


Abstract
Many cellular proteins assemble into macromolecular protein complexes. The identification of protein–protein interactions and quantification of their stoichiometry is therefore crucial to understand the molecular function of protein complexes. Determining the stoichiometry of protein complexes is usually achieved by mass spectrometry-based methods that rely on introducing stable isotope-labeled reference peptides into the sample of interest. However, these approaches are laborious and not suitable for high-throughput screenings. Here, we describe a robust and easy to implement label-free relative quantification approach that combines the detection of high-confidence protein–protein interactions with an accurate determination of the stoichiometry of the identified protein–protein interactions in a single experiment. We applied this method to two chromatin-associated protein complexes for which the stoichiometry thus far remained elusive: the MBD3/NuRD and PRC2 complex. For each of these complexes, we accurately determined the stoichiometry of the core subunits while at the same time identifying novel interactors and their stoichiometry.
INTRODUCTION
Many cellular proteins assemble into protein complexes consisting of stable core subunits as well as dynamic and substoichiometric but functionally relevant secondary interactors. During the last decade, mass-spectrometry has proven itself as a powerful tool to identify protein–protein interactions. The first qualitative, systems-wide protein–protein interaction landscapes were generated in yeast using TAP-tagging approaches (1,2). In recent years, quantitative mass spectrometry-based proteomics approaches have been developed and these can be used to determine cellular protein–protein interactions with high confidence when performing single affinity purifications from crude lysates. Since mass spectrometry is not inherently quantitative, most methods rely on the introduction of stable isotopes in the specific pull-down and the control. This allows a pair-wise, quantitative comparison of peptides between the two samples and enables discrimination of highly abundant background proteins from specific interactors (3). Recently, novel label-free quantification (LFQ) algorithms leading to comparable although slightly less-accurate results have been implemented (4–6).
Each of the above-mentioned methods can be used to identify specific protein–protein interactions, but they do not reveal any information about the stoichiometry of the interactions. This would require an estimation of the relative abundance of all the proteins co-purified specifically during affinity enrichment. In recent years, several groups have developed absolute quantification strategies that mostly rely on introducing isotope-labeled reference peptides after affinity purification (7–9). These labeled reference peptides have to be synthesized and this can be quite costly. Furthermore, designing the appropriate reference peptides is in many cases not trivial. Therefore, these methods have not yet been applied in a high-throughput and comprehensive manner. As an alternative to isotope-labeled reference peptides, label-free absolute quantification methods have been developed, such as emPAI, APEX and intensity-based absolute quantification (iBAQ) (10–12). In iBAQ, the sum of intensities of all tryptic peptides for each protein is divided by the number of theoretically observable peptides. The resulting iBAQ intensities provide an accurate determination of the relative abundance of all proteins identified in a sample.
Here, we show that iBAQ, in combination with LFQ of single affinity enrichments, enables accurate determination of the stoichiometry of detected statistically significant interactions. We benchmarked the method using a complex for which the stoichiometry was determined previously using labeled reference peptides. The approach was then used to determine the stoichiometry of two chromatin-associated protein complexes: MBD3/NuRD and PRC2. We show that the MBD3/NuRD complex contains six molecules of RbAp48/46 per complex, a trimer of MTA1/2/3, a GATA2a/2b dimer, a DOC-1 dimer and only one HDAC1/2 and CHD3/4 molecule per complex. The PRC2 complex contains a monomer of each of its three core subunits Ezh2, EED and Suz12 and we identify C17orf96 and C10orf12 as two novel substoichiometric PRC2 interactors. The method described in this study is simple, robust and generic and can be applied to determine the stoichiometry of all cellular protein–protein interactions.
MATERIALS AND METHODS
Bacterial artificial chromosomes lines and cell culture
To ensure (near) endogenous transgenic protein expression, the proteins of interest were GFP-tagged using BAC-TransGeneOmics (13). Briefly, recombineered bacterial artificial chromosomes (BACs) were transfected in HeLa cells and stably integrated transgenes were selected for using media containing 400 µg/µl geneticin (G418, Gibco). The HeLa BAC-GFP lines and HeLa wild-type cells were cultured in high-glucose Dulbecco’s modified Eagle medium (Invitrogen) supplemented with 10% (vol/vol) fetal bovine serum (FBS; Invitrogen) and 100 U/ml penicillin and streptomycin (Invitrogen).
