
From their study of T7 RNA polymerase and promoter interaction, Patel and co-workers (29) have suggested that the closed complex may contain bent or distorted DNA. Others have also suggested the possibility that the bending of the DNA may play crucial role in recognition and isomerization of closed complexes (30). Thus, we have attempted to find out if the DNA in the closed complex is distorted. CD spectra of DNA is a sensitive monitor of DNA distortion (31). Protein CD spectra in this wavelength region is orders of magnitude weaker than the DNA CD spectra and for most purpose can be neglected in comparison with that of the DNA (32). Figure 4A shows the CD spectra of the galP1 promoter DNA (oligonucleotide duplex P1) in the absence of RNA polymerase and in its presence (two concentrations) at 4°C. CD spectra of the DNA in the presence of RNA polymerase is distinctly different from the spectra of the DNA alone, suggesting significant distortion of the promoter DNA in the closed complex. To obtain a more quantitative assessment of the nature of DNA distortion, we have attempted to measure FRET from two ends of the promoter DNA duplex in the closed complex. The promoter DNA that we have used so far contains 63 bp and hence are ∼225 Å long. It is difficult to obtain a FRET pair that can measure FRET at such a long distance.
Figure 4.

(A) Circular dichroism spectra of galP1+P2−P3+ DNA (oligo duplex P1) with and without E. coli RNA polymerase (black line), 400 nM RNA polymerase (green line) and 600 nM RNA polymerase (blue line) at 4°C. (B) Fluorescence resonance energy transfer between two ends of a 21-mer DNA (P1–21) encompassing the extended −10 elements of galP1 promoter (+1 to −20; galP1+P2−) at 4°C. The two spectra shown are excitation spectra in which the peaks at 590 nm have been normalized. Donor only spectra and buffer have been subtracted from the excitation spectra of donor + acceptor and acceptor only, respectively.
Thus we have used the 21-mer duplex DNA described above (P1–21), containing the region +1 to −20 bp of galP1. The two strands of this shorter DNA (containing hexyl amine at the 5′-end) are separately labeled with donor (Alexa 488) and acceptor (Alexa 594) FRET pairs and annealed. The FRET was measured by comparing the excitation spectra of the donor and the acceptor labeled promoter DNA (FD+A) with that of a duplex DNA in which only the acceptor was labeled (FA) (Figure 4B). FD+A/FA is a function of the FRET efficiency (33). The calculated FRET efficiency of the free DNA was 0.155 which translates to a distance of 79.6 Å. The distance between the two ends of the 21-mer DNA should be ∼72 Å; if one assumes that the hexyl amine linker stretches for another few Ås, the FRET data are in excellent agreement with the calculated distance. When, the FRET was measured in the closed complex (Figure 4B), the FRET efficiency increased to 0.19, possibly indicating somewhat closer approach of the two ends. The distortion observed in the CD spectra and the change in efficiency seen in the FRET experiments argues in favor of a modestly distorted DNA in the closed complex.
Modeling of the closed complex
Two different modes of interaction of RNA polymerase with two promoters belonging to two different classes were inferred from the single-base pair substitution studies. In order to find out plausible spatial relationships of the promoter DNA and the σ70 subunit, we have attempted to model the closed complex for two different classes of promoters by flexible docking program HADDOCK. To test whether the RNA polymerase binds through the major groove, we have titrated a pre-formed galP1:RNA polymerase closed complex at 4°C with netropsin, a minor groove binding antibiotic (34). Supplementary Figure S2 shows the change in anisotropy of the galP1 closed complex as a function of netropsin concentration. The anisotropy value decreases modestly as a function of netropsin concentration but quickly levels off. The final anisotropy value is much higher than that of the free promoter DNA even at 200 µM concentration of netropsin. Clearly netropsin is not a competitor for the RNA polymerase binding site on the DNA. We thus conclude that RNA polymerase does not interact with the promoter DNA (−10 hexamer) in galP1 through the minor groove. Similar results were obtained for the λ-PR promoter. We have put constraints by inserting known experimental proximity relations to constrain many possible structures. The details of the procedure and the constraints used are given in ‘Materials and Methods’ section. The two complexes are shown in Figure 5A. To model the complexes, a bend in the promoter DNA was introduced by changing the roll, twist and slide parameters of the TA and TG steps in the −10 hexamer. The interacting part of the σ70 is docked on to the promoter DNA after defining the active residues in the HADDOCK program. Both the complexes interact through the major groove of the promoter, consistent with the netropsin titration. Orientation of the recognition helix (residues 428–445) are different in the two complexes, resulting in somewhat different interaction pattern of the residue 437. The details of the interaction is shown in Figure 5B.
