


Abstract
Protein complexes directing messenger RNA (mRNA) degradation are present in all kingdoms of life. In Escherichia coli, mRNA degradation is performed by an RNA degradosome organized by the major ribonuclease RNase E. In bacteria lacking RNase E, the existence of a functional RNA degradosome is still an open question. Here, we report that in the bacterial pathogen Helicobacter pylori, RNA degradation is directed by a minimal RNA degradosome consisting of Hp-RNase J and the only DExD-box RNA helicase of H. pylori, RhpA. We show that the protein complex promotes faster degradation of double-stranded RNA in vitro in comparison with Hp-RNase J alone. The ATPase activity of RhpA is stimulated in the presence of Hp-RNase J, demonstrating that the catalytic capacity of both partners is enhanced upon interaction. Remarkably, both proteins are associated with translating ribosomes and not with individual 30S and 50S subunits. Moreover, Hp-RNase J is not recruited to ribosomes to perform rRNA maturation. Together, our findings imply that in H. pylori, the mRNA-degrading machinery is associated with the translation apparatus, a situation till now thought to be restricted to eukaryotes and archaea.
INTRODUCTION
The interplay between messenger RNA (mRNA) translation and degradation has been recognized as one of the key determinants in the control of gene expression. Although the translational machinery is highly conserved in bacteria, the regulation of mRNA stability involves diverse mechanisms. The well-established Escherichia coli paradigm in which the endoribonuclease RNase E plays a major role in RNA processing and degradation [reviewed in (1)] has been recently supplemented by new insights into the RNA maturation and decay mechanisms in a Gram-positive bacterium, Bacillus subtilis (2). RNase E is not conserved in B. subtilis and is functionally replaced by two essential ribonucleases RNase J1 and RNase Y. Both enzymes were first characterized as endoribonucleases specific for single-stranded RNA (ssRNA) (3,4) and orthologs of these proteins are widely distributed in bacteria. However, RNase J1 was later shown to also have 5′–3′ exoribonuclease activity (5), previously thought to be restricted to eukaryotes. RNase J1 thus combines two modes of RNA degradation, endo- and exonucleolytic, in one enzyme (6,7).
In E. coli and some other bacteria, RNase E operates in a complex of enzymes termed the RNA degradosome [reviewed in (8)]. Functional studies of the E. coli degradosome demonstrated that one of its major components, the RNA-helicase RhlB, facilitates degradation of structured RNA by RNase E and by the 3′–5′ exoribonuclease polynucleotide phosphorylase (PNPase). As RNase E and PNPase are ssRNA-specific endo- and exoribonucleases, respectively, the combination of their activities with the double-stranded RNA (dsRNA) unwinding activity of an RNA helicase provides an efficient means of eliminating mRNAs destined for degradation. However, despite extensive studies and the reported importance of the 5′-end structure and phosphorylation status for mRNA stability [for a review, see (1)], events leading to the initiation of mRNA cleavage in E. coli remain obscure. For a subset of mRNAs, it has been demonstrated that cessation of translation decreases the mRNA stability [reviewed in (9)], suggesting a close link between translation and mRNA degradation in some cases. However, E. coli RNase E has been found to be localized primarily to the inner membrane (10), leading the authors to propose compartmentalized RNA processing and degradation and leaving open the question of a possible direct coordination between translation and mRNA decay in bacteria.
Although the function and localization of the RNase E-based degradosome have been extensively studied in E. coli, no evidence for a functional degradosome in bacteria lacking RNase E has been provided so far. Recently, contradictory results on proteins interacting with B. subtilis RNases have been reported. It was shown that RNase Y physically interacts with CshA, one of the four RNA helicases of B. subtilis, but this interaction was exclusively observed after formaldehyde-mediated cross-linking of proteins (11). This suggested the existence of an RNase Y-based degradosome in B. subtilis. A previous bacterial two-hybrid analysis by the same group proposed the additional association of PNPase, RNase J1 and its paralog J2, enolase and phosphofructokinase in an RNase Y-based degradosome complex (12). Using bacterial two-hybrid, a similar degradosome was reported in Staphylococcus aureus (13). In both studies, indications for in vivo interactions under non-cross-linked conditions were missing and the functional relevance of such interactions has not been explored.
The involvement of the B. subtilis RNase J1 and J2 in a degradosome complex has been challenged by another study in which it appears that RNase J1 of B. subtilis forms a complex exclusively with its paralog RNase J2 (14). Moreover, the association of RNase Y with RNase J1 and J2 is difficult to reconcile with the data on subcellular localization of these two proteins, where RNase J1 is cytoplasmic and RNase Y is associated with the cellular membrane (15). Thus, no evidence for a functional RNase J-based degradosome was available when we started our study.
In the present work, we addressed the issue of a functional RNase J-based degradosome and its cellular localization in the gastric pathogen Helicobacter pylori, a Gram-negative epsilon-proteobacterium. H. pylori is a major pathogen that exclusively colonizes the human stomach and is found in half of the world’s population (16). Infection by H. pylori is associated with the development of gastritis, peptic ulcers and adenocarcinoma (17). Helicobacter pylori survival in the acidic environment of the stomach relies on its major virulence factor, urease (18) that catalyzes the hydrolysis of urea to the buffering compounds, ammonia and carbon dioxide (19). Consistent with its restricted gastric niche, H. pylori has a small genome of 1.6 Mb and a reduced functional redundancy (20). Compared with E. coli and B. subtilis (2.5-fold larger genomes), each possessing 17 identified RNases, H. pylori presents a remarkably low number of predicted conserved RNases. Its genome carries no gene encoding an ortholog of RNase E, but contains ORFs coding for a predicted RNase J [hp1430 in strain 26695, (20)], RNase Y (hp0760), as well as six other RNases (RNase III, RNase P, RNase R, PNPase, RNase HI and HII). Moreover, H. pylori has only one predicted DExD-box RNA helicase (hp0247), whereas E. coli and B. subtilis have five and four RNA helicases of this family, respectively. Several studies evoked post-transcriptional regulation of gene expression in H. pylori, in particular, in its complex response to acidity (21–23) but surprisingly, this aspect of gene regulation has never been explored in H. pylori. It was suggested that the urease operon is regulated post-transcriptionally but no further analysis of such regulation was reported (24). Thus, given the low redundancy in the available enzymatic activities related to the control of mRNA decay, H. pylori is an excellent model in which to study mechanisms of post-transcriptional regulation of gene expression.
Here we present results showing that the only RNA helicase of H. pylori (RhpA) forms a complex with Hp-RNase J in vivo suggesting the existence of a novel type of RNA degradosome. We demonstrate functional significance of this interaction by in vitro activity tests for both partners. Most importantly, the two proteins co-localize to translating ribosomes, which taking into account that Hp-RNase J does not mature ribosomal RNA, suggests that the mRNA-degrading machinery in H. pylori is coupled with translation.
MATERIALS AND METHODS
Bacterial strains
Helicobacter pylori strain 26695 (20) was used as in previous studies (25) for tandem affinity purification (TAP) taking Hp-RNase J and RhpA as baits (Supplementary Table S1). For all other experiments, we used H. pylori strain B128 (26) in which the inducible plasmids could be introduced and stably maintained (Supplementary Table S1). For the details of bacterial growth conditions, see Supplementary Methods. Plasmids expressing the individual hexahistidine-tagged versions of Hp-RNase J, ΔN-Hp-RNase J, and RhpA were introduced into E. coli strain BL21-CodonPlus®-RIL (Stratagene) (Supplementary Tables S1 and S2). Escherichia coli strain BL21-AI™ (Invitrogen) was used as a recipient for the co-expression of hexahistidine-tagged full-length or truncated Hp-RNase J together with GST-RhpA.
Construction of plasmids and strains
Plasmids pILL2301 and pPH132 carrying around 500-bp 3′-end sequences of rnj and rhpA fused to the TAP tag sequence and the corresponding downstream regions of rnj and rhpA were constructed as in (25). These plasmids were used to obtain strains UPH298 and UPH691 expressing from the chromosomes Hp-RNase J-TAP and RhpA-TAP, respectively, fused to the TAP tag at their C-terminus. For details and primer sequences, see Supplementary Methods and Supplementary Table S3. Plasmids and corresponding strains used to produce H. pylori proteins in E. coli are provided in Supplementary Tables S1 and S2.
Plasmids replicating in H. pylori were derived from the E. coli/H. pylori shuttle vector pILL2157 (27) (for details, see Supplementary Data). To construct the H. pylori strains allowing the controlled expression of Hp-RNase J or ΔN-RNase J, the full-length or truncated rnj gene was cloned into pILL2157 under the control of an IPTG-inducible promoter resulting in plasmids pPH134 or pPH135, respectively. They were introduced into H. pylori strain B128 by mobilization, as described by Backert et al. (28). Subsequently, the chromosomal copy of rnj was replaced by a non-polar kanamycin resistance cassette using plasmid pPH138 [as in (29)] generating strains UPH738 and UPH739. For details of pPH138 construction and for construction of ΔrhpA mutant (UPH740) see Supplementary Data.