Nuclear extract isolation and GFP pull-down
Nuclear extracts from BAC-GFP and wild-type HeLa cells were generated as described (14). Briefly, cells were incubated in hypotonic buffer after harvesting and homogenized using a type B (tight) pestle in the presence of 0.15% NP-40 (Roche) and complete protease inhibitors (Roche). The nuclei were pelleted by centrifugation and incubated with lysis buffer (420 mM NaCl, 0.1% NP-40 and complete protease inhibitors) for 1 h to extract nuclear proteins. The nuclear extract was obtained by a final centrifugation step at 20 000g for 30 min at 4°C.
The BAC-GFP HeLa and HeLa WT nuclear extracts were subjected to GFP-affinity enrichment using GFP nanotrap beads (Chromotek). As a second control, BAC-GFP HeLa nuclear extracts were incubated with beads lacking the GFP nanotrap (Chromotek). For each pull-down, 1 mg of nuclear extract was incubated with 7.5–10 µl beads in incubation buffer [300 mM NaCl, 0.10% NP-40, 0.5 mM DDT, 20 mM HEPES–KOH (pH 7.9)] containing ethidium bromide at a final concentration of 50 µg/µl. Ethidium bromide is added to the reaction to prevent indirect, DNA-mediated interactions. Beads were then washed two times with incubation buffer containing 0.5% NP-40, two times with PBS containing 0.5% NP-40 and finally two times with PBS.
Sample preparation and mass spectrometry
Precipitated proteins were subjected to on-bead trypsin digestion as described previously (15). In short, 50 µl of elution buffer (2 M Urea, 10 mM DTT and 50 mM Tris–HCl pH 7.5) was added to the beads in order to partially denature the proteins. After incubation, for 20 min at RT in a thermoshaker, the supernatant was collected in a separate tube and iodoacetamide (IAA, Sigma) was added to a final concentration of 50 mM. The beads were then incubated with 50 µl of elution buffer containing 50 mM IAA instead of DTT for 5 min at RT. Proteins on the beads were then partially digested from the beads by adding 0.25 µg trypsin (Promega) for 1 h at RT in a thermoshaker. The supernatant was then collected and added to the first supernatant. A total of 0.1 µg of fresh trypsin was added and proteins were digested overnight at RT. Tryptic peptides were finally acidified and desalted using Stagetips (16) prior to mass spec analyses.
After elution from the Stagetips, the tryptic peptides were applied to online nanoLC-MS/MS, using a 120-min gradient from 7% until 32% acetonitril followed by step-wise increases up to 95% acetonitril. Mass spectra were recorded on a LTQ-Orbitrap-Velos mass spectrometer (Thermo Fisher Scientific), selecting the 15 most intense precursor ions of every full scan for fragmentation.
Data analysis
Raw data were analyzed by MaxQuant (version 1.2.2.5) (17) using standard settings with the additional options match between runs, LFQ and iBAQ selected. The generated ‘proteingroups.txt’ table was filtered for contaminants, reverse hits, number of unique peptides (>0) and number of peptides (>1) in Perseus (from MaxQuant package) or R.
For interactor identification, t-test-based statistics was applied on LFQ as described earlier (5). First, the logarithm (log 2) of the LFQ values were taken, resulting in a Gaussian distribution of the data (Supplementary Figure S1). This allowed imputation of missing values by normal distribution (width = 0.3, shift = 1.8), assuming these proteins were close to the detection limit. Statistical outliers for the GFP pull-down of the BAC HeLa compared to HeLa WT were then determined using two-tailed t-test. Multiple testing correction was applied by using a permutation-based false discovery rate (FDR) method in Perseus.
