

Abstract
NSC (neural stem cells)/NPC (neural progenitor cells) are multipotent and self-renew throughout adulthood in the SVZ (subventricular zone) of the mammalian CNS (central nervous system). These cells are considered interesting targets for CNS neurodegenerative disorder cell therapies, and understanding their behaviour in vitro is crucial if they are to be cultured prior to transplantation. We cultured the SVZ tissue belonging to newborn rats under the form of NS (neurospheres) to evaluate the effects of Tf (transferrin) on cell proliferation. The NS were heterogeneous in terms of the NSC/NPC markers GFAP (glial fibrillary acidic protein), Nestin and Sox2 and the OL (oligodendrocyte) progenitor markers NG2 (nerve/glia antigen 2) and PDGFRα (platelet-derived growth factor receptor α). The results of this study indicate that aTf (apoTransferrin) is able to increase cell proliferation of SVZ-derived cells in vitro, and that these effects were mediated at least in part by the TfRc1 (Tf receptor 1). Since OPCs (oligodendrocyte progenitor cells) represent a significant proportion of the proliferating cells in the SVZ-derived primary cultures, we used the immature OL cell line N20.1 to show that Tf was able to augment the proliferation rate of OPC, either by adding aTf to the culture medium or by overexpressing rat Tf in situ. The culture medium supplemented with ferric iron, together with aTf, increased the DNA content, while ferrous iron did not. The present work provides data that could have a potential application in human cell replacement therapies for neurodegenerative disease and/or CNS injury that require the use of in vitro amplified NPCs.
Keywords: nerve/glia antigen 2 (NG2), oligodendrocyte, platelet-derived growth factor receptor α (PDGFRα), progenitor, proliferation, transferrin
Abbreviations: aTf, apoTransferrin; bFGF, basic fibroblast growth factor; BrdU, bromodeoxyuridine; CNS, central nervous system; CSF, cerebrospinal fluid; DMEM, Dulbecco’s modified Eagle’s medium; EGF, epidermal growth factor; FCS, fetal calf serum; GFAP, glial fibrillary acidic protein; ICC, immunocytochemistry; NG2, nerve/glia antigen 2; NPC, neural progenitor cell; NS, neurosphere; NSC, neural stem cell; OL, oligodendrocyte; OPC, oligodendrocyte progenitor cell; OS, oligosphere; PDGFRα, platelet-derived growth factor receptor α; pExpTf, pExpressTf; PFA, paraformaldehyde; PO, polyornithine; RT–PCR, reverse transcription–PCR; SVZ, subventricular zone; Tf, transferrin; TfRc, Tf receptor; Tf-TR, Texas Red-labelled Tf; WB, Western blotting
INTRODUCTION
The developing and adult CNS (central nervous system) of higher vertebrates contains NPCs (neural progenitor cells) that preserve the capacity to self-renew throughout most of the adult life (Eriksson et al., 1998). Their most relevant properties reside in their unique ability to commit and mature into any of the different CNS neural cell lineages, including neurons and macroglia (Gould, 2007). One of the largest niches in the CNS harbouring these cells is the SVZ (subventricular zone), although other regions within the brain contain NPCs as well, such as the spinal cord subependymal zone, the hippocampus dentate gyrus subgranular zone and the previously described sub-cortical zone (Merkle and Alvarez-Buylla, 2006; Kriegstein and Alvarez-Buylla, 2009; Miller and Gauthier-Fisher, 2009).
The SVZ niche has been extensively studied in the past few years and evidence shows that the cells residing in it respond to injury (Picard-Riéra et al., 2002; Nait-Oumesmar et al., 2007; Jablonska et al., 2010). The fact that SVZ NPCs actively engage in brain repair make them promising targets for the design of neurodegenerative disease therapies (Lois and Alvarez-Buylla, 1993; Kokovay et al., 2009; Zhao et al., 2008). Particularly, cells expressing the NG2 (nerve/glia antigen 2) chondroitin sulfate proteoglycan are in part derived from the SVZ and are potential candidates that have been shown to function as precursors for OL (oligodendrocyte) regeneration in demyelinating diseases (Tripathi et al., 2010; Richardson et al., 2011). Progenitor proliferation and differentiation are required during oligodendrogenesis and both processes seem to depend on an adequate iron supply (Schonberg and McTigue, 2009).
Mammalian iron metabolism is tightly regulated (Wang and Pantopoulos, 2011) and involves, among many key regulators, Tf (transferrin) and TfRc1 (Tf receptor) 1 and TfRc2 (Dautry-Varsat, 1986; Anderson and Vulpe, 2009). In the brain, TfRc1 is mainly detected in neurons and endothelial cells. It is also expressed in oligodendroglial precursors and is down-regulated in mature OL (Leitner and Connor, 2012). TfRc2 has also been detected in the brain (Kawabata et al., 1999). On the other hand, Tf is mainly synthesized in the liver and secreted into the blood stream from where it is able to transcytose across the blood–brain barrier into the CNS (Broadwell et al., 1996); however, the amount that effectively reaches the brain parenchyma is insufficient to satisfy the brain's iron metabolic demands. Therefore, most of the Tf found in the CNS is endogenously synthesized by OL and choroid plexus cells, and is required for iron mobilization within the brain parenchyma and CSF (cerebrospinal fluid) (Espinosa de los Monteros et al., 1988; Morris et al., 1992; Connor and Menzies, 1995; Moos et al., 2007).