Figure 5.

(A) Docking of the interacting part of σ70 to the λ-PR promoter (left panel) and the galP1 promoter (right panel). (B) Enlarged pictures showing possible interactions in the respective panels.
The most striking feature of the results presented in this article is that the stability of the closed complex is not significantly compromised by any single-base pair substitution in the −10 region. Only a few-fold effect is seen for the −10 and −8 bp in the galP1 and −7 and −12 in the λ-PR promoter. The magnitude of the effect is smaller than that of several other DNA binding proteins where single-base pair substitution effects are known. In λ-CI and λ-Cro, for example, substitutions of bases that form direct hydrogen bonds with the protein side chains lead to 102- to 103-fold change in the dissociation constants (17,35). A likely explanation of the weak base substitution effect observed in this study is that the RNA polymerase reads out the sequence dependent conformation of the promoters rather than the base sequence itself. Using different techniques, similar weak base-specific effect in promoter recognition has been noticed in other studies before. Gross and co-workers (36) made several constructs of sigma factors lacking the inhibitory region 1.1. In sigma factors, the region 1.1 prevents free sigma factors from binding to promoters. Gross and co-workers had observed that Δ1.1 sigma factors bind promoters tightly, presumably in a closed complex like fashion as sigma factors without the core enzyme are unlikely to proceed to the open complex. They also observed that these constructs bound non-specific DNA with high affinity as well. The observed promoter versus non-specific DNA discrimination by the Δ1.1 sigma factors alone was ∼5-fold. The discrimination of promoter versus non-specific DNA in the closed complex observed in this study is of similar magnitude, suggesting that promoter versus non-specific DNA discrimination in the closed complex is not very large. This is consistent with the hypothesis that RNA polymerase recognizes the base sequence dependent conformation of the promoters. Feklistov and Darst (3) also suggested that the recognition of the −10 element in the closed complex is complicated. They suggested that specificity of recognition of the −10 element is a result of coupling of non-specific recognition of the duplex state and specific recognition of the flipped bases. Our results are consistent with this hypothesis and in physiological temperatures, these two steps may indeed be strongly coupled resulting in transient nature of the duplex state in the closed complex. However, at least in the present study, at low temperatures, the duplex state seems to be uncoupled from the base-flipped state. This is evident from lack of substantial effect of −11 substitutions on the dissociation constants.
Indirect readout of sequence dependent conformation of the DNA has been noticed for several protein–DNA complexes (37,38). Indirect readout may also be accompanied by significant distortion of the DNA (39). The DNA CD and FRET studies indicate a modest distortion of the DNA. At this stage the exact nature of the DNA distortion is not known; however, we note that there are more than one TA or TG steps in the promoters (depending on the promoter sequence) which can easily adopt a non-canonical conformation (15). We speculate that the binding of the RNA polymerase to a promoter causes the duplex DNA to distort. It is interesting to note that two relatively major base substitution effect observed here (−12 at λ-PR and −10 at galP1) results in loss of TA or TG step. It has been suggested previously that upon binding of RNA polymerase to the promoter DNA, −11 A base flips out initiating the strand separation. It has been proposed that the interaction of flipped out −11 A base with aromatic residues in the sigma factors stabilize the flipped conformation (3,4,40). The putative DNA distortion observed here may also aid the flipping out of −11 A by destabilizing the DNA duplex. Previously, a hypothesis has been forwarded in which the DNA distortion is seen to play a crucial role in initiating the strand separation (30).