To determine the stoichiometry of the identified complexes, we compared the relative abundance of the identified interactors as measured by the iBAQ intensities. The background binding level of proteins as measured by the iBAQ intensity in the different control samples were subtracted from the BAC HeLa GFP pulldown iBAQ intensity. Next, these relative abundance values were scaled to the obtained abundance of the bait protein which was set to 1.
RESULTS
Recently, iBAQ has been described, which, in combination with a spike-in of a protein standard mixture with known molar concentration, converts peptide intensities measured by mass spectrometry into absolute protein amounts (11). In a recently published comparison of different absolute quantification methods, iBAQ was shown to be most accurate (18). We reasoned that iBAQ, when applied to label-free single-step affinity enrichment experiments without a protein standard spike-in, could be used to determine the stoichiometry of detected protein–protein interactions. To this end, we devised an experimental setup based on a recently published label-free single GFP-affinity enrichment method called QUBIC (5). In this method, genes of interest are tagged with GFP at near endogenous levels using BAC-transgenomics (13). Nuclear extracts are generated from these cells, as well as from wild-type HeLa cells. These lysates are then subjected to single step GFP-affinity enrichment (ipGFP) in triplicate. In addition to making use of nuclear extracts from wild-type HeLa cells as a control (cWT), nuclear extracts from the GFP-tagged cell line are incubated with beads that do not contain GFP binder (cBEAD). Thus, nine pull-downs are performed in total, three specific pull-downs targeting the GFP-tagged protein and six control pull-downs (Figure 1A). The precipitated proteins are then subjected to on-bead trypsin digestion after which peptide mixtures are analyzed by nanoLC-MS/MS on an Orbitrap-Velos mass spectrometer. After raw data processing using MaxQuant, the obtained label-free (LFQ) intensities are used to determine statistically enriched proteins in the GFP-BAC IP as described previously (5) (Figure 1B). Next, iBAQ intensities for statistically enriched proteins are calculated in each of the nine pull-downs. The iBAQ values obtained in the six control samples (cBEAD and cWT) indicate background binding. These iBAQ intensities are therefore subtracted from the iBAQ intensity in the GFP pull-downs [triplicate iBAQ (ipGFP-cBEAD); x-axis and triplicate iBAQ (ipGFP-cWT); y–axis] (Figure 1C). The resulting corrected iBAQ intensity for the GFP-tagged protein is set to 1 and the iBAQ values of the interacting proteins with their SD are scaled accordingly. This finally results in a stoichiometry determination of all the interactors relative to the bait protein.
Figure 1.

Stoichiometry of protein complexes revealed by label-free quantitative proteomics. (A) Nuclear extracts from BAC-GFP transgenic cell lines and HeLa WT cells are subjected to single-step GFP-affinity enrichment using GFP trap beads in triplicate (ipGFP and cWT, respectively). As an additional control, BAC-GFP cell line nuclear extract is incubated with beads lacking GFP binder in triplicate (cBEAD). Thus, nine separate pull-downs are performed and each of these is separately analyzed by LC-MS/MS. (B) Statistically enriched proteins in the GFP-BAC IP are identified by permutation-based FDR-corrected t-test. The LFQ intensity of the GFP pull-down over the control is plotted against the–log 10 (P-value) and the red line indicates the permutation-based FDR threshold. The proteins in the upper right corner represent the bait and its interactors. (C) Stoichiometry of the interactors is determined by calculating the iBAQ intensities. Since the iBAQ values obtained in the control samples (cBEAD and cWT) indicate background binding, these iBAQ intensities are subtracted from the iBAQ intensity in the GFP pull-down [iBAQ (ipGFP-cBEAD); x-axis and iBAQ (ipGFP-cWT); y-axis]. The remaining values were scaled according to the abundance of the bait protein, resulting in the stoichiometry of the interactors relative to the bait.