Previous reports regarding Tf effects on the CNS demonstrate this glycoprotein is able to increase myelin content during the rat's early postnatal development (Escobar Cabrera et al., 1994; 1997; Marta et al., 2000), and suggest Tf is an OL pro-maturating factor. Nonetheless, recent data support the trophic effects of Tf on rat SVZ cells in vivo, where an aTf (apoTransferrin) intracranial injection into the ventricles of rats after hypoxia/ischaemia increases the expression of the PCNA (proliferation cell nuclear antigen; Guardia Clausi et al., 2010). Although these in vivo findings clearly show that Tf is able to modulate both cell proliferation and differentiation, they also suggest that Tf effects are mainly dependent on the identity of the cells it targets.
The NS (neurospheres) culture assay has been widely used as an in vitro model to study the progression of undifferentiated cells to OL. We have recently examined the effects of aTf treatment on young rat SVZ-derived NS and found that during oligodendrogenesis aTf was able to control cell proliferation, lineage commitment or cell differentiation depending on the time point at which the treatments were carried out (Silvestroff et al., 2012). Therefore, in this report, we evaluated if the early event associated with the activation of NPC proliferation was conserved in neonatal SVZ tissue cultures and further investigated the molecular mechanisms by which Tf is able to stimulate cell proliferation.
We determined that the increase in NS proliferation rate was associated with the increment in NS size, and this effect was mediated by the incorporation of Tf into cells through TfR1. Since OPC (oligodendrocyte progenitor cell) represented the highest proportion of proliferating cells in the NS, we used the OL cell line N20.1 and confirmed Tf had similar effects in this culture system. We conclude OPC is responsible for the increase in NS size after Tf treatment. Furthermore, Tf could be used to augment OPC numbers for future cell replacement therapies, where NPC require in vitro expansion in a serum-free culture medium.
MATERIALS AND METHODS
Animals
All animal procedures used in this study were performed following the guidelines established by Buenos Aires University School of Pharmacy and Biochemistry. Albino Wistar rats (Rattus norvegicus) were housed under 12 h light/12 h dark cycles. They were fed rodent pellet chow and water ad libitum.
The culture medium
DMEM/F12 (Dulbecco's modified Eagle medium/nutrient mixture F-12; Gibco®) culture media was prepared with slight modifications. We added 1.2 g of sodium bicarbonate (Calbiochem) and 4 g of anhydrous glucose (Sigma–Aldrich®) per litre. Sodium penicillin G and streptomycin sulfate were added to a final concentration of 20 units/ml and 20 μg/ml respectively, and the pH was adjusted to 7.4. For SVZ-derived NS cultures, B27 supplement (Gibco®) was added to the medium at a final concentration of 2% (DMEM/F12-B27). The B27 supplement used to prepare the culture medium contains unknown-to-the-buyer Tf concentrations (Brewer et al., 1993). The proliferation medium was prepared by supplementing the DMEM/F12-B27 media with the mitogens bFGF (basic fibroblast growth factor; 20 ng/ml) and EGF (epidermal growth factor, 20 ng/ml) (Peprotech®). For N20.1 cells, we used DMEM/F12 supplemented with 2% FCS (fetal calf serum).
SVZ-derived free-floating NS cultures
Two- to four-day-old male and female rats were killed and brains were removed. For SVZ-derived NS cultures, the periventricular tissue was removed from the brains and mechanically dissociated to a single cell suspension with a 1 ml automatic pipette. Cells were expanded in proliferating medium in a humidified incubator at 36°C and 5% CO2. Growth factors (bFGF and EGF) were added to the medium every 2 days. Proliferating cells grew into free-floating aggregates known as NS, which began appearing as from the third day in vitro. NS were allowed to expand for 6 days before dissociation. To dissociate whole NS into a single cell suspension, NS were allowed to settle for 10 min at room temperature (20°C), and were then mechanically dissociated to a single cell suspension by pipetting them up and down 15 times with a 1 ml automatic pipette. Finally, the cell suspension was resuspended in fresh proliferating medium. Alternatively, NS were dissociated using the Neurocult® Chemical Dissociation Kit protocol (Stem Cell Technologies). The cell suspensions were either used to regenerate a new passage of NS, or were directly sewed on to an adherent surface: a Petri dish or a glass coverslip. For whole NS analysis, NS were plated on PO (polyornithine)-coated coverslips for at least 4 h and then fixed. For individual cell analyses, dissociated NS were plated over night in a 100 μl volume of culture medium on PO-coated coverslips within a 24-well plate. Once individual cells were attached, the wells were completed with 400 μl of fresh proliferating medium. For the culture treatments with aTf, the NS-derived cells were incubated for 6 days in the presence of mitogens.