Tandem affinity purification
Two liters of cultures (OD 0.5–0.8) of strains UPH298 and UPH691 that express TAP-tagged Hp-RNase J and TAP-tagged RhpA, respectively, were used for TAP. The protocol was essentially the same as in (25) with the exception of the cell lysis step, which was as follows. Bacteria were pelleted, resuspended in 10 ml of lysis buffer (100 mM HEPES pH 7.4, 100 mM KCl, 8% glycerol), centrifuged and the pellet was resuspended in a minimal volume of lysis buffer containing Complete® Protease Inhibitor Cocktail (Roche). Suspensions were frozen in drops in liquid nitrogen and stored at −80°C. Cells were lysed by grinding the frozen drops in a cryo-grinder (Freezer/Mill® 6770 SPEX SamplePrep) and the powder was used for the TAP of the tagged proteins as in previous study (25). Purified proteins were Trichloroacetic acid (TCA)-precipitated and separated by PAGE on a gradient gel (BioRad). Finally, each protein band was extracted from the gel and analyzed by Matrix-assisted laser desorption/ionization (MALDI) Mass spectrometry following the procedure described by Stingl et al. (25).
Purification of recombinant proteins from E. coli
The details of the purification of individual His-tagged H. pylori proteins are provided in Supplementary Data. Proteins were concentrated by ultrafiltration when appropriate and stored in aliquots at −80°C. Purified His-Hp-RNase J and His-RhpA were also used to produce antibodies, that were subsequently purified.
Co-purification of Hp-RNase J and RhpA from E. coli on glutathione Sepharose or Ni2+-NTA columns was performed from 100 ml of induced cultures as described in Supplementary Data. Purified proteins were concentrated and stored in 10% glycerol at −80°C.
Helicobactor pylori total RNA preparation and northern blotting
Total RNA was extracted from 10 ml of H. pylori cultures at OD600 0.5–0.7 by three cycles of phenol/chloroform extraction. For details, see Supplementary Data.
For northern blotting, 3 µg of RNA were separated on an agarose–formaldehyde gel and transferred to Hybond N+ (Amersham Biosciences) membrane by passive transfer overnight in 10x saline-sodium citrate (SSC) pH 7 buffer. Transferred RNA was fixed to the membrane by UV irradiation for 2 min. The membrane was blocked for 45 min at 65°C with ULTRAhyb® Hybridization Buffer (Ambion), then 5 µl of 33P-labeled riboprobe were added and the membrane was further incubated for 45 min at the same temperature. After three washes for 10 min at 65°C with 2x SSC 0.2 % SDS, the membrane was exposed to a phosphorimager screen (KODAK) and scanned with Pharos FX Molecular Imager (Bio-Rad).
In vitro transcription
ureI and 5S riboprobes used for northern blotting and seRNA72 and asRNA66 substrates for the Hp-RNase J activity tests were generated by T7 RNA polymerase from PCR templates as described previously (30). For primers, see Supplementary Table S3. seRNA72 DNA template was transcribed in vitro in the presence of α-[33P]-UTP (Perkin Elmer) and with 8-fold excess of Guanosine monophosphate (GMP) over Guanosine triphosphate (GTP) to obtain uniformly labeled RNA that was 5′-monophosphorylated (30). The asRNA66 was 5′-triphosphorylated and unlabeled.
Primer extension experiment
Primer extension assay for rRNAs was performed as described previously (31). Primers used in the reverse transcription and sequencing PCR reactions were labeled with 6-carboxyfluorescein (FAM). The 5′-end sequencing of rRNAs was performed with Thermo Sequenase™ Cycle Sequencing Kit (USB) using PCR fragments covering ∼100 bp of the predicted 5′-ends of rRNA and ∼300 bp of the upstream regions (for primers, see Supplementary Table S3). Resulting fluorescently labeled DNA products of PCR and reverse transcription reactions were separated on 5% acrylamide 7M urea sequencing gel and visualized with Pharos FX Molecular Imager (Bio-Rad).
Hp-RNase J activity tests
The RNA substrate chosen for the Hp-RNase J in vitro activity tests was (i) an RNA fragment comprising 72 nt at the 5′-end of rnj (seRNA72) and (ii) its antisense counterpart (asRNA66) that was complementary to 66 nt of seRNA72. To obtain dsRNA, seRNA72 was mixed with a slight excess of asRNA66 (in 1:1.3 ratio), heated for 5 min at 90°C and chilled on ice. To reconstitute the Hp-RNase J–RhpA complex, equimolar amounts of each enzyme (measured per monomer) were mixed and incubated at 37°C for 5 min. Five pmol of RNA was incubated at 37°C with 3.5 pmol of Hp-RNase J or of the Hp-RNase J–RhpA complex comprising 3.5 pmol of each protein, in a final volume of 5 µl with 20 mM HEPES pH 7.8, 8 mM MgCl2, 100 mM NaCl, 0.1 mM DTT and 3 mM ATP during the indicated time points. Reactions were stopped by the addition of 5 µl RNA sample loading buffer (SIGMA) and loaded on a 5% acrylamide/7 M urea gel.
ATPase activity tests for RhpA
The ATPase activity of RhpA was assayed using a pyruvate kinase–lactate dehydrogenase method based on the measurement of NADH oxidation, coupled with ATP hydrolysis (32). For details see Supplementary Data. Yeast RNA (Ambion) was added to a final concentration of 100 µg/ml when indicated. Reactions were initiated by the addition of (i) 30 or 60 pmol of RhpA, (ii) an equimolar mixture of RhpA and Hp-RNase J or ΔN-Hp-RNase J (to final 0.06 or 0.12 µM of each protein) or (iii) the same amounts of Ded1, a yeast DEAD-box helicase as a control. NADH oxidation was monitored at 340 nm and ATP hydrolysis rate was calculated from linear NADH oxidation plots assuming NADH extinction coefficient of 6300 M−1 cm−1.
Polysome profile on sucrose gradient
A quantity of 250 ml of exponentially growing H. pylori cells were incubated at 4°C during 10 min with 100 µg/ml chloramphenicol to avoid polysome run-off. Then bacteria were pelleted and washed with 10 ml breaking buffer (10 mM Tris–HCl pH 7.4, 50 mM KCl, 7.5 mM MgCl2), containing 100 µg/ml chloramphenicol and Complete® Protease Inhibitor Cocktail (Roche). The breaking buffer used in the ‘low magnesium’ experiment contained 10 mM Tris–HCl pH 7.4, 50 mM KCl, 1 mM MgCl2 and 20 mM EDTA. The pellet was resuspended in 200 µl breaking buffer and cells were lysed by vortexing with beads in FastPrep Instrument (MPbio). The extract was cleared by centrifugation and the supernatant (corresponding to 20 OD 260 nm) was layered on a 10–50% or 10–40 % sucrose gradient. After centrifugation at 4°C at 190 000g for 2 h and 45 min in SW41-Ti rotor, fractions were collected with ISCO fraction collector (Pharmacia). The absorbance at 254 nm was monitored with a Dual Path UV-2 spectrophotometer (Pharmacia). Proteins were precipitated with TCA and loaded on 10% acrylamide gels for western blotting revealed with Hp-RNase J and RhpA-specific antibodies. After protein transfer, amido-black staining of the membranes served us to verify that the loading was comparable for the different strains analyzed: wild-type, ΔrhpA and RNase J-depleted mutants.
Western blotting
Western blotting was performed as described before (29). Polyclonal rabbit antibodies specific to Hp-RNase J and RhpA were used at a 1/5000 dilution. Secondary goat anti-rabbit–Horse-Raddish-Peroxidase conjugate antibodies (SIGMA) were used at a 1/10 000 dilution. Blots were revealed with ECL SuperSignal® West Femto kit (Thermo Scientific) and visualized with ChemiDoc (Bio-Rad).
RESULTS
Essentiality of Hp-RNase J for H. pylori growth and construction of conditional mutants
The available genomes of H. pylori and the other Helicobacter species of the epsilon proteobacteria carry a single gene predicted to code for an ortholog of RNase J. This contrasts with B. subtilis and the majority of Firmicutes that express at least two paralogs, RNase J1 and J2. In H. pylori strain B128 (26), gene hpB128_16g80 encodes a protein of 77.5 kDa that presents 39 and 34% identity with RNase J1 and RNase J2 of B. subtilis, respectively (Figure 1 and Supplementary Figure S1). Similar to RNases J1 and J2, it belongs to the β-CASP family of metallo-β-lactamases. Importantly, all functionally significant amino acids that contribute to zinc ion and phosphate coordination in RNase J1 (33) are conserved in HPB128_16g80, suggesting that this protein is a ribonuclease with properties similar to RNase J1 of B. subtilis. Therefore, we will refer to it as H. pylori RNase J (Hp-RNase J) and to the corresponding gene as rnj. An interesting feature of Hp-RNase J is a 132 amino acid N-terminal extension, conserved in the H. pylori species, but not in its Firmicutes orthologs (Figure 1 and Supplementary Figure S1) and for which we found no homologous structures using the HHpred algorithm. It is present in RNase J of all the available sequenced Helicobacter species, although with limited sequence similarities.
Figure 1.