Benchmarking the method: the spliceosomal PRP19/CDC5L complex
The spliceosomal PRP19/CDC5L complex was chosen to benchmark our method since the stoichiometry of this complex has been determined previously using isotope-labeled reference peptides (7). This protein complex is known to harbor four salt-stable core subunits: PRP19, CDC5L, Spf27 and PLRG1 in an apparent stoichiometry of ∼4:2:1:1, respectively. In addition, the complex contains three additional interactors, namely AD-002, CTNNBL1 and HSP70 (7,19). We generated a BAC-GFP transgenic cell line that expresses full-length CDC5L with a C-terminal GFP tag at near endogenous levels. Affinity purifications of CDC5L–GFP resulted in the identification of approximately 50 interacting proteins, including all previously described PRP19/CDC5L protein complex subunits (permutation-based FDR t-test; P = 0.0001 and S0 = 0.4) (Figure 2A). These 50 proteins were subsequently analyzed to determine their stoichiometry relative to CDC5L as described earlier. We obtained 12 interactors showing a stoichiometry relative to CDC5L of >0.05 (Figure 2B and C). Consistent with previous data, the known four core subunits of the complex show the highest stoichiometry: PRP19 2.08, Spf27 0.62 and PLRG1 0.37. Given the fact that CDC5L forms a dimer, this results in a stoichiometry of: PRP19 4.16, CDC5L 2, Spf27 1.24 and PLRG1 0.72. These data are in very good agreement with the previously published data from Schmidt et al. (7). Besides these core complex subunits, we determined the stoichiometry of two PRP19/CDC5L interactors (AD-002 and HSP70) to be 0.36 (=0.72) and 0.20 (=0.40), respectively. Schmidt and co-workers did not determine the stoichiometry of AD-002 in their complex purifications since they were not able to synthesize suitable reference peptides for this protein. Finally, the CDC5L interacting protein CTNNBL1 showed a stoichiometry of 0.06 (=0.12) in our pull-down, whereas Schmidt et al. determined this protein to have a stoichiometry of 0.5 relative to CDC5L. Since CTNNBL1 is not part of the salt stable core PRP19/CDC5L complex, this protein–protein interaction most likely did not remain stable in our GFP-pull-down (pull-down and washes in the presence of 300 mM NaCl). In summary, these results show that our LFQ approach is yielding data of similar quality compared to reference peptide-based methods but at a fraction of the time and effort and in a comprehensive and unbiased manner.
Figure 2.
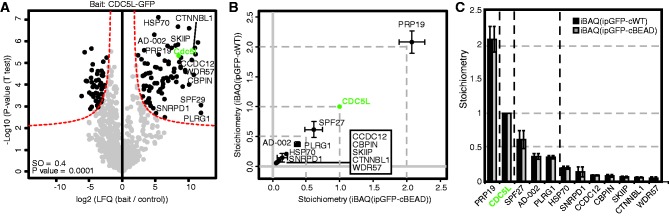
Benchmarking the method using the PRP19/CDC5L spliceosomal complex. (A) Significant interactors of CDC5L–GFP are identified by permutation-based FDR-corrected t-test (threshold: P = 0.0001 and S0 = 0.4). The LFQ intensity of the GFP pull-down over the control is plotted against the −log 10 (P-value). The red line indicates the permutation-based FDR threshold. (B and C) Stoichiometry determination for the 11 most abundant and statistically significant CDC5L–GFP interactors. The abundance of the interactors in the specific pull-down (iBAQ value) was corrected for the obtained abundance in the control pull-downs: iBAQ (ipGFP-cBEAD) [x-axis (b) or grey bar(c)] and iBAQ (ipGFP-cWT) [y-axis (b) or black bar (c)] pull-down. The remaining values were scaled according to the abundance of the bait protein (CDC5L–GFP, green), which was set to 1. Error bars indicate the SD in the triplicate pull-downs.