The N20.1 cell line
The N20.1 oligodendroglial cell line used to evaluate Tf effects on cell proliferation was a gift from Dr Campagnoni's laboratory. The cell line generation has been described by Foster et al. (1993) and Verity et al. (1993). The cells were generated from mouse OL cultures, and were immortalized by infecting them with a viral vector that expresses the simian virus large T antigen. The simian virus large T antigen is capable of maintaining an immortalized phenotype at a proliferation-permissive temperature (34°C). At higher temperatures (39°C), the thermo-labile antigen is degraded, forcing the N20.1 cells to exit the cell cycle and initiate their maturation process. We used a constant cell culture temperature of 36°C at all times, as a compromise temperature between the 37°C needed for the SVZ-primary NS cultures and the proliferation-permissive temperature needed for this immortalized cell line. Under these conditions, these cells continue to enter the cell cycle and remain as immature OL progenitors. The N20.1 cells were grown attached to plastic flasks in 2% FCS-supplemented DMEM/F12 medium. The cell line was also cultured as free floating spheres in non-adhesive plastic Petri dishes. The N20.1 spheres were called OS (oligospheres), to differentiate them from SVZ-derived NS.
aTf treatments
A human aTf (Sigma–Aldrich®) sterile 50×stock solution (5 mg/ml) was used to treat the SVZ-derived primary cultures and the N20.1 cell line at a final concentration of 100 μg/ml. The culture medium was replaced every 2 days during treatment. All the aTf treatments were performed in parallel with control cultures (CTL) lacking aTf supplementation. For the SVZ-derived primary cultures, attached cells were treated with aTf for 6 days, while N20.1 cells were exposed to aTf for 2 or 6 days according to the assay performed. For Tf uptake studies, attached cells were exposed to Tf-TR (Texas Red-labelled Tf; Invitrogen™) at a 100 μg/ml final concentration for 24 h prior to fixation. For the treatment of free-floating NS with the Tf receptor blocking antibody (clone 42/6), the antibody concentration was set at 5 μg/ml in the culture medium according to data published by Trowbridge and Lopez (1982).
Tf overexpression in the N20.1 cell line
Cells were plated overnight before the transfection. The cells were transfected with circular pExpTf (pExpressTf; Open Biosystems) to overexpress the secretable form of Tf by using Lipofectamine™ 2000 (Invitrogen) for 4 h. The pExpTf vector carries a rat Tf cDNA obtained from rat testis mRNA (NCBI accession number BC087021). The control cells were transfected with a circular pExpress vector lacking Tf cDNA (pExp). For BrdU (bromodeoxyuridine) incorporation assays, cells were transfected with either vector (pExp or pExpTf) and cultured for 2 days after the transfection protocol. For DNA quantitative analysis, the cells were cultured for 6 days after the transfection protocol. In this last case, the cells were transfected with linear plasmid vectors. We used a pExpTf vector digested within the Tf coding sequence as a negative control. Both linear vectors were treated with calf intestine ALP (alkaline phosphatase) to avoid any re-ligation. Cells were analysed 6 days post transfection to decrease the probability of plasmid loss.
Culture medium supplementation with ferric citrate or ferrous sulfate
For studies of iron supplementation in the culture medium of the N20.1 cell line, we calculated the basal ferric and ferrous ion status of the DMEM/F12 culture medium to be 0.125 μM and 1.5 μM respectively. The 2% FCS was analysed for iron content and showed to be 0.35 μM. For an aTf concentration of 100 μg/ml in the culture treatments, we estimated it equalled 1.25 μM if we considered Tf had a molecular mass of 80 kDa. Since aTf has two binding sites for iron, we explored the effects of ferric citrate and ferrous sulfate at 2.5 μM, a concentration that equalled the amount of binding sites on the added aTf. A 1000-fold lower concentration of 2.5 nM for each iron source was added to the experimental design as a low dose condition.
Fluorescent DNA quantitative analysis
DNA was quantified following the technique described by Gallagher (2000). After the N20.1 cells were treated for 6 days, either with aTf in the culture medium or by overexpressing Tf, the wells were rinsed twice with pre-warmed PBS and incubated in TNE (10 mM Tris base, 1 mM EDTA, 0.2 M NaCl and 100 ng/ml Hoechst 33258, pH 7.4). The fluorescence was measured using the FlexStation 3 (Molecular Devices) or the Victor™ X Multilabel plate reader (PerkinElmer). The λex was set at 355 nm and the λem at 460 nm.