RNase J depletion in H. pylori strain B128. (A) Schematic representation of the RNase J proteins from B. subtilis and H. pylori. Catalytic domains are shown with hatched boxes; the N-terminal 132 amino acid extension characteristic of Hp-RNase J is represented by a gray box. (B) Growth curves of Hp-RNase J and ΔN-Hp-RNase J depleted and induced strains. Strains UPH738 and UPH739 with controlled expression of rnj (Pi-rnj) or ΔN-rnj (Pi-ΔN-rnj) were grown in the presence (+IPTG) or the absence (−IPTG) of the inducer, wild-type B128 was used as a control for normal growth. (C) Western blot showing relative levels of Hp-RNase J and ΔN-Hp-RNase J in the depleted and induced strains. Cell extracts were prepared from H. pylori cultures at the OD600 0.5–0.7. Equivalents of 0.125 OD600 were loaded on two gels one of which was stained with coomassie and served as a loading control (lower panel). Upper panel shows western blot revealed with anti-Hp-RNase J specific antibodies.
Our efforts to delete rnj in several H. pylori strains proved unsuccessful, suggesting that Hp-RNase J is essential. Therefore, we constructed an H. pylori strain derived from B128 that carried a stable plasmid expressing Hp-RNase J under the control of an Isopropyl β-D-1-thiogalactopyranoside (IPTG)-inducible promoter and in which the chromosomal rnj copy was deleted (strain UPH738, Supplementary Table S1). In the absence of the inducer, the constructed strain showed only slight growth retardation, the residual growth being due to the leakiness of ureI promoter used for conditional expression (27) (Figure 1B). To explore the role of the 132 amino acids-long N-terminal extension of Hp-RNase J, the same strategy was used to construct a strain expressing Hp-RNase J deleted of the corresponding N-terminal region, ΔN-Hp-RNase J (Figure 1 and Supplementary Figure S1) resulting in strain UPH739 (Supplementary Table S1). In the absence of the inducer, the growth defect of this strain was significantly stronger than that of the strain depleted for the full-length protein (Figure 1B). Western blotting with Hp-RNase J-specific antibodies showed that the Hp-RNase J levels were decreased ∼3-fold in strain UPH738 grown without the inducer compared with the wild-type strain (Figure 1C). The truncated ΔN-Hp-RNase J protein was barely detected in the induced strain UPH739, consequently, we were not able to detect it under the conditions of depletion (Figure 1C). This suggests that the deletion of the N-terminus compromises the stability of Hp-RNase J in vivo. A band of lower Molecular weight (MW) was detected by western blot for extracts of strains expressing either the wild type or the ΔN-Hp-RNase J proteins, suggesting the existence of a post-translational modification of these proteins.
Taken together, our results strongly suggest that the Hp-RNase J is an essential enzyme and that the deletion of its N-terminus compromises its stability in vivo. To further analyze the Hp-RNase J targets, we obtained two conditional mutants allowing the depletion of the Hp-RNase J level, the mutant depleted of the truncated protein being strongly deficient in Hp-RNase J amounts and hence in overall RNase J activity.
Hp-RNase J controls the degradation of the ureI transcript and is not required for the final maturation of ribosomal RNAs
To find targets of Hp-RNase J in vivo, we chose to examine the mRNA of ureI that is one of the accessory genes of the urease operon. The ureI gene encodes a membrane channel responsible for the import of urea (34,35). It was previously proposed that the ureI transcript level is regulated by an unknown RNase (24). Therefore, we judged it to be a good candidate for a Hp-RNase J target. Total RNA was isolated from the Hp-RNase J and ΔN-Hp-RNase J-depleted strains at defined time intervals after addition of the transcription inhibitor rifampicin. Northern blotting with an ureI riboprobe showed that in the wild-type strain bearing an empty vector, the ureI transcript half-life was comparable with the one reported previously (Figure 2A, upper panel) (24). Depletion of the full-length Hp-RNase J had an indirect positive transcriptional effect and led to a 2-fold increase in the transcript half-life in comparison with the induced strain or the wild-type strain. However, the ureI mRNA was stabilized to a much greater extent in the ΔN-Hp-RNase J-depleted strain in comparison with the wild-type strain. This indicates that Hp-RNase J is involved in the decay of the ureI transcript in vivo and confirms that the RNase J amounts and overall activity is sufficiently decreased in the ΔN-Hp-RNase J-depleted strain to look for other bona fide targets (manuscript in preparation).
Figure 2.

RNase J activity in vivo. (A) Northern blot of total RNA isolated from the same cell cultures as in Figure 1. Three µg of total RNA was resolved in a 1% agarose-formaldehyde gel, transferred to a HYBOND N+ membrane (GE Healthcare) and hybridized with a ureI RNA probe uniformly labeled with 33P. mRNA half-lives are given under the northern blot. The 5S RNA probe served as a loading control (lower panel). (B) Primer extension of rRNA in the ΔN-Hp-RNase J-depleted and induced strain (− or + IPTG, respectively) compared with the wild-type. Total RNA were prepared from the same cultures as in Figure 1B, at OD 0.5–0.7. Sequencing reactions and 5′-end mapping for each rRNA are shown to the left of each panel.
We wondered whether Hp-RNase J was also required for the final maturation of ribosomal RNAs (rRNAs), as was shown for its examined bacterial orthologs (36–38). Primer extension analysis of rRNAs isolated from the strains depleted of the truncated Hp-RNase J was performed and compared with the wild-type and the strain in which ΔN-Hp-RNase J was induced (Figure 2B). No difference in the 5′-end lengths of any of the three rRNAs was observed in the strain depleted of truncated Hp-RNase J compared with the wild-type or induced strain. This shows that Hp-RNase J is not involved in the final maturation of the 5′-ends of rRNA in H. pylori.
Hp-RNase J and DExD-box RNA helicase RhpA are reciprocally co-purified from H. pylori by tandem affinity purification
In an attempt to find interacting protein partners of Hp-RNase J, we used the tandem affinity purification (TAP) approach that we had established in H. pylori (25). A tag sequence encoding protein A and the calmodulin-binding domain was introduced in frame at the 3′-end of the chromosomal copy of rnj, ensuring physiological expression levels of the tagged Hp-RNase J in H. pylori. The resulting strain displayed no growth retardation, which, given the essential nature of Hp-RNase J, indicated that the tag did not interfere with the function of the ribonuclease. After affinity purification in two steps under native conditions, the purified proteins were identified by mass-spectrometry (Supplementary Table S4). Interestingly, the only DExD-box RNA-helicase of H. pylori (HP0247 for strain 26695) was reproducibly detected among the co-purified proteins suggesting that the two proteins interact in vivo (Figure 3A). HP0247 shares about 40% sequence identity with each of the E. coli RNA helicases and 46% with B. subtilis RNA helicase CshA (YdbR). Motifs involved in RNA binding and ATP hydrolysis are conserved in HP0247 (Supplementary Figure S2), therefore this protein was designated RhpA for RNA-helicase of H. pylori.
Figure 3.

Tandem affinity purification of (A) Hp-RNase J and (B) DExD-box RNA helicase (RhpA) of H. pylori. Major associated proteins are indicated. The complete lists of co-purified proteins and mass-spectrometry data are presented in Supplementary Tables S4 and S5.
In addition to the recurring contaminants, TAP of Hp-RNase J also revealed interactions with the β–β′ subunit of RNA polymerase and with 15 ribosomal proteins that exceeded the expected level of contamination that we have previously seen for H. pylori TAP [see Supplementary Data of (25)]. This pointed to the possibility of an association of Hp-RNase J with ribosomes that we investigated further.
To confirm the in vivo interaction of RhpA with Hp-RNase J, we examined whether Hp-RNase J co-purified from H. pylori when the helicase is used as a bait. An H. pylori strain expressing a chromosomal tagged version of RhpA was constructed and used for TAP. Mass-spectrometry identification of the purified proteins showed that Hp-RNase J was reproducibly co-purified with RhpA (Figure 3B and Supplementary Table S5). As for Hp-RNase J, 11 ribosomal proteins were associated with this purified complex. It is worth noting that in both TAP experiments, the partner was purified in sub-stoichiometric amounts, a situation often observed with TAP, except for very stable complexes. Interestingly, neither RNase Y (HP0760) nor PNPase (HP1213) were detected in Hp-RNase J or RhpA TAP experiments. Thus, the reciprocal co-purification of Hp-RNase J and RhpA by TAP strongly suggests that the two proteins interact in H. pylori in vivo.
Hp-RNase J and RhpA co-purify as a complex when co-expressed in E. coli
Escherichia coli expression system was used to further characterize the interaction between Hp-RNase J and RhpA and to evaluate the importance of the N-terminal 132 amino acid extension of Hp-RNase J in this interaction. The following recombinant proteins were all tagged at the N-terminus and expressed in E. coli either alone or in pairs: RhpA tagged with glutathione S-transferase (GST), GST-RhpA full-length Hp-RNase J and its truncated version—with hexahistidine (His-Hp-RNase J and His-ΔN-Hp-RNase J, respectively).