Stoichiometry determination of chromatin-associated protein complexes
MBD3/NuRD
Next, we applied our method to chromatin-associated protein complexes for which the stoichiometry thus-far remained elusive. The first complex we studied is the MBD3/NuRD protein complex, which contains ATP-dependent chromatin remodeling and histone deacetylase activity (20,21). Purification of MBD3-GFP and LFQ resulted in the identification of 12 significant interactors, many of which are known MBD3/NuRD core subunits (Figure 3A) (21,22). iBAQ-based stoichiometry determination revealed a stable core complex with the following stoichiometry: 6×RbAp48/46, 3×MTA1/2/3, 2×DOC-1, 2×GATAD2a/b, 1×MBD3, 1×CHD3/4 and 1×HDAC1/2 (Figure 3B and C). RbAp48 and RbAp46 are highly similar in their amino acid sequence and therefore share a large number of tryptic peptides. It is therefore not possible to specify the individual iBAQ intensity for each of these proteins. To overcome this problem, the iBAQ intensities for RbAp48 and RbAp46 are collapsed into a single value which accurately reflects their combined stoichiometry. In a similar logic, the iBAQ intensities for MTA1/2/3, GATAD2a/b, CHD3/4 and HDAC1/2 are also collapsed into four stoichiometry values (Figure 3B and C). We used western blotting to show that the MBD3 protein is monomeric (Figure 3D) and DOC-1 was recently shown to form dimers (23), thus revealing that the observed stoichiometry is indicative of the amount of subunit molecules per MBD3/NuRD complex. Finally, we identified two novel zinc finger proteins (ZMYND8 and ZNF592) as potential substoichiometric interactors of the MBD3–NuRD complex. Each of these proteins has a stoichiometry of ∼0.01. These substoichiometric NuRD interacting proteins may serve to recruit the NuRD complex to a specific subset of target genes in the genome and are therefore expected to be of lower stoichiometry compared to the core subunits.
Figure 3.

Stoichiometry of NuRD revealed by label-free quantitative proteomics. (A) Identification of specific interactors of MBD3-GFP by a permutation-based FDR-corrected t-test (threshold: P = 0.01 and S0 = 0.5). Layout as in Figure 2A. (B and C) Stoichiometry determination of the statistically significant interactors of MBD3-GFP. Layout as in (Figure 2B and C). (D) Western-blot analysis of MBD3-GFP affinity purification reveals that MBD3-GFP does not form multimers, as no endogenous MBD3 was detected in the GFP affinity purification. Nuclear extracts from MBD3-GFP and WT HeLa cells were subjected to GFP-affinity enrichment and probed with an antibody against GFP (upper panel) as well as an endogenous antibody to MBD3 (lower panel).
PRC2
The second protein complex we focused on is PRC2, a well-studied and important Polycomb complex that methylates histone H3 on lysine 27 and represses transcription (24). PRC2 consists of three core subunits: EED, Ezh2 and Suz12, which were, among other proteins, identified in our EED-GFP purification (Figure 4A). Quantification of EED-GFP interactors revealed that the PRC2 core complex assembles in a 1:1:1 stoichiometry (Figure 4B and C). Co-immunoprecipitation experiments revealed that EED does not form multimers (Figure 4D). Thus, we conclude that the core PRC2 complex contains one molecule of EED, Ezh1/2 and Suz12. Besides known substoichiometric interactors of PRC2, such as RbAp48/46 (0.60), PCL1/2/3 (0.35), AEBP2 (0.20) and Jarid2 (0.12), we also identified a novel substoichiometric interactor called c17orf96 (0.47). The stoichiometry of this protein relative to the core complex is higher than most other known PRC2 interactors, indicating that this may be an interaction of substantial importance. A second previously unknown substoichiometric PRC2 interactor is C10orf12. This protein has a stoichiometry of ∼0.06 relative to the core subunits.
Figure 4.

Stoichiometry of PRC2 revealed by label-free quantitative proteomics. (A) Identification of specific interactors of EED-GFP by a permutation-based FDR-corrected t-test (threshold: P = 0.01 and S0 = 0.5). Layout as in Figure 2A. (B and C) Stoichiometry determination of the statistically significant interactors of EED-GFP. Layout as in (Figure 2B and C). (D) Western-blot analysis of EED-GFP affinity purification reveals that EED-GFP does not form multimers, since no endogenous EED is detected in the GFP-EED affinity purification. Nuclear extracts from EED-GFP and WT HeLa cells were subjected to GFP-affinity enrichment and probed with an antibody against GFP (upper panel) as well as an endogenous antibody to EED (lower panel).