ICC (immunocytochemistry)
A 4% (w/v) PFA (paraformaldehyde)–PBS solution was used for cell fixation of attached cultures. Fixed cells were blocked overnight in 5% FCS. Primary antibodies were incubated overnight at 4°C (Table 1). Coverslips were rinsed in PBS, and secondary fluorochrome-conjugated antibodies were incubated for 2 h at room temperature. All antibodies were prepared in a 1% FCS, 0.1% Triton X-100, PBS solution. Secondary antibodies were diluted together with Hoechst 33258 nuclear dye. The fluorochromes conjugated to the secondary antibodies were the following: DyLight 488, DyLight 594, Cy2, Cy3 and FITC (Jackson ImmunoResearch Inc.). Finally, coverslips were rinsed in deionized water and mounted on glass slides with Mowiol® 4-88 anti-fade mounting solution (Calbiochem). For BrdU incorporation analysis by ICC in N20.1 cells or on attached dissociated NS cells, 10 μM BrdU (Sigma–Aldrich®) was added to culture media for 4 h or 24 h before fixation respectively. BrdU incorporation on whole NS was performed by exposing cultures to BrdU for 4 h before fixation. All BrdU-treated cells were fixed in 4% PFA for 20 min at room temperature. For BrdU immunodetection, the cells were rinsed in PBS after fixation and exposed to 2 M HCl for 20 min at 37°C, neutralized with PBS, and blocked overnight in a 5% FCS-PBS solution. After the FCS blocking step, the ICC procedure with primary and secondary antibodies continued as described above.
Table 1. Primary antibodies used for ICC and WB.
| Antigen (Clone) | Immunogen | Species | Isotype, clonality* | Brand | Dilution for ICC (WB) |
|---|---|---|---|---|---|
| Acetylated α-tubulin (6-11B-1) | Outer arm of Strongylocentrotus purpuratus | Mouse | IgG2b, monoclonal | Sigma–Aldrich® | 1:1000 |
| Activated caspase III | Peptide RGTELDCGIETD of human activated caspase III with added KLH amino acids | Rabbit | Monoclonal† | Cell Signaling Technologies | 1:100 |
| BrdU (BMC 9318) | BrdU conjugated to BSA | Mouse | IgG1, monoclonal | Roche | 1:100 |
| GFAP | Amino acids 1–428 of GFAP from bovine spinal cord | Chicken | IgY, polyclonal | Neuromics | 1:500 |
| GFAP-Cy3 (G-A-5) | GFAP from pig spinal cord | Mouse | IgG1, monoclonal | Sigma–Aldrich® | 1:1000 |
| Human Tf | Human seric Tf synthetic peptide | Chicken | IgY, polyclonal | GeneTex | 1:100 |
| MBP | Amino acids 1 to 154 from rat MBP N-terminal fragment | Rabbit | Polyclonal | Dr A. Campagnoni Lab (UCLA, USA) | 1:500 |
| Mouse Tf (I-20) | N-terminal peptide of mouse Tf | Mouse | Polyclonal | Santa Cruz Biotechnologies | 1:50 |
| Nestin | Peptide containing peptide sequence shared between human and rat Nestin | Chicken | Polyclonal | Neuromics | 1:100 |
| NG2 | NG2 proteoglycan chondrotin sulfate purified from rat | Rabbit | IgG, polyclonal | Millipore | 1:200 |
| Olig2 (211F1.1) | Human recombinant Olig2 | Mouse | IgG2a, k, monoclonal | Millipore | 1:100 |
| PDGFRα | Mouse recombinant PDGFRα | Goat | Polyclonal | Neuromics | 1:100 |
| Rat Tf | Unknown | Rabbit | Polyclonal | Dr. Zakin laboratory (Institute Pasteur, France) | 1:100 (1:2500) |
| Sox2 | Synthetic peptide sequence containing amino acids 300 to C-terminal end of human Sox2 with added KLH amino acids | Rabbit | IgG, Polyclonal | Abcam | 1:100 |
| TfRc1 or CD71 (42/6) | Human purified TfRc1 | Mouse | IgA, monoclonal | Millipore | 1:20 |
| TfRc1 or CD71 (OX-26) | Phytohemagglutinine-activated lymph node cells | Mouse | IgG2a,k, monoclonal | BD Pharmingen™ | 1:50 |
*The polyclonal antibodies with no assigned isotype indicate they contained more than one isotype or the isotype was not specified by the manufacturer.
†We did not have access to data regarding the antibody isotype.