A key question for the future is why RNA polymerase prefers an indirect readout of the sequence in the closed complex? One distinct possibility lies in the fact that direct hydrogen bonding with the base pairs may not be suitable for subsequent steps in the open-complex formation pathway. Structure of the open complex suggests large conformational change in the DNA and the protein from a putative closed complex form (41). Strong hydrogen bond formation may require breakage of these bonds in subsequent steps as well, which may increase the activation energies of the steps down-stream. Indirect readout may be a compromise between the requirement of the steps down-stream and initial recognition of the promoters.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Tables 1–11 and Supplementary Figures 1–3.
FUNDING
Council of Scientific and Industrial Research (India); JC Bose Fellowship (Department of Science and Technology, Government of India) (to S.R.). Funding for open access charge: Council of Scientific and Industrial Research, Government of India.
Conflict of interest statement. None declared.
Supplementary Material
REFERENCES
- 1.Record M, Reznikoff WS, Craig ML, McQuade KL, Schlax PJ. Escherichia coli RNA polymerase (Es70), promoters, and the kinetics of the steps of transcription initiation. In: Neidhardt FC, et al., editors. Escherichia coli and Salmonella Cellular and Molecular Biology. Washington DC: ASM Press; 1996. pp. 792–821. [Google Scholar]
- 2.deHaseth PL, Zupancic ML, Record MT., Jr RNA polymerase-promoter interactions: the comings and goings of RNA polymerase. J. Bacteriol. 1998;180:3019–3025. doi: 10.1128/jb.180.12.3019-3025.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Feklistov A, Darst SA. Structural basis for promoter −10 element recognition by the bacterial RNA polymerase σ subunit. Cell. 2011;147:1257–1269. doi: 10.1016/j.cell.2011.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lim HM, Lee HJ, Roy S, Adhya S. A “master” in base unpairing during isomerization of a promoter upon RNA polymerase binding. Proc. Natl Acad. Sci. USA. 2001;98:14849. doi: 10.1073/pnas.261517398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cook VM, deHaseth PL. Strand opening-deficient Escherichia coli RNA polymerase facilitates investigation of closed complexes with promoter DNA effects of DNA sequence and temperature. J. Biol. Chem. 2007;282:21319–21326. doi: 10.1074/jbc.M702232200. [DOI] [PubMed] [Google Scholar]
- 6.Lundblad JR, Laurance M, Goodman RH. Fluorescence polarization analysis of protein-DNA and protein-protein interactions. Mol. Endocrinol. 1996;10:607–612. doi: 10.1210/mend.10.6.8776720. [DOI] [PubMed] [Google Scholar]
- 7.Kovacic R. The 0 degree C closed complexes between Escherichia coli RNA polymerase and two promoters, T7-A3 and lacUV5. J. Biol. Chem. 1987;262:13654–13661. [PubMed] [Google Scholar]
- 8.Roy S, Semsey S, Liu M, Gussin GN, Adhya S. GalR represses galP1 by inhibiting the rate-determining open complex formation through RNA polymerase contact: a GalR negative control mutant. J. Mol. Biol. 2004;344:609–618. doi: 10.1016/j.jmb.2004.09.070. [DOI] [PubMed] [Google Scholar]
- 9.Zhi H, Yang W, Jun Jin D. Escherichia coli proteins eluted from mono Q chromatography, a final step during RNA polymerase purification procedure. Methods Enzymol. 2003;370:291–300. doi: 10.1016/s0076-6879(03)70026-3. [DOI] [PubMed] [Google Scholar]
- 10.Dominguez C, Boelens R, Bonvin AMJJ. HADDOCK: a protein-protein docking approach based on biochemical and/or biophysical information. J. Am. Chem. Soc. 2003;125:1731–1737. doi: 10.1021/ja026939x. [DOI] [PubMed] [Google Scholar]