DISCUSSION
In this study, we have described a novel method that can be used to determine the stoichiometry of protein–protein interactions using label-free quantitative mass-spectrometry-based proteomics. Since this method does not rely on labeled reference peptides, a priori knowledge of interaction partners is not required. Thus, in a single experiment, known and novel protein–protein interactions are detected and for these interactions the stoichiometry is immediately determined. We first benchmarked our method using a protein complex for which the stoichiometry was determined previously using labeled reference peptides (PRP19/CDC5l) and we show that our method is equally accurate. We then went on to determine the stoichiometry of two important and well-studied chromatin-associated proteins complexes: MBD3/NuRD and PRC2. Strikingly, despite being studied for over a decade, the stoichiometry of these two complexes thus far remained elusive.
The stoichiometry analysis of the MBD3/NuRD complex revealed that the DOC-1 protein is present within the complex as a stoichiometric dimer. Given its small size of 12 kDa, DOC-1 has most likely been overseen in many previous gel-based characterizations of the NuRD complex. Our results now unambiguously reveal that DOC-1 indeed is a core NuRD complex subunit, as we have suggested previously (21,22). One surprising observation is the fact that there is only one HDAC1/2 molecule per MBD3/NuRD complex. Previously, it was always assumed that there is one HDAC1 and one HDAC2 molecule present in each NuRD complex. We cannot exclude the possibility that a fraction of HDAC1 and/or HDAC2 get removed from the complex during affinity purification but in this case it would be unlikely to yield a stoichiometry of exactly 1 relative to MBD3. Furthermore, affinity purification of DOC-1-GFP from HeLa cells revealed a stoichiometry between RbAp48/46, MTA1/2/3 and HDAC1/2 of 6:3:1 (data not shown), which is in perfect agreement with the stoichiometry of these proteins in the MBD3-GFP pull-down (Figure 3). Nevertheless, cross-linking approaches to ‘freeze’ cellular protein–protein interactions could be applied to further investigate whether HDAC1 and HDAC2 molecules are partially displaced from the core NuRD complex during the affinity purification. The same holds true for the substoichiometric interactions that we observe in our experiments. Our method does not reveal whether these interactions are indeed substoichiometric or whether they get partially washed away during affinity purification. The dynamic nature of certain interactions could also be further investigated using stable isotope labeling and mixing of light and heavy lysates prior to the pull-down (25).
Purification of GFP-EED from HeLa cells followed by quantitative mass spectrometry revealed that the core PRC2 complex consisting of EED, Suz12 and Ezh1/2 assembles in a 1:1:1 stoichiometry with each subunit being present as a monomer. Consistent with previous observations, a number of proteins interact with PRC2 in a substoichiometric manner, such as AEBP2, Jarid2, RbAp48/46 and PCL1/2/3. In addition, we identified two novel uncharacterized proteins as substoichiometric EED interactors: C17orf96 and C10orf12. Based on our results, it is impossible to distinguish whether these observed interactions all occur simultaneously or whether there are PRC2 subcomplexes each containing one or more of the substoichiometric interactors as a stoichiometric component. Reciprocal tagging and purification of PRC2 interactors can be performed to address this question.
Given the extremely high sensitivity and sequencing speed of modern mass spectrometers, gel-free single step affinity purifications will yield both core protein complex subunits as well as transient, substoichiometric protein–protein interactions. Our method is a powerful tool to efficiently discriminate these different interactions, which is crucial to understand the biology of the complexes. Furthermore, our affinity enrichments yielded high peptide counts for the bait and its interactors (37 measured peptides on an average), resulting in high-accuracy measurements which enabled us to determine monomers or multimers of proteins within a complex, thus providing highly valuable information from a structural perspective. The method can also be applied to study the dynamics of protein complexes, for example during stem cell differentiation or throughout the cell cycle. Finally, the method is easily scalable for high-throughput approaches and should be applicable to already existing large-scale label-free protein–protein interaction data sets. The method can be adapted to any label-free single affinity enrichment workflow; we therefore expect it to become widely used in the chromatin and transcription field and beyond to determine protein–protein interactions and their stoichiometry.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Table 1 and Supplementary Figure 1.