ICC and cell number quantification
For Figures 2(P), 3(L), 3(P), 4(D), 4(H) and 6(J), cell proportions were quantified from microscope images according to the immunodetection of two different cell markers. The two cell markers simultaneously analysed on a given cell preparation were identified using green or red fluorochrome-conjugated secondary antibodies. The images acquired with the different microscope filters for a given field were merged to obtain a single three coloured image, where blue always corresponded to the total cell nuclei that incorporated the Hoechst dye. Cells expressing each one or both markers were manually quantified and normalized to the total amount of Hoechst+ nuclei in the field and expressed as the means±S.E.M. Number of cells lacking the expression of both markers was calculated. For the BrdU+ nuclei counting in whole (non-dissociated) NS, an average of 4 (±1) epi-fluorescence microscopy images were taken of the BrdU+ nuclei at different focal planes within each NS (as shown in Supplementary Figure S4 at http://www.asnneuro.org/an/005/an005e107add.htm). The number of images varied according to the size of the NS. The BrdU+ nuclei were counted on the first image and labelled, after which these digital labels were merged on the second image. Then, the BrdU+ nuclei were counted and labelled on this second image. We avoided counting BrdU+ nuclei that already had a digital label (brought on from the previous image). The labels of the second image were added to the labels of the previous image and altogether merged on the third image. The procedure was repeated for each additional image of a given NS to prevent the counting of BrdU+ nuclei more than once. The resulting number of BrdU+ labelled cells was finally normalized to the NS size. The NS size (volume) was calculated from the NS radius (Supplementary Figure S4A). At least 80 NS were counted from each condition from two independent experiments.
Figure 2. Sox2 and Olig2 expression in the SVZ and in NS cultures.

For all images, Sox2 is labelled in green and Olig2 is labelled in red. (A–D) Sox2 and Olig2 immunohistochemical analysis in coronal brain sections belonging to neonatal rat brains. (E–H) Sox2 and Olig2 expression analysis in the SVZ wall of coronal brain sections belonging to neonatal rat brains. (I–L) Sox2 and Olig2 expression analysis in non-dissociated NS. (K) A cell that co-expresses both Sox2 and Olig2 is indicated with an arrow, while a Sox2+/Olig2− cell is indicated with a white arrowhead and a Sox2−/Olig2+ cell is indicated with an empty arrowhead. (M–P) Quantitative analysis of the cell proportions expressing either Sox2 or Olig2. (O) Nuclei expressing both Olig2 and Sox2 are indicated with empty arrowheads, while Sox2+/Olig2− nuclei are indicated with white arrowheads and a Sox2−/Olig2+ nucleus is indicated with an arrow. Bars in (P) represent means±S.E.M. for each cell population. The scale bar (A) represents 250 μm for (A)–(D), scale bar in (E) represents 10 μm for (E)–(H), and scale bar in (I) represents 100 μm for (I)–(O).
Figure 3. NPC markers in SVZ-derived NS cultures.
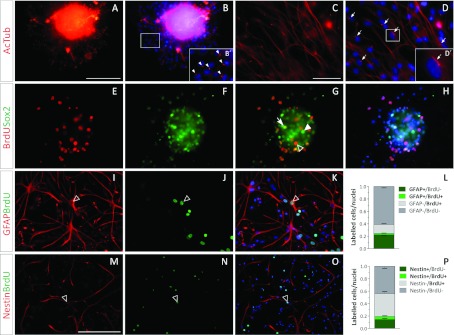
(A–D) Acetylated tubulin (red) in whole NS (A, B) and in dissociated NS cultures (C, D). The insets in (B) and (D) are shown at a higher magnification in (B′) and (D′) respectively. Acetylated tubulin+ cilia are indicated with arrowheads in (B′) and with arrows in (D). (E–H) BrdU incorporation is shown in red and Sox2 expression is shown in green. (G) A BrdU and Sox2 double-labelled nucleus is indicated with an arrow, while Sox2+/BrdU− and Sox2−/Brdu+ nuclei are indicated with a filled arrowhead and an empty arrowhead respectively. Images (E) and (F) are merged with the corresponding Hoechst-stained nuclei image in (H). (I–K) GFAP+ immunostained filaments are shown in red and BrdU+ nuclei are shown in green. The proportions of GFAP and BrdU-labelled cells were quantified and are shown in (L). (M–O) Nestin+ immunostained filaments are shown in red and BrdU+ nuclei are shown in green. The proportions of Nestin and BrdU-labelled cells were quantified and are shown in (P). The empty arrowheads in (I)–(K) and (M–O), indicate a GFAP+/BrdU+ and a Nestin+/BrdU+ cell respectively. The blue colour in the images belongs to the Hoechst nuclear dye. The scale bar in (A) represents 100 μm in (A), (B), (E)–(K), the scale bar in (C) represents 250 μm for (C), (D), and the scale bar in (M) equals 200 μm in (M–O). The bars in graphs (L) and (P) represent means±S.E.M. for each cell population.
Figure 4. Glia and OPC-related markers in SVZ-derived NS cultures.
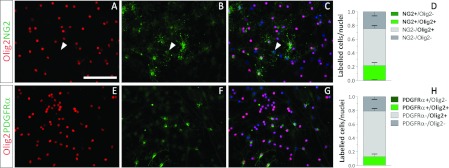
(A–D) NG2 (green) and Olig2 (red) expression analysis. An NG2+/Olig2− cell is indicated with a white arrow head (A–C). (E–H) Olig2 (red) and PDGFRα (green) expression analysis. The blue colour in the images indicates the Hoechst nuclear dye. The scale bar in (A) represents 100 μm in (A–C) and (E–G). Scale bars in graphs (D) and (H) represent means±S.E.M. for each cell population.