When a glutathione column was used for purification of extracts from an E. coli strain co-expressing GST-RhpA and the full-length His-Hp-RNase J, two major protein bands were obtained corresponding to RhpA (as expected) and its interaction partner Hp-RNase J as identified by mass-spectrometry (Figure 4A, panel 1 and Supplementary Table S6). When a Ni2+-NTA column was used for purification of His-Hp-RNase J from the same strain, the same two major proteins corresponding to Hp-RNase J (as expected) and RhpA were purified (Figure 4B, panel 1). Control purification of His-Hp-RNase J on a glutathione column and of GST-RhpA on a Ni2+-NTA column demonstrated that protein co-purification was a consequence of complex formation rather than non-specific interaction of the associated proteins with the column matrices (Figure 4A and B, panels 3). Co-expression and glutathione column purification of Hp-RNase J with GST alone showed that the GST tag does not interfere with the co-purification (Figure 4A, panel 4). Treatment of the extracts with RNase A did not affect the co-purification of the two proteins either, suggesting that their interaction is direct and is not mediated by RNA (Supplementary Figure S3). Interestingly, the N-terminal extension of Hp-RNase J appeared to be non-essential for the complex formation as ΔN-Hp-RNase J was co-purified with RhpA and vice versa on the glutathione and Ni2+-NTA columns, respectively (Figure 4A and B, panels 2). DnaK and GroEL protein chaperons of E. coli (marked with asterisks) are contaminants that co-purify with the GST-RhpA/ΔN-Hp-RNase J complex (Figure 4A, panels 2 and Supplementary Table S6). In summary, when co-expressed in E. coli, Hp-RNase J and RhpA form a complex that can be purified using a tag on either of the two proteins, confirming that they are physically associated. The N-terminal extension of Hp-RNase J seems to be dispensable for this interaction.
Figure 4.

Co-expression in E. coli and purification of GST-RhpA and His-tagged Hp-RNase J. GST-RhpA (filled triangle) and His-tagged full-length or truncated Hp-RNase J (empty triangle) were co-expressed in E. coli and purified on (A) glutathione column and (B) Ni2+-NTA column. Panel 1 (A and B)—strain UPH574 expressing GST-RhpA + His-Hp-RNase J (RNJ); panel 2 (A and B) —strain UPH598 expressing GST-RhpA + His-ΔN-Hp-RNase J; panel A3—strain UPH600 expressing His-Hp-RNase J alone, panel B3—strain UPH597 expressing GST-RhpA alone; panel A4—strain UPH780 expressing GST and His-Hp-RNase J. The single asterisk and double asterisk denote DnaK and GroEL, respectively. Strains are listed in Supplementary Table S1. Mass spectrometry data are presented in Supplementary Table S6. CL, cleared lysate; P, purified protein.
RhpA assists Hp-RNase J in the degradation of dsRNA in vitro
To explore the functionality of the minimal RNA degradosome complex, we tested whether the enzymatic activity of each of the two proteins is enhanced in the complex in comparison with the individual proteins. First, we tested whether RhpA assists Hp-RNase J in the degradation of dsRNA in vitro. Hp-RNase J activity assay was designed to estimate the rate of dsRNA degradation by the reconstituted Hp-RNase J/RhpA complex compared with Hp-RNase J alone. The substrate of Hp-RNase J was an RNA fragment corresponding to 72 nt at the 5′-end of the rnj coding region (sense RNA72 or seRNA72). It was synthesized by in vitro transcription with T7 RNA polymerase and was uniformly labeled with α-33P-UTP. 5′-monophosphate was introduced to facilitate seRNA72 degradation by RNase J in exonucleolytic mode by analogy to its B. subtilis ortholog (5). Its antisense counterpart (asRNA66) was an unlabeled 66-nt RNA that was shorter than the sense RNA to leave an overhang of 6 nt at the 5′-end of seRNA72 in the RNA duplex (Figure 5A). This overhang was conceived to serve as a ramp to improve the initiation of the dsRNA degradation by Hp-RNase J and to ensure easier access of RhpA to its dsRNA substrate as reported for some other RNA helicases. The dsRNA was obtained by hybridizing seRNA72 with a slight excess of asRNA66 to avoid remaining single-stranded seRNA72, this was confirmed by native acrylamide gel electrophoresis of the products.
Figure 5.

Effect of RhpA on the dsRNA-degrading activity of Hp-RNase J in vitro. Panel A—dsRNA design; seRNA72 was monophosphorylated and uniformly labeled with α-33P-UTP, asRNA66 was triphosphorylated and unlabeled. Panel B—Degradation of seRNA72 by Hp-RNase J (RNJ) in vitro. (I) seRNA72 was incubated with Hp-RNase J; (II) seRNA72/asRNA66 and Hp-RNase J; (III) seRNA72/asRNA66 and Hp-RNase J/RhpA complex; (IV) seRNA72/asRNA66 and RhpA; (V) seRNA72 and ΔN-Hp-RNase J. Reactions were incubated for indicated time points after the addition of the enzymes. −E: reactions incubated without enzyme. Reaction products were separated on a 5% acrylamide/7M urea gel.
As shown in Figure 5B, (I), purified recombinant Hp-RNase J rapidly degraded the seRNA72 substrate to mononucleotides without the accumulation of intermediates, thus acting in an exonucleolytic mode. Recombinant Hp-RNase J was also able to cleave a structured RNA substrate, the tmRNA precursor, in an endonucleolytic mode (unpublished data). This indicates that Hp-RNase J acts both as an endo- and exoribonuclease as its B. subtilis ortholog (3,5).
After hybridization to asRNA66, seRNA72 was degraded at a slower rate when incubated with Hp-RNase J alone [Figure 5B (II)]. Partial RNA degradation in this case, as judged by the accumulation of UMP, is probably due to the minor RNA-unwinding activity of Hp-RNase J itself. In contrast, seRNA72 was rapidly degraded by the reconstituted Hp-RNase J/RhpA complex [Figure 5B (III)]. As a control, RhpA alone did not degrade seRNA72/asRNA66 duplex [Figure 5B (IV)]. ΔN-Hp-RNase J presented a dramatic decrease of activity [Figure 5B, compare (V) with (I)] confirming the absence of contaminating RNase in the protein preparations and suggesting that the N-terminal extension is important for the full activity of Hp-RNase J.
These experiments show that RhpA assists Hp-RNase J in the degradation of dsRNA in vitro. This is consistent with the predicted RNA helicase activity of RhpA that unwinds the dsRNA helix to give RNase J a direct access to its ssRNA substrate. Therefore, the above observations provide an in vitro demonstration of the importance of the concerted action of Hp-RNase J and RhpA for the degradation of structured RNA.
The ATPase activity of RhpA is stimulated upon interaction with the full-length and truncated Hp-RNase J
DExD-box RNA helicases are known to have RNA-stimulated ATPase activity and are thought to use the free energy of ATP hydrolysis in the recycling process during unwinding of structured RNA. This ATPase activity has been shown in some cases to be stimulated by the interaction with protein partners (8,39). Therefore, we examined the impact of the interaction of Hp-RNase J with RhpA on the ATPase activity of RhpA. The in vitro ATPase activity of RhpA was assayed either without or with purified recombinant His-Hp-RNase J or His-ΔN-Hp-RNase J.
Interestingly, RhpA displayed only background ATPase activity alone even in the presence of RNA (Table 1), while it was stimulated 5- to 6-fold when incubated with either Hp-RNase J or ΔN-RNase J. BSA was used to ensure that the ATPase activity of RhpA was independent of the protein environment and the Ded1 helicase of yeast was used as a positive control of the assay. Important controls included tests with Hp-RNase J and ΔN-Hp-RNase J alone confirming the absence of contaminating ATPase activity in the RNase preparations.
Table 1.
ATPase activity of RhpA in the presence or absence of Hp-RNase J
| Proteins | RNA, μg/ml |
|
|---|---|---|
| 0 | 100 | |
| RhpA | 0.47 ± 0.27* | 0.28 ± 0.28 |
| RhpA + BSA | nd | 0.35 ± 0.33 |
| RhpA + RNase J | 0.47 ± 0.34 | 2.81 ± 0.15 |
| RhpA + ΔN-RNaseJ | 0.90 ± 0.11 | 2.44 ± 0.81 |
| RNaseJ | nd | 0.57 ± 0.55 |
| ΔN-RNaseJ | nd | 0.20 ± 0.20 |
| Ded1 | 2.52 ± 0.14 | 18.12 ± 0.66 |
ATPase activity is presented in moles of ATP hydrolysed per minute per mole of the enzyme. Protein mixtures were at equimolar amounts. Total RNA from yeast (Ambion) was used at 100 µg/ml. Ded1 helicase from yeast was used as a reference. Mean values and standard deviations were calculated from at least two independent experiments. nd, non-determined.
Altogether, these results demonstrate that the interaction between RhpA and Hp-RNase J induces allosteric changes in RhpA that lead to the stimulation of the ATPase activity. Truncated Hp-RNase J is still able to activate RhpA, confirming that it is not impaired in its capacity to interact with RhpA and indicating that the N-terminal extension of Hp-RNase J is not involved in stimulating ATPase activity.