FUNDING
European Community’s Seventh Framework Programme [FP7/2007-2013, work of I.P. in the laboratory of A.A.H.]; MitoSys [grant agreement 241548]; Netherlands Organisation for Scientific Research [NWO VIDI, work in the Vermeulen lab]; Dutch Cancer Society and European Community’s Seventh Framework Programme Project ‘4DCellFate’. Funding for open access charge: NWO-VIDI.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Nina Hubner, Marc Timmers, Henk Stunnenberg and members of the Vermeulen lab for critical reading of the manuscript. We thank Roberto Bonasio and Danny Reinberg for providing EED antibody and Matthias Selbach for advice. Furthermore, A.H.H. acknowledges support by the Deutsche Forschungsgemeinschaft (DFG) Gottfried-Wilhelm-Leibniz Prize, which was awarded to him in 2011.
REFERENCES
- 1.Krogan NJ, Cagney G, Yu H, Zhong G, Guo X, Ignatchenko A, Li J, Pu S, Datta N, Tikuisis AP, et al. Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature. 2006;440:637–643. doi: 10.1038/nature04670. [DOI] [PubMed] [Google Scholar]
- 2.Gavin AC, Aloy P, Grandi P, Krause R, Boesche M, Marzioch M, Rau C, Jensen LJ, Bastuck S, Dumpelfeld B, et al. Proteome survey reveals modularity of the yeast cell machinery. Nature. 2006;440:631–636. doi: 10.1038/nature04532. [DOI] [PubMed] [Google Scholar]
- 3.Vermeulen M, Hubner NC, Mann M. High confidence determination of specific protein-protein interactions using quantitative mass spectrometry. Curr. Opin. Biotechnol. 2008;19:331–337. doi: 10.1016/j.copbio.2008.06.001. [DOI] [PubMed] [Google Scholar]
- 4.Sowa ME, Bennett EJ, Gygi SP, Harper JW. Defining the human deubiquitinating enzyme interaction landscape. Cell. 2009;138:389–403. doi: 10.1016/j.cell.2009.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hubner NC, Bird AW, Cox J, Splettstoesser B, Bandilla P, Poser I, Hyman A, Mann M. Quantitative proteomics combined with BAC TransgeneOmics reveals in vivo protein interactions. J. Cell Biol. 2010;189:739–754. doi: 10.1083/jcb.200911091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rinner O, Mueller LN, Hubalek M, Muller M, Gstaiger M, Aebersold R. An integrated mass spectrometric and computational framework for the analysis of protein interaction networks. Nat. Biotechnol. 2007;25:345–352. doi: 10.1038/nbt1289. [DOI] [PubMed] [Google Scholar]
- 7.Schmidt C, Lenz C, Grote M, Luhrmann R, Urlaub H. Determination of protein stoichiometry within protein complexes using absolute quantification and multiple reaction monitoring. Anal. Chem. 2010;82:2784–2796. doi: 10.1021/ac902710k. [DOI] [PubMed] [Google Scholar]
- 8.Bennett EJ, Rush J, Gygi SP, Harper JW. Dynamics of cullin-RING ubiquitin ligase network revealed by systematic quantitative proteomics. Cell. 2010;143:951–965. doi: 10.1016/j.cell.2010.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wepf A, Glatter T, Schmidt A, Aebersold R, Gstaiger M. Quantitative interaction proteomics using mass spectrometry. Nat. Methods. 2009;6:203–205. doi: 10.1038/nmeth.1302. [DOI] [PubMed] [Google Scholar]
- 10.Ishihama Y, Oda Y, Tabata T, Sato T, Nagasu T, Rappsilber J, Mann M. Exponentially modified protein abundance index (emPAI) for estimation of absolute protein amount in proteomics by the number of sequenced peptides per protein. Mol. Cell. Proteomics. 2005;4:1265–1272. doi: 10.1074/mcp.M500061-MCP200. [DOI] [PubMed] [Google Scholar]