Figure 6. NG2 and PDGFR expression in the SVZ and in different culture systems.

(A–C) Confocal images of an NG2 (green) cell that incorporates Tf-TR (red) in the CC of a neonatal rat brain section after a 4 h Tf-TR injection. (D, E) Immunocytochemical analysis of NG2, PDGFRα and BrdU-labelled cells in dissociated NS cultures. (G–I) Immunocytochemical analysis of NG2, PDGFRα and BrdU-labelled cells in the N20.1 cell line. (D, G) NG2+ and BrdU+ cells are labelled in red and green respectively. (E, H) PDGFRα+ and BrdU+ cells are labelled in green and red respectively. (F, I) NG2+ and PDGFRα+ cells are labelled in red and green respectively. (F) NG2/PDGFRα double-labelled cells are indicated with arrows, NG2+/PDGFRα− cells are indicated with empty arrowheads, and NG2−/PDGFRα+ cells are indicated with filled arrowheads. (I) Areas with strong NG2 expression are indicated with white arrowheads. (J) Proportions of BrdU+ nuclei that express either NG2 or PDGFRα in dissociated NS cells after a 24 h BrdU pulse. (H) Cell proportions in NS and N20.1 cells that express either NG2 (light grey boxes), PDGFRα (dark green boxes) or both markers (light green boxes). (I) Proliferation rates of NG2+ and PDGFRα+ cell populations in the N20.1 cell line after a 4 h BrdU pulse. Scale bar in (A) equals 5 μm in (A–C), scale bar (F) equals 100 μm in (D–F), and scale bar in (I) equals 50 μm in (G–I).
Tf-TR intraventricular injections
Rat pups (P2–4) were anaesthetized by hypothermia and subjected to an intraventricular injection of 1 μl of sterile Tf-TR (100 μg/ml). A 1 cm-long longitudinal and superficial incision was performed with a scalpel over the cranium skin. A 30 gauge needle was used to gently prick the cranium bones at 1 mm (posterior) and 1 mm (lateral) from the Bregma to ease the access of the injection pipette needle. The rats were then injected with the Tf-TR solution by inserting the injection needle 4 mm deep into the CNS lateral ventricle.
Immunohistochemistry
The brains of the P2–4 rat pups were removed and rinsed in PBS, and then fixed in a 4% (w/v) PFA overnight at 4°C. Whole brains were sequentially soaked for 24 h in 15% and 30% sucrose solutions. Finally, the brains were frozen at −80°C. To obtain 30 μm-thick coronal sections, the brains were processed using a Leica CM 1850 cryostat. The brain sections were collected in a 50% glycerol solution and kept at −20°C. For the histochemistry procedure, brain slices were rinsed thrice in PBS, mounted on to gelatin-treated glass slides and air dried. The sections were blocked in 5% FCS–PBS solution for 2 h and then sequentially exposed to the primary and secondary antibodies, as described for the ICC procedure. Finally, the sections were mounted with Mowiol mounting solution.
SDS/PAGE and Western blotting
For cultured-cell protein analysis, adherent cells were washed twice with pre-warmed 0.1 M PBS at 37°C, scrapped off the Petri dishes and lysed in TOTEX buffer [20 nM Hepes, pH 7.9, 350 nM NaCl, 20% glycerol, 1% Igepal®, 1 nM MgCl2, 0.5 nM EGTA, 0.1 nM, 10 μg/ml leupeptine, 10 μg/ml pepstatine, 0.5 nM DTT (dithiothreitol) and 0.5 nM PMSF] on ice and kept at −20°C until processed. Whenever culture media were replaced on adherent cultures, samples of the old culture media were retrieved for protein analysis. These culture media aliquots were centrifuged for 10 min at 6700 g to remove any cell debris, and supernatants were recovered for storage at −20°C. For 35-day-old rat brain tissue samples, animals were anaesthetized and perfused with PBS to remove blood from vasculature before sample collection. The rat brains were then homogenized in a volume of PBS equal to the brain weight. The CSF samples were obtained from non-perfused rats and were centrifuged. CSF sample containing a red blood cell pellet were discarded due to serum-Tf contamination.