Hp-RNase J and RhpA co-localize to translating ribosomes
The TAP of Hp-RNase J and RhpA revealed numerous associated ribosomal proteins. Therefore, we examined whether this could be indicative of an association between the Hp-RNase J/RhpA complex and the ribosomes. Centrifugation of H. pylori cell extracts through a sucrose gradient resulted in typical sedimentation profiles (Figure 6A). Analysis of the collected fractions by western blotting showed that both Hp-RNase J and RhpA are present in the polysome fractions and that they are mostly co-localized in agreement with their physical interaction (Figure 6A). Most interestingly, they were not detected in the 30S and 50S fractions but rather in fractions corresponding to fully assembled 70S ribosomes and to polysomes. The amounts of RhpA were greater in 70S fraction in comparison with the polysomes, whereas Hp-RNase J was distributed rather evenly between 70S and polysome fractions. This indicates that only part of the ribosome-associated RhpA is involved in the interaction with Hp-RNase J and suggests that apart from being involved in mRNA degradation in association with Hp-RNase J, RhpA is also involved in other translation-related processes.
Figure 6.

Co-localization of RhpA and Hp-RNase J to the ribosomes. (A) Upper panel: polysome profile for H. pylori B128 cell extracts on a 10–50% sucrose gradient. Peaks corresponding to 30S, 50S subunits and 70S ribosomes are indicated. Numbers correspond to the collected fractions. Lower panel: western blot revealed with RhpA- and subsequently with Hp-RNase J-specific antibodies showing the distribution of RhpA and Hp-RNase J in the ribosomal fractions. Empty triangle: Hp-RNase J, filled triangle: RhpA. (B) Upper panel: polysome profile of H. pylori B128 cell extract on 10–40% sucrose gradient in the presence of 1 mM MgCl2 and 20 mM EDTA. Peaks corresponding to the dissociated 30S and 50S subunits are indicated. Lower panel: western blots showing the distribution of RhpA (filled triangle) and Hp-RNase J (empty triangle) in the collected fractions. (C) polysome profiles (10–50% sucrose gradient) and distribution of Hp-RNase J in the ΔrhpA mutant (strain UPH740). (D) Polysome profiles (10–40% sucrose gradient) and distribution of Hp-RNase J and of RhpA in the strain depleted of Hp-RNase J (UPH738, no IPTG).
To confirm the physical association of the complex with ribosomes, we performed cell lysis and sucrose gradient centrifugation in the presence of 1 mM MgCl2 and 20 mM EDTA (Figure 6B). The low magnesium concentration is known to lead to the dissociation of assembled ribosomes into individual subunits (40). Under these conditions, Hp-RNase J and RhpA were co-localized to isolated 30S and 50S subunits. This confirmed that both enzymes are physically associated with ribosomes and do not co-sediment independently of ribosomes in the sucrose gradients.
To find out whether Hp-RNase J and RhpA are associated with ribosomes independently of each other or require the presence of the other partner for the association, we performed sucrose gradient centrifugation with cellular extracts of ΔrhpA strain (UPH740) and the strain depleted of Hp-RNase J (UPH738). As shown in Figure 6C and D, neither rhpA deletion nor Hp-RNase J depletion had an impact on the association of the other partner with the ribosomes suggesting that both proteins interact with the ribosomes independently. Thus, Hp-RNase J/RhpA degradosome complex is localized to the translating ribosomes and both proteins seem to be associated with the ribosomes independently of each other.
Ribosomal localization of Bacillus subtilis RNase J1 differs from that of Hp-RNase J
Previous observations suggested that RNase J1 of B. subtilis (Bsu-RNase J1) co-localizes with ribosomes (15). However no data on its distribution in polysome gradients were available. Therefore, we performed 10–40 % sucrose gradient centrifugation with the lysate of B. subtilis strain W168 and determined the localization of Bsu-RNase J1 in the collected fractions by western blotting with anti-Bsu-RNase J1 antibodies. Bsu-RNase J1 was present in all ribosomal fractions: the 30S and 50S subunits, 70S ribosomes and polysomes (Figure 7A). In contrast, in the same 10–40% sucrose gradient of H. pylori extracts, Hp-RNase J was only present in the fractions corresponding to 70S ribosomes and polysomes (Figure 7B). Thus, whereas Bsu-RNase J1 is associated both with individual ribosomal subunits and translating ribosomes, Hp-RNase J is only associated with translating ribosomes.
Figure 7.
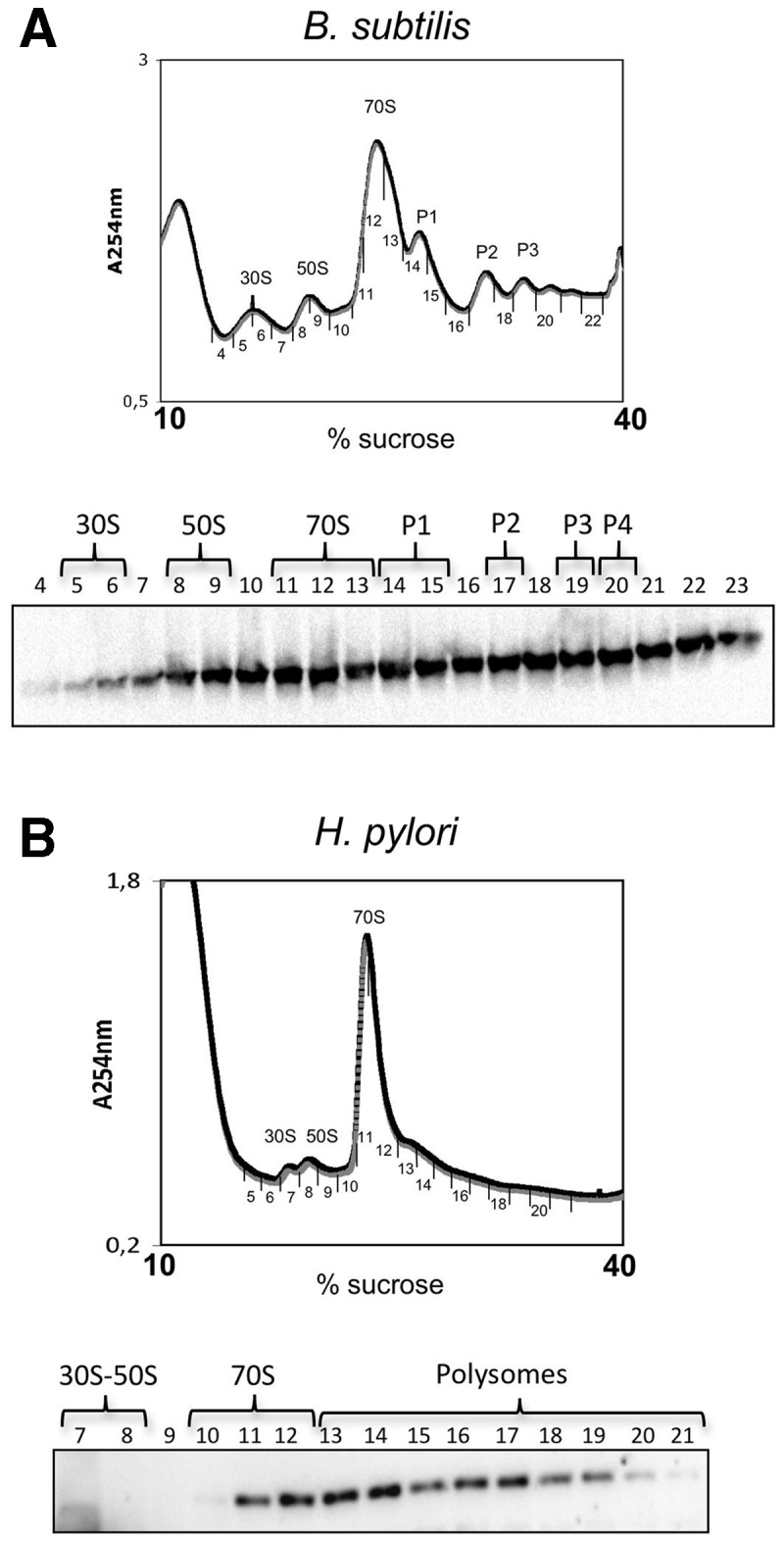
Comparison of RNase J distribution between ribosomal fractions in B. subtilis and H. pylori. (A) Upper panel: polysome profile of B. subtilis cell extracts on a 10–40% sucrose gradient. Peaks corresponding to ribosome fractions are indicated. Lower panel: western blot with Bsu-RNase J1-specific antibodies revealing the presence of Bsu-RNase J1 in the collected fractions. (B) Polysome profile of H. pylori cell extract on 10–40% sucrose gradient and distribution of Hp-RNase J in the collected fractions.
DISCUSSION
Since the discovery of the E. coli RNase E-based degradosome in 1994 (41,42), similar RNA-degrading protein complexes have been found in other bacterial species possessing RNase E (43–46). The functional significance of such RNA-degrading machineries lies in the association of the RNA decay-initiating endoribonuclease RNase E and the 3′–5′ directional exoribonuclease PNPase with an RNA helicase that unwinds structured RNAs to make them accessible for the degradation by RNase E and PNPase. Therefore, the association of endo-/exoribonuclease RNase J with an RNA helicase would result in a minimal complex that is highly efficient in mRNA degradation. We discovered the interaction between H. pylori RNase J and its only DExD-box RNA helicase RhpA using the TAP technique (25). After identification of RhpA among proteins that co-purified with Hp-RNase J, we were also able to detect RNase J when RhpA was used as a bait. The TAP procedure has been reported to give the least false positive results of all the methods for detection of protein–protein interactions, making it a highly reliable tool to discover protein complexes. Notably, this procedure does not include a protein cross-linking step. Therefore, our finding of a reciprocal interaction is a very strong indication of the existence of the Hp-RNase J/RhpA complex in H. pylori. As previously reported, the TAP procedure does not allow to precisely infer the stoichiometry of the proteins within the complex with the exception of very stable complexes, which is probably not the case here. However, co-purification of Hp-RNase J and RhpA from E. coli suggests a stoichiometry of the two proteins ranging from 1:1 to 2:1. Further experiments are needed to define this parameter in H. pylori.