- 11.De Vries SJ, Van Dijk M, Bonvin AMJJ. The HADDOCK web server for data-driven biomolecular docking. Nat. Protocol. 2010;5:883–897. doi: 10.1038/nprot.2010.32. [DOI] [PubMed] [Google Scholar]
- 12.Vassylyev DG, Sekine S, Laptenko O, Lee J, Vassylyeva MN, Borukhov S, Yokoyama S. Crystal structure of a bacterial RNA polymerase holoenzyme at 2.6 A resolution. Nature. 2002;417:712–719. doi: 10.1038/nature752. [DOI] [PubMed] [Google Scholar]
- 13.Van Dijk M, Bonvin AMJJ. 3D-DART: a DNA structure modelling server. Nucleic Acids Res. 2009;37:W235–W239. doi: 10.1093/nar/gkp287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Travers AA. DNA bending and kinking–sequence dependence and function. Curr. Opin. Struc. Biol. 1991;1:114–122. [Google Scholar]
- 15.Travers AA. The structural basis of DNA flexibility. Philos. Trans. R. Soc. A. 2004;362:1423–1438. doi: 10.1098/rsta.2004.1390. [DOI] [PubMed] [Google Scholar]
- 16.Burns HD, Belyaeva TA, Busby S, Minchin SD. Temperature-dependence of open-complex formation at two Escherichia coli promoters with extended-10 sequences. Biochem J. 1996;317:305–311. doi: 10.1042/bj3170305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Takeda Y, Sarai A, Rivera VM. Analysis of the sequence-specific interactions between Cro repressor and operator DNA by systematic base substitution experiments. Proc. Natl Acad. Sci. USA. 1989;86:439–443. doi: 10.1073/pnas.86.2.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Melancon P, Burgess RR, Record MT., Jr Direct evidence for the preferential binding of Escherichia coli RNA polymerase holoenzyme to the ends of deoxyribonucleic acid restriction fragments. Biochemistry. 1983;22:5169–5176. doi: 10.1021/bi00291a017. [DOI] [PubMed] [Google Scholar]
- 19.Ross W, Gourse RL. Analysis of RNA polymerase-promoter complex formation. Methods. 2009;47:13–24. doi: 10.1016/j.ymeth.2008.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shultzaberger RK, Chen Z, Lewis KA, Schneider TD. Anatomy of Escherichia coli σ70 promoters. Nucleic Acids Res. 2007;35:771–788. doi: 10.1093/nar/gkl956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Burr T, Mitchell J, Kolb A, Minchin S, Busby S. DNA sequence elements located immediately upstream of the −10 hexamer in Escherichia coli promoters: a systematic study. Nucleic Acids Res. 2000;28:1864–1870. doi: 10.1093/nar/28.9.1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Choy HE, Adhya S. Control of gal transcription through DNA looping: inhibition of the initial transcribing complex. Proc. Natl Acad. Sci. USA. 1992;89:11264–11268. doi: 10.1073/pnas.89.23.11264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Djordjevic M. Redefining Escherichia coli σ70 promoter elements: −15 motif as a complement of the −10 motif. J. Bacteriol. 2011;193:6305–6314. doi: 10.1128/JB.05947-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ponnambalam S, Spassky A, Busby S. Studies with the Escherichia coli galactose operon regulatory region carrying a point mutation that simultaneously inactivates the two overlapping promoters Interactions with RNA polymerase and the cyclic AMP receptor protein. FEBS Lett. 1987;219:189–196. doi: 10.1016/0014-5793(87)81214-0. [DOI] [PubMed] [Google Scholar]
- 25.Malhotra A, Severinova E, Darst SA. Crystal structure of a [sigma] 70 subunit fragment from E. coli RNA polymerase. Cell. 1996;87:127–136. doi: 10.1016/s0092-8674(00)81329-x. [DOI] [PubMed] [Google Scholar]