The proteins belonging to the different samples were quantified by the Bradford assay and electrophoresed in denaturing SDS polyacrylamide 12% gels. Proteins were then transferred on to a methanol-activated Immobylon™ PVDF membrane and blocked in a 5% (w/v) non-fat dried skimmed milk powder, PBS solution for 2 h at room temperature. Membranes were then incubated with primary antibodies overnight at 4°C (Table 1). After rinsing in a 0.1% Tween 20, PBS solution, membranes were incubated for 2 h at room temperature with HRP (horseradish peroxidase)-conjugated secondary antibodies, and then rinsed in a 0.1% Tween 20, PBS solution. Primary and secondary antibodies were diluted in a 1% FCS, 0.1% Tween 20, PBS solution. Protein immunodetection was performed with a 0.1% 3,3′-DAB (diaminobenzidine), 0.1% NiSO4, 0.1 M sodium acetate buffered solution (pH 5), with freshly added H2O2 (1 μl/ml). After colour development, membranes were rinsed in PBS, air dried and scanned. Immunolabelled bands on membranes were quantified with ScionImage software. In the case of Tf detection in whole P35 and P4 rat brains, the Tf immunopositve band was corrected by the dilution factor of the samples. For Coomassie Blue stained gels, proteins were electrophoresed in polyacrylamide gels and directly stained with the dye (Coomassie Brilliant Blue R 0.25% in methanol:glacial acetic acid:water 45:7:48) overnight at room temperature, and discoloured at room temperature in methanol:glacial acetic acid:water 45:7:48 until gel background was transparent.
RT–PCR (reverse transcription–PCR)
For the isolation of RNA samples from brain tissues we used the Trizol® Reagent (Invitrogen), and followed the protocol provided by the manufacturer. The retro-transcription and PCRs were performed using MMLV (Moloney murine leukaemia virus)–RT and Go-Taq from Promega, and the resulting PCR products were run in 2% agarose gels. The primer sequences used for the PCRs are listed in Table 2.
Table 2. PCR primer sequences.
| Gene | NCBI GenBank® ID | Protein | Sequence (5′→3′)* | Primer | Exon | Brand |
|---|---|---|---|---|---|---|
| Gapdh | NM_017008.4 | Glyceraldehyde-3-phosphate dehydrogenase | TTAGCCCCCCTGGCCAAGG | Forward | 5 | IDT® |
| CTTACTCCTTGGAGGCCATG | Reverse | 8 | IDT® | |||
| Trf | NM_001013110.1 | Transferrin | TTCTCATGCTGTTGTGGCTC | Forward | 7 | IDT® |
| CAGGTCACAGAGGGTGGAAT | Reverse | 13 | IDT® | |||
| Tfrc1 | NM_022712.1 | Transferrin receptor type 1 | GCGAAGTCCAGTGTGGGAACAGGT | Forward | 10 | IDT® |
| GTAGCACGGAAGTAGTCTCCACGAG | Reverse | 17 | IDT® | |||
| Tfrc2 | NM_001105916.1 | Transferrin receptor type 2 (α and β)* | GCTTAGGCTCCTGTCCTGG | Forward | 4 | Sigma–Aldrich® |
| GTAGCTGCTCTTGGACGCTC | Reverse | 7 | Sigma–Aldrich® |
*The TfRc2 primers were designed to obtain a single 483 bp band for both α and β transcripts of the gene.
Free-floating NS radius size measurement
To evaluate aTf effects on NS formation, NS primary cultures were mechanically dissociated to single cells. Identical aliquots of the cell suspension were amplified in control proliferating medium in the absence (CTL) or in the presence of aTf. Free-floating NS growing in flasks were photographed. Radius of NS was measured from digital images at different time points using Image Pro® Plus 5.1 software.
Microscopy image quantification and statistical analysis
Image acquisition was performed with an Olympus BX50 epi-fluorescence microscope and images were analysed with the Image-Pro® Plus Software or the Olympus Stream Image Analysis Software. For confocal microscopy, we used an Olympus FV1000 confocal microscope and images were analysed using the Fluoview software (2.0a version). Densitometric analysis of Western blot membranes and Coomassie Blue-stained gels was performed with Scion Image software. The statistical analysis of data obtained from Western blot, ELISA or microscopy image data was performed with the GraphPad Prism® using a Student's t test, one-way ANOVA or two-way ANOVA. The error bars in all graphs represent the means±S.E.M.
RESULTS
The culture medium analysis
We decided to evaluate if Tf enhanced cell division in the neonatal rat brain-derived NS assay, since Tf has been described as a key regulator of cell cycle. To avoid the use of FCS-supplemented medium and the possible variations in the culture medium composition, we used B27-supplemented DMEM/F12 (DMEM/F12-B27) to culture SVZ-derived cells. Although the B27-supplemented culture medium in which these primary cultures developed contained Tf, the manufacturers did not share information regarding its concentration or the species it comes from or if it was a recombinant molecule. Nonetheless, we analysed the DMEM/F12-B27 culture medium by WB (Western blotting) and observed a low Tf detection. Since the anti-Tf antibody used for the WB was raised against rat Tf, it could have had a low affinity for the B27 Tf, if the latter belonged to another species different from rat. We performed an SDS/PAGE and Coomassie Blue staining analysis of the culture media proteins and confirmed that the B27-supplement provided negligible amounts of Tf to the culture medium compared with the aTf-supplemented condition (aTf 100 μg/ml) we wanted to test (Supplementary Figures S1A and S1B at http://www.asnneuro.org/an/005/an005e107add.htm).