Although an interaction between B. subtilis RNA helicase CshA and RNase Y was reported by Lehnik-Habrink et al. (11), we did not find RNase Y among the H. pylori proteins that co-purified with RhpA. Helicobacter pylori PNPase did not co-purify with RhpA either, suggesting that the helicase does not participate in other RNA-degrading complexes in H. pylori. Nevertheless, our results cannot strictly exclude that other minor components are part of the H. pylori degradosome. However, the fact that Hp-RNase J carries both endo- and exoribonuclease activities might explain the absence of PNPase in the H. pylori degradosome, the latter being present as an exoribonucleolytic partner of RNase E in the RNA degradosome of E. coli.
The E. coli RNA degradosome is organized by the C-terminal non-catalytic and non-structured part of RNase E (47,48) with each degradosome component (RNA helicase RhlB, PNPase and enolase) having a separate binding site on this scaffold. Whereas it is not essential for RNase E activity, the presence of the C-terminal half accelerates mRNA degradation by RNase E in vivo (49). An interesting feature of RNase J of H. pylori is its N-terminal extension of 132 amino acids that evokes the C-terminal degradosome scaffold of RNase E. However, in contrast to the C-terminal half of RNase E, which is naturally unstructured, the N-terminal extension of Hp-RNase J is likely to be folded into a globular tertiary structure as judged by GlobPlot structure prediction analysis. In addition, unlike the C-terminal half of RNase E, this domain of Hp-RNase J is essential for its activity in vitro, suggesting that either it contributes to the correct folding of the active site and/or carries RNA-binding activity. Finally, the N-terminal extension of Hp-RNase J does not participate in the interaction of Hp-RNase J with RhpA as demonstrated by (i) the co-purification of RhpA with both the full-length and truncated form of Hp-RNase J from E. coli and (ii) the induction of ATPase activity of RhpA by both forms of Hp-RNase J. This implies that the interaction between RhpA and Hp-RNase J occurs via a surface distant from the N-terminus of Hp-RNase J.
The induction of ATPase activity of RhpA by Hp-RNase J suggests that the two proteins form a complex that is functionally altered in comparison with the individual enzymes. This evokes the activation of RhlB of E. coli upon interaction with RNase E (47), and is remarkable as there is no sequence conservation between the two RNases. Therefore, the interaction between RhpA and Hp-RNase J seems to have different structural basis than that between RNase E and RhlB of E. coli. However, it results in a similar increase in the catalytic capacity of the complex and strongly supports the existence of a functional RNase J-based degradosome in H. pylori. This conclusion is also supported by in vitro RNase activity tests, which clearly demonstrate that dsRNA is degraded faster by the Hp-RNase J/RhpA complex than by Hp-RNase J alone. This suggests that RhpA assists Hp-RNase J in degradation of structured RNA by unwinding the RNA duplex and provides further evidence for mutual activation of catalytic capacities of the two proteins upon interaction.
The association of a 5′–3′ RNA-degrading activity with translating ribosomes has recently been demonstrated for yeast (50), and was suspected in bacteria (9). Our finding that RNase J and RhpA are co-localized to the polysomes is therefore a first demonstration of such an association in bacteria. First, it corroborates our data suggesting that the two proteins interact in vivo. Second, it suggests that the Hp-RNase J/RhpA degradosome plays a major role in mRNA decay in H. pylori in conjunction with translation.
Interestingly, both proteins seem to interact with ribosomes independently of each other and of mRNA, because the interaction is maintained after ribosome subunits dissociation in the presence of EDTA. Further experiments will be needed to identify the specific ribosomal proteins involved. It is possible that a fraction of RhpA interacts with ribosomes independently and another fraction is associated with them via Hp-RNase J, taking into account (i) the higher amounts of RhpA detected on 70S ribosome, as compared with those of Hp-RNase J in the same fractions and (ii) the decreased amounts of this protein in the RNase J-depleted strain compared with the wild-type. This raises the possibility that, apart from assisting Hp-RNase J in mRNA degradation, RhpA is also involved either in the final steps of ribosomal assembly or in translation initiation, as has been shown for some eukaryotic DEAD-box proteins (51,52). As RhpA is the only RNA helicase of H. pylori, it is likely that it plays multiple roles in the cellular metabolism.
Using primer extension assays, we showed that Hp-RNase J is not recruited to the ribosomes to perform final rRNA maturation. In contrast to their H. pylori ortholog, RNases J of B. subtilis, Sinorhizobium meliloti and Mycobacterium smegmatis were shown to be required for the last steps of rRNA maturation (36–38). As a consequence, the ribosomal cellular localization of RNase J1 in B. subtilis (15) and the fact that it was purified from a high-salt ribosome wash (3) have been attributed to its role in the maturation of rRNA. The fact that Bsu-RNase J1 co-sediments with 30S, 50S, 70S and polysomes in sucrose gradients suggests that it is associated both with newly assembled and translating ribosomes (Figure 7A). Such localization is consistent with the observation that final maturation of rRNA begins upon assembly of individual ribosomal subunits and continues on polysomes (36). Bsu-RNase J1 has been shown to play a major role in mRNA decay in B. subtilis (53,54). Thus, it cannot be excluded that B. subtilis RNase J1 remains attached to the ribosome after rRNA maturation and plays a further role in the coordinated degradation of a subset of mRNAs in conjunction with translation. In contrast to its B. subtilis homolog, Hp-RNase J is associated with 70S ribosomes and polysomes and not with 30S or 50S subunits. Together with the finding that it does not mature rRNA, this strongly suggests that the major function of the Hp-RNase J degradosome in H. pylori is most probably mRNA degradation in association with the translation machinery.
Interestingly, apart from the large number of ribosomal proteins, TAP for Hp-RNase J has also identified the β–β′ subunit of RNA polymerase as an interacting partner (Supplementary Table S4). Although this interaction might be indirect, it correlates with known coupling of transcription and translation and further supports the conclusion of the association of Hp-RNase J with the gene expression machinery. The functional consequences of such an association for a given mRNA need to be investigated, in particular, with respect to the determinants of the initiation of mRNA degradation. The accumulated evidence of the importance of the structure of the mRNA 5′-end and the accessibility of the ribosomal binding site for mRNA stability in B. subtilis (55) implies that the decision about mRNA degradation is controlled by a competition between the ribosomes and RNase J1 for the access to the messenger. The experimental confirmation of such a competition comes from the example of an RNase J1 target in B. subtilis, the hbs mRNA. Indeed, this mRNA seems to be partially protected from RNase J1 degradation by ribosomes initiating translation (56). The association of Hp-RNase J with ribosomes may appear to contradict this notion. However, it is possible that steric effects prevent the accessibility of mRNA for degradation by RNase J once translation is initiated. Biochemical approaches are needed to elucidate the signals required for the initiation of mRNA decay in this context that could potentially be determined by the endonucleolytic attack of RNase J on mRNA. Structural studies have proposed that the internal access of RNase J to the message in the endonucleolytic mode has to be preceded by the opening of the RNA-binding channel between β-CASP and metallo-β-lactamase domains of RNase J (6). Thus, it is plausible that the initiation of RNA decay is regulated by the ribosome-dependent modulation of relative positions of these key domains of RNase J.
In this study, we discovered and characterized a novel type of bacterial RNA degradosome comprising Hp-RNase J and the only DExD-box RNA helicase RhpA of H. pylori. This was the first detailed characterization of such a complex emphasizing functional significance of the association of the two RNA-specific activities. We found that RhpA assists RNase J in the degradation of the dsRNA in vitro and RNase J in turn stimulates the energy-providing ATPase activity of the helicase. Our finding that the complex is associated with translating ribosomes implies that part of the RNA-degrading machinery in the H. pylori is coupled with translation, a situation never before reported in bacteria.
NOTE ADDED IN PROOF
Interestingly, a very recent paper by Tsai et al. (2012) in Nucleic Acids Res., doi:10.1093/nar/gks739 reports the recognition of the 70S ribosome and polysome by the RNA degradosome in E. coli.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Methods, Supplementary Tables 1–6, Supplementary Figures 1–3 and Supplementary Reference [57].