- 26.Kenney TJ, Moran CP., Jr Genetic evidence for interaction of sigma A with two promoters in Bacillus subtilis. J. Bacteriol. 1991;173:3282–3290. doi: 10.1128/jb.173.11.3282-3290.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fenton MS, Lee SJ, Gralla JD. Escherichia coli promoter opening and− 10 recognition: mutational analysis of σ70. EMBO J. 2000;19:1130–1137. doi: 10.1093/emboj/19.5.1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hellman LM, Fried MG. Electrophoretic mobility shift assay (EMSA) for detecting protein–nucleic acid interactions. Nat. Protoc. 2007;2:1849–1861. doi: 10.1038/nprot.2007.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tang GQ, Patel SS. T7 RNA polymerase-induced bending of promoter DNA is coupled to DNA opening. Biochemistry. 2006;45:4936–4946. doi: 10.1021/bi0522910. [DOI] [PubMed] [Google Scholar]
- 30.Coulombe B, Burton ZF. DNA bending and wrapping around RNA polymerase: a “revolutionary” model describing transcriptional mechanisms. Microbiol. Mol. Biol. Rev. 1999;63:457–478. doi: 10.1128/mmbr.63.2.457-478.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bandyopadhyay S, Mukhopadhyay C, Roy S. Dimer-dimer interfaces of the lambda-repressor are different in liganded and free states. Biochemistry. 1996;35:5033–5040. doi: 10.1021/bi952123f. [DOI] [PubMed] [Google Scholar]
- 32.Mandal AK, Bhattacharyya A, Bhattacharyya S, Bhattacharyya T, Roy S. A cognate tRNA specific conformational change in glutaminyl-tRNA synthetase and its implication for specificity. Protein Sci. 1998;7:1046–1051. doi: 10.1002/pro.5560070422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cantor CR, Schimmel PR. Biophysical Chemistry: Techniques for the Study of Biological Structure and Function. San Fransisco: WH Freeman & Co; 1980. [Google Scholar]
- 34.Baguley B. Nonintercalative DNA-binding antitumour compounds. Mol. Cell. Biochem. 1982;43:167–181. doi: 10.1007/BF00223008. [DOI] [PubMed] [Google Scholar]
- 35.Sarai A, Takeda Y. Lambda repressor recognizes the approximately 2-fold symmetric half-operator sequences asymmetrically. Proc. Natl Acad. Sci. USA. 1989;86:6513–6517. doi: 10.1073/pnas.86.17.6513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dombroski AJ, Walter WA, Record MT, Jr, Siegele DA, Gross CA. Polypeptides containing highly conserved regions of transcription initiation factor sigma 70 exhibit specificity of binding to promoter DNA. Cell. 1992;70:501–512. doi: 10.1016/0092-8674(92)90174-b. [DOI] [PubMed] [Google Scholar]
- 37.Rice PA, Yang S, Mizuuchi K, Nash HA. Crystal structure of an IHF-DNA complex: a protein-induced DNA U-turn. Cell. 1996;87:1295–1306. doi: 10.1016/s0092-8674(00)81824-3. [DOI] [PubMed] [Google Scholar]
- 38.Otwinowski Z, Schevitz R, Zhang R, Lawson C, Joachimiak A, Marmorstein R, Luisi B, Sigler P. Crystal structure of trp represser/operator complex at atomic resolution. Nature. 1988;335:321–329. doi: 10.1038/335321a0. [DOI] [PubMed] [Google Scholar]
- 39.Aeling KA, Opel ML, Steffen NR, Tretyachenko-Ladokhina V, Hatfield GW, Lathrop RH, Senear DF. Indirect recognition in sequence-specific DNA binding by Escherichia coli integration host factor. J. Biol. Chem. 2006;281:39236–39248. doi: 10.1074/jbc.M606363200. [DOI] [PubMed] [Google Scholar]
- 40.Schroeder LA, Gries TJ, Saecker RM, Record MT, Jr, Harris ME, deHaseth PL. Evidence for a tyrosine–adenine-stacking interaction and for a short-lived open intermediate subsequent to initial binding of Escherichia coli RNA polymerase to promoter DNA. J. Mol. Biol. 2009;385:339–349. doi: 10.1016/j.jmb.2008.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hudson BP, Quispe J, Lara-González S, Kim Y, Berman HM, Arnold E, Ebright RH, Lawson CL. Three-dimensional EM structure of an intact activator-dependent transcription initiation complex. Proc. Natl Acad. Sci. USA. 2009;106:19830–19835. doi: 10.1073/pnas.0908782106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.