The SVZ-derived NS culture system
In the absence of growth factors in the culture media, NS were still able to develop although they reached smaller sizes after 6 days in culture compared with the bFGF/EGF-supplemented condition (Supplementary Figures S2A–S2D at http://www.asnneuro.org/an/005/an005e107add.htm). These results indicate that cells in this system are still capable of proliferating in vitro in the absence of exogenously added growth factors, suggesting the possibility of growth factor(s) being endogenously synthesized in these cultures. Nonetheless, NS formation was performed from here on in the presence of the growth factors EGF and bFGF to enhance cell amplification in vitro. We termed this a proliferating culture condition, since it was more permissive to cell division than the condition lacking exogenously added growth factors.
The basal proliferation rate of SVZ-derived cells in vitro was analysed after mechanical cell dissociation and seeding on PO-coated coverslips. As BrdU exposure time increased, the proportion of BrdU+ nuclei increased as well (Supplementary Figure S2E). However, due to cell heterogeneity in this kind of culture, the cell cycle time of the culture as a whole is not a representative measure of the cell cycle of each different cell type derived from the SVZ. Nonetheless, for dissociated NS cultures, the BrdU pulse was then set at 24 h.
Tf and TfRc expression in the CNS and cell cultures
We were interested in comparing the Tf content in the CSF, since this fluid bathes the enterity of the lateral ventricle walls and is in intimate contact with the SVZ niche. Since CSF without plasma contamination was only obtainable from rats from a young adolescent age, we compared the Tf content of the CSF belonging to P35 rats with the whole brain and plasma of age-matched animals, and with the Tf content in whole brain samples and serum belonging to newborn rats (P4). The Tf protein expression was analysed and semi-quantified by WB, and its presence was confirmed in high amounts in P35 and P4 rat serum (Figure 1A). Tf concentrations in whole brain homogenates (P35 and P4) and CSF samples (P35) were notoriously lower than that observed in serum samples but nonetheless detectable. The aTf concentration used to supplement the NS culture medium (100 μg/ml) was similar to the Tf concentrations detected in the CSF and brain tissue. The WB membranes, which were used to build Figure 1(A), are shown in Supplementary Figure S3 (at http://www.asnneuro.org/an/005/an005e107add.htm).
Figure 1. Tf and TfRc expression analysis in the CNS and in cell cultures.

(A) The Tf protein content was quantified in serum, whole brain homogenates and the CSF of at least two 35-day-old rats (P35) and at least two 4-day-old rats (P4) by WB, and compared with a culture medium sample supplemented with aTf (100 μg/ml). The absorbance of the WB bands was semi-quantified using a calibration curve containing different aTf concentrations, as indicated in Supplementary Figure S3. (B) RT–PCRs were performed on RNA isolated from P35 and P4 rats using primers for Tf, TfRc1, TfRc2 and GAPDH (glyceraldehyde-3-phosphate dehydrogenase). Testis, liver and choroid plexus were obtained from P35 rats, while liver, choroid plexus and SVZ tissue samples belonged to P4 rats. The NS samples were obtained from culturing the SVZ of P4 rats, and N20.1 indicates samples obtained from the immature OL cell line, N20.1. Bars in (A) represent means±S.E.M.
To confirm that Tf was synthesized in the rat CNS, the Tf expression was analysed together with TfRc by RT–PCR in tissues belonging to P35 and neonatal P4 rats (Figure 1B). The RNA belonging to P35 rat testis, liver and choroid plexus were used as positive controls, since all these samples expressed Tf and both TfRc 1 and 2. TfRc2 showed the presence of a major transcript, and a faint band corresponding to a larger transcript. The liver, choroid plexus and the SVZ tissues belonging to P4 rats showed Tf and TfRc1 expression, similar to those detected in samples belonging to the P35 rats. In the case of TfRc2, a larger transcript was detected in neonatal liver, choroid plexus and SVZ samples together with the classic isoforms, although at higher levels compared with the P35 rats. Furthermore, the proportions of both TfRc2 isoforms were different depending on the neonatal tissue analysed. These results indicate that besides the detection of various TfRc2 isoforms, other than the already described α and β isoforms, they were differentially regulated. The NS obtained from the SVZ of P4 rats contained transcripts belonging to Tf and to both TfRc1 and TfRc2. The N20.1 cell line belonging to immortalized mouse OL progenitors contained transcripts belonging to Tf and to both TfRc 1 and TfRc2, with a single PCR amplicon for each primer set.
Sox2 and Olig2 expression in vivo and in vitro
The SVZ of P2–4 rats was analysed in vivo, where we searched for cells expressing the NPC transcription factor Sox2 and the glial transcription factor Olig2. Although Sox2 expression was expected to be restricted to the SVZ, Sox2+ nuclei were identified in the CC (corpus callosum) and the CP (caudate putamen) as well (Figure 2A). Nonetheless, the greatest concentration of Sox2-expressing cells was located in the SVZ, confirming that