FUNDING
The Agence National de la Recherche [ANR 09 BLAN 0287 01, PyloRNA to H.D.R., F.D. and Y.R.]; Erasmus fellowship (to A.S.); Institut Pasteur for the Bourse Roux research fellowship (to Y.R.). Funding for open access charge: Institut Pasteur.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to Dr Josette Banroque for a kind gift of purified Ded1 used as a control in ATPase assay and to Dr Ciaran Condon for B. subtilis strain and anti-Bsu-RNase J1 antibodies. We thank Drs Ciaran Condon and Isabelle Iost for critical reading of the manuscript. We are also thankful to all the members of the Unité Pathogenèse de Helicobacter for fruitful discussions and to Andrea Sirianni for his experimental help. We also appreciate the expertise and help of Drs Micheline Fromont-Racine, Aurélie Galopier and Alain Jacquier with sucrose gradients.
REFERENCES
- 1.Carpousis AJ, Luisi BF, McDowall KJ. Endonucleolytic initiation of mRNA decay in Escherichia coli. Prog. Mol. Biol. Transl. Sci. 2009;85:91–135. doi: 10.1016/S0079-6603(08)00803-9. [DOI] [PubMed] [Google Scholar]
- 2.Condon C, Bechhofer DH. Regulated RNA stability in the Gram positives. Curr. Opin. Microbiol. 2011;14:148–154. doi: 10.1016/j.mib.2011.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Even S, Pellegrini O, Zig L, Labas V, Vinh J, Brechemmier-Baey D, Putzer H. Ribonucleases J1 and J2: two novel endoribonucleases in B. subtilis with functional homology to E. coli RNase E. Nucleic Acids Res. 2005;33:2141–2152. doi: 10.1093/nar/gki505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shahbabian K, Jamalli A, Zig L, Putzer H. RNase Y, a novel endoribonuclease, initiates riboswitch turnover in Bacillus subtilis. EMBO J. 2009;28:3523–3533. doi: 10.1038/emboj.2009.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mathy N, Benard L, Pellegrini O, Daou R, Wen T, Condon C. 5′-to-3′ exoribonuclease activity in bacteria: role of RNase J1 in rRNA maturation and 5′ stability of mRNA. Cell. 2007;129:681–692. doi: 10.1016/j.cell.2007.02.051. [DOI] [PubMed] [Google Scholar]
- 6.Dorleans A, Li de la Sierra-Gallay I, Piton J, Zig L, Gilet L, Putzer H, Condon C. Molecular basis for the recognition and cleavage of RNA by the bifunctional 5′-3′ exo/endoribonuclease RNase J. Structure. 2011;19:1252–1261. doi: 10.1016/j.str.2011.06.018. [DOI] [PubMed] [Google Scholar]
- 7.Newman JA, Hewitt L, Rodrigues C, Solovyova A, Harwood CR, Lewis RJ. Unusual, dual endo- and exonuclease activity in the degradosome explained by crystal structure analysis of RNase J1. Structure. 2011;19:1241–1251. doi: 10.1016/j.str.2011.06.017. [DOI] [PubMed] [Google Scholar]
- 8.Carpousis AJ. The RNA degradosome of Escherichia coli: an mRNA-degrading machine assembled on RNase E. Annu. Rev. Microbiol. 2007;61:71–87. doi: 10.1146/annurev.micro.61.080706.093440. [DOI] [PubMed] [Google Scholar]
- 9.Dreyfus M. Killer and protective ribosomes. Prog. Mol. Biol. Transl. Sci. 2009;85:423–466. doi: 10.1016/S0079-6603(08)00811-8. [DOI] [PubMed] [Google Scholar]
- 10.Khemici V, Poljak L, Luisi BF, Carpousis AJ. The RNase E of Escherichia coli is a membrane-binding protein. Mol. Microbiol. 2008;70:799–813. doi: 10.1111/j.1365-2958.2008.06454.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lehnik-Habrink M, Pfortner H, Rempeters L, Pietack N, Herzberg C, Stulke J. The RNA degradosome in Bacillus subtilis: identification of CshA as the major RNA helicase in the multiprotein complex. Mol. Microbiol. 2010;77:958–971. doi: 10.1111/j.1365-2958.2010.07264.x. [DOI] [PubMed] [Google Scholar]
- 12.Commichau FM, Rothe FM, Herzberg C, Wagner E, Hellwig D, Lehnik-Habrink M, Hammer E, Volker U, Stulke J. Novel activities of glycolytic enzymes in Bacillus subtilis: interactions with essential proteins involved in mRNA processing. Mol. Cell Proteomics. 2009;8:1350–1360. doi: 10.1074/mcp.M800546-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roux CM, DeMuth JP, Dunman PM. Characterization of components of the Staphylococcus aureus mRNA degradosome holoenzyme-like complex. J. Bacteriol. 2011;193:5520–5526. doi: 10.1128/JB.05485-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mathy N, Hebert A, Mervelet P, Benard L, Dorleans A, Li de la Sierra-Gallay I, Noirot P, Putzer H, Condon C. Bacillus subtilis ribonucleases J1 and J2 form a complex with altered enzyme behaviour. Mol. Microbiol. 2010;75:489–498. doi: 10.1111/j.1365-2958.2009.07004.x. [DOI] [PubMed] [Google Scholar]
- 15.Hunt A, Rawlins JP, Thomaides HB, Errington J. Functional analysis of 11 putative essential genes in Bacillus subtilis. Microbiology. 2006;152:2895–2907. doi: 10.1099/mic.0.29152-0. [DOI] [PubMed] [Google Scholar]
- 16.Atherton JC. The pathogenesis of Helicobacter pylori-induced gastro-duodenal diseases. Annu. Rev. Pathol. 2006;1:63–96. doi: 10.1146/annurev.pathol.1.110304.100125. [DOI] [PubMed] [Google Scholar]
- 17.van Amsterdam K, van Vliet AH, Kusters JG, van der Ende A. Of microbe and man: determinants of Helicobacter pylori-related diseases. FEMS Microbiol. Rev. 2006;30:131–156. doi: 10.1111/j.1574-6976.2005.00006.x. [DOI] [PubMed] [Google Scholar]
- 18.Eaton KA, Brooks CL, Morgan DR, Krakowka S. Essential role of urease in pathogenesis of gastritis induced by Helicobacter pylori in gnotobiotic piglets. Infect. Immun. 1991;59:2470–2475. doi: 10.1128/iai.59.7.2470-2475.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burne RA, Chen YY. Bacterial ureases in infectious diseases. Microbes Infect. 2000;2:533–542. doi: 10.1016/s1286-4579(00)00312-9. [DOI] [PubMed] [Google Scholar]
- 20.Tomb JF, White O, Kerlavage AR, Clayton RA, Sutton GG, Fleischmann RD, Ketchum KA, Klenk HP, Gill S, Dougherty BA, et al. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature. 1997;388:539–547. doi: 10.1038/41483. [DOI] [PubMed] [Google Scholar]
- 21.Merrell DS, Goodrich ML, Otto G, Tompkins LS, Falkow S. pH-regulated gene expression of the gastric pathogen Helicobacter pylori. Infect. Immun. 2003;71:3529–3539. doi: 10.1128/IAI.71.6.3529-3539.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bury-Mone S, Thiberge JM, Contreras M, Maitournam A, Labigne A, De Reuse H. Responsiveness to acidity via metal ion regulators mediates virulence in the gastric pathogen Helicobacter pylori. Mol. Microbiol. 2004;53:623–638. doi: 10.1111/j.1365-2958.2004.04137.x. [DOI] [PubMed] [Google Scholar]
- 23.Sharma CM, Hoffmann S, Darfeuille F, Reignier J, Findeiss S, Sittka A, Chabas S, Reiche K, Hackermuller J, Reinhardt R, et al. The primary transcriptome of the major human pathogen Helicobacter pylori. Nature. 2010;464:250–255. doi: 10.1038/nature08756. [DOI] [PubMed] [Google Scholar]
- 24.Akada JK, Shirai M, Takeuchi H, Tsuda M, Nakazawa T. Identification of the urease operon in Helicobacter pylori and its control by mRNA decay in response to pH. Mol. Microbiol. 2000;36:1071–1084. doi: 10.1046/j.1365-2958.2000.01918.x. [DOI] [PubMed] [Google Scholar]
- 25.Stingl K, Schauer K, Ecobichon C, Labigne A, Lenormand P, Rousselle JC, Namane A, de Reuse H. In vivo interactome of Helicobacter pylori urease revealed by tandem affinity purification. Mol. Cell Proteomics. 2008;7:2429–2441. doi: 10.1074/mcp.M800160-MCP200. [DOI] [PubMed] [Google Scholar]
- 26.McClain MS, Shaffer CL, Israel DA, Peek RM, Jr, Cover TL. Genome sequence analysis of Helicobacter pylori strains associated with gastric ulceration and gastric cancer. BMC Genomics. 2009;10:3. doi: 10.1186/1471-2164-10-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boneca IG, Ecobichon C, Chaput C, Mathieu A, Guadagnini S, Prevost MC, Colland F, Labigne A, de Reuse H. Development of inducible systems to engineer conditional mutants of essential genes of Helicobacter pylori. Appl. Environ. Microbiol. 2008;74:2095–2102. doi: 10.1128/AEM.01348-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Backert S, Kwok T, Konig W. Conjugative plasmid DNA transfer in Helicobacter pylori mediated by chromosomally encoded relaxase and TraG-like proteins. Microbiology. 2005;151:3493–3503. doi: 10.1099/mic.0.28250-0. [DOI] [PubMed] [Google Scholar]