- 11.Schwanhausser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, Selbach M. Global quantification of mammalian gene expression control. Nature. 2011;473:337–342. doi: 10.1038/nature10098. [DOI] [PubMed] [Google Scholar]
- 12.Lu P, Vogel C, Wang R, Yao X, Marcotte EM. Absolute protein expression profiling estimates the relative contributions of transcriptional and translational regulation. Nat. Biotechnol. 2007;25:117–124. doi: 10.1038/nbt1270. [DOI] [PubMed] [Google Scholar]
- 13.Poser I, Sarov M, Hutchins JR, Heriche JK, Toyoda Y, Pozniakovsky A, Weigl D, Nitzsche A, Hegemann B, Bird AW, et al. BAC TransgeneOmics: a high-throughput method for exploration of protein function in mammals. Nat. Methods. 2008;5:409–415. doi: 10.1038/nmeth.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dignam JD, Lebovitz RM, Roeder RG. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 1983;11:1475–1489. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hubner NC, Mann M. Extracting gene function from protein-protein interactions using Quantitative BAC InteraCtomics (QUBIC) Methods. 2011;53:453–459. doi: 10.1016/j.ymeth.2010.12.016. [DOI] [PubMed] [Google Scholar]
- 16.Rappsilber J, Mann M, Ishihama Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2007;2:1896–1906. doi: 10.1038/nprot.2007.261. [DOI] [PubMed] [Google Scholar]
- 17.Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 18.Arike L, Valgepea K, Peil L, Nahku R, Adamberg K, Vilu R. Comparison and applications of label-free absolute proteome quantification methods on Escherichia coli. J. Proteomics. 2012;75:5437–5448. doi: 10.1016/j.jprot.2012.06.020. [DOI] [PubMed] [Google Scholar]
- 19.Grote M, Wolf E, Will CL, Lemm I, Agafonov DE, Schomburg A, Fischle W, Urlaub H, Luhrmann R. Molecular architecture of the human Prp19/CDC5L complex. Mol. Cell. Biol. 2010;30:2105–2119. doi: 10.1128/MCB.01505-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lai AY, Wade PA. Cancer biology and NuRD: a multifaceted chromatin remodelling complex. Nat. Rev. Cancer. 2011;11:588–596. doi: 10.1038/nrc3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Le Guezennec X, Vermeulen M, Brinkman AB, Hoeijmakers WA, Cohen A, Lasonder E, Stunnenberg HG. MBD2/NuRD and MBD3/NuRD, two distinct complexes with different biochemical and functional properties. Mol. Cell. Biol. 2006;26:843–851. doi: 10.1128/MCB.26.3.843-851.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Spruijt CG, Bartels SJ, Brinkman AB, Tjeertes JV, Poser I, Stunnenberg HG, Vermeulen M. CDK2AP1/DOC-1 is a bona fide subunit of the Mi-2/NuRD complex. Mol. Biosyst. 2010;6:1700–1706. doi: 10.1039/c004108d. [DOI] [PubMed] [Google Scholar]
- 23.Ertekin A, Aramini JM, Rossi P, Leonard PG, Janjua H, Xiao R, Maglaqui M, Lee HW, Prestegard JH, Montelione GT. Human cyclin dependent kinase 2 associated protein 1 is dimeric in its disulfide-reduced state, with natively disordered N-terminal region. J. Biol. Chem. 2012;287:16541–16549. doi: 10.1074/jbc.M112.343863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469:343–349. doi: 10.1038/nature09784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mousson F, Kolkman A, Pijnappel WW, Timmers HT, Heck AJ. Quantitative proteomics reveals regulation of dynamic components within TATA-binding protein (TBP) transcription complexes. Mol. Cell. Proteomics. 2008;7:845–852. doi: 10.1074/mcp.M700306-MCP200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.