- 29.Thibonnier M, Aubert S, Ecobichon C, De Reuse H. Study of the functionality of the Helicobacter pylori trans-translation components SmpB and SsrA in an heterologous system. BMC Microbiol. 2010;10:91. doi: 10.1186/1471-2180-10-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Condon C, Pellegrini O, Mathy N, Benard L, Redko Y, Oussenko IA, Deikus G, Bechhofer DH. Assay of Bacillus subtilis ribonucleases in vitro. Methods Enzymol. 2008;447:277–308. doi: 10.1016/S0076-6879(08)02215-5. [DOI] [PubMed] [Google Scholar]
- 31.Redko Y, Condon C. Ribosomal protein L3 bound to 23S precursor rRNA stimulates its maturation by Mini-III ribonuclease. Mol. Microbiol. 2009;71:1145–1154. doi: 10.1111/j.1365-2958.2008.06591.x. [DOI] [PubMed] [Google Scholar]
- 32.Iost I, Dreyfus M, Linder P. Ded1p, a DEAD-box protein required for translation initiation in Saccharomyces cerevisiae, is an RNA helicase. J. Biol. Chem. 1999;274: 17677–17683. doi: 10.1074/jbc.274.25.17677. [DOI] [PubMed] [Google Scholar]
- 33.Li de la Sierra-Gallay I, Zig L, Jamalli A, Putzer H. Structural insights into the dual activity of RNase J. Nat. Struct. Mol. Biol. 2008;15:206–212. doi: 10.1038/nsmb.1376. [DOI] [PubMed] [Google Scholar]
- 34.Weeks DL, Eskandari S, Scott DR, Sachs G. A H+-gated urea channel: the link between Helicobacter pylori urease and gastric colonization. Science. 2000;287:482–485. doi: 10.1126/science.287.5452.482. [DOI] [PubMed] [Google Scholar]
- 35.Skouloubris S, Thiberge JM, Labigne A, De Reuse H. The Helicobacter pylori UreI protein is not involved in urease activity but is essential for bacterial survival in vivo. Infect. Immun. 1998;66:4517–4521. doi: 10.1128/iai.66.9.4517-4521.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Britton RA, Wen T, Schaefer L, Pellegrini O, Uicker WC, Mathy N, Tobin C, Daou R, Szyk J, Condon C. Maturation of the 5′ end of Bacillus subtilis 16S rRNA by the essential ribonuclease YkqC/RNase J1. Mol. Microbiol. 2007;63:127–138. doi: 10.1111/j.1365-2958.2006.05499.x. [DOI] [PubMed] [Google Scholar]
- 37.Madhugiri R, Evguenieva-Hackenberg E. RNase J is involved in the 5′-end maturation of 16S rRNA and 23S rRNA in Sinorhizobium meliloti. FEBS Lett. 2009;583:2339–2342. doi: 10.1016/j.febslet.2009.06.026. [DOI] [PubMed] [Google Scholar]
- 38.Taverniti V, Forti F, Ghisotti D, Putzer H. Mycobacterium smegmatis RNase J is a 5′-3′ exo-/endoribonuclease and both RNase J and RNase E are involved in ribosomal RNA maturation. Mol. Microbiol. 2011;82:1260–1276. doi: 10.1111/j.1365-2958.2011.07888.x. [DOI] [PubMed] [Google Scholar]
- 39.Bleichert F, Baserga SJ. The long unwinding road of RNA helicases. Mol. Cell. 2007;27:339–352. doi: 10.1016/j.molcel.2007.07.014. [DOI] [PubMed] [Google Scholar]
- 40.Sharrock WJ, Rabinowitz JC. Fractionation of ribosomal particles from Bacillus subtilis. Methods Enzymol. 1979;59:371–382. doi: 10.1016/0076-6879(79)59098-3. [DOI] [PubMed] [Google Scholar]
- 41.Py B, Causton H, Mudd EA, Higgins CF. A protein complex mediating mRNA degradation in Escherichia coli. Mol. Microbiol. 1994;14:717–729. doi: 10.1111/j.1365-2958.1994.tb01309.x. [DOI] [PubMed] [Google Scholar]
- 42.Carpousis AJ, Van Houwe G, Ehretsmann C, Krisch HM. Copurification of E. coli RNAase E and PNPase: evidence for a specific association between two enzymes important in RNA processing and degradation. Cell. 1994;76:889–900. doi: 10.1016/0092-8674(94)90363-8. [DOI] [PubMed] [Google Scholar]
- 43.Jager S, Fuhrmann O, Heck C, Hebermehl M, Schiltz E, Rauhut R, Klug G. An mRNA degrading complex in Rhodobacter capsulatus. Nucleic Acids Res. 2001;29:4581–4588. doi: 10.1093/nar/29.22.4581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Purusharth RI, Klein F, Sulthana S, Jager S, Jagannadham MV, Evguenieva-Hackenberg E, Ray MK, Klug G. Exoribonuclease R interacts with endoribonuclease E and an RNA helicase in the psychrotrophic bacterium Pseudomonas syringae Lz4W. J. Biol. Chem. 2005;280:14572–14578. doi: 10.1074/jbc.M413507200. [DOI] [PubMed] [Google Scholar]
- 45.Hardwick SW, Chan VS, Broadhurst RW, Luisi BF. An RNA degradosome assembly in Caulobacter crescentus. Nucleic Acids Res. 2010;39:1449–1459. doi: 10.1093/nar/gkq928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ait-Bara S, Carpousis AJ. Characterization of the RNA degradosome of Pseudoalteromonas haloplanktis: conservation of the RNase E-RhlB interaction in the gammaproteobacteria. J. Bacteriol. 2010;192:5413–5423. doi: 10.1128/JB.00592-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vanzo NF, Li YS, Py B, Blum E, Higgins CF, Raynal LC, Krisch HM, Carpousis AJ. Ribonuclease E organizes the protein interactions in the Escherichia coli RNA degradosome. Genes Dev. 1998;12:2770–2781. doi: 10.1101/gad.12.17.2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kaberdin VR, Miczak A, Jakobsen JS, Lin-Chao S, McDowall KJ, von Gabain A. The endoribonucleolytic N-terminal half of Escherichia coli RNase E is evolutionarily conserved in Synechocystis sp. and other bacteria but not the C-terminal half, which is sufficient for degradosome assembly. Proc. Natl Acad. Sci. USA. 1998;95:11637–11642. doi: 10.1073/pnas.95.20.11637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lopez PJ, Marchand I, Joyce SA, Dreyfus M. The C-terminal half of RNase E, which organizes the Escherichia coli degradosome, participates in mRNA degradation but not rRNA processing in vivo. Mol. Microbiol. 1999;33:188–199. doi: 10.1046/j.1365-2958.1999.01465.x. [DOI] [PubMed] [Google Scholar]
- 50.Hu W, Sweet TJ, Chamnongpol S, Baker KE, Coller J. Co-translational mRNA decay in Saccharomyces cerevisiae. Nature. 2009;461:225–229. doi: 10.1038/nature08265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chuang RY, Weaver PL, Liu Z, Chang TH. Requirement of the DEAD-Box protein ded1p for messenger RNA translation. Science. 1997;275:1468–1471. doi: 10.1126/science.275.5305.1468. [DOI] [PubMed] [Google Scholar]
- 52.Rogers GW, Jr, Komar AA, Merrick WC. eIF4A: the godfather of the DEAD box helicases. Prog. Nucleic Acid Res. Mol. Biol. 2002;72:307–331. doi: 10.1016/s0079-6603(02)72073-4. [DOI] [PubMed] [Google Scholar]
- 53.Mader U, Zig L, Kretschmer J, Homuth G, Putzer H. mRNA processing by RNases J1 and J2 affects Bacillus subtilis gene expression on a global scale. Mol. Microbiol. 2008;70:183–196. doi: 10.1111/j.1365-2958.2008.06400.x. [DOI] [PubMed] [Google Scholar]
- 54.Durand S, Gilet L, Bessieres P, Nicolas P, Condon C. Three essential ribonucleases-RNase Y, J1, and III-control the abundance of a majority of Bacillus subtilis mRNAs. PLoS Genet. 2012;8:e1002520. doi: 10.1371/journal.pgen.1002520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bechhofer DH. Messenger RNA decay and maturation in Bacillus subtilis. Prog. Mol. Biol. Transl. Sci. 2009;85:231–273. doi: 10.1016/S0079-6603(08)00806-4. [DOI] [PubMed] [Google Scholar]
- 56.Daou-Chabo R, Mathy N, Benard L, Condon C. Ribosomes initiating translation of the hbs mRNA protect it from 5′-to-3′ exoribonucleolytic degradation by RNase J1. Mol. Microbiol. 2009;71:1538–1550. doi: 10.1111/j.1365-2958.2009.06620.x. [DOI] [PubMed] [Google Scholar]
- 57.Muller C, Bahlawane C, Aubert S, Delay CM, Schauer K, Michaud-Soret I, De Reuse H. Hierarchical regulation of the NikR-mediated nickel response in Helicobacter pylori. Nucleic Acids Res. 2011;39:7564–7575. doi: 10.1093/nar/gkr460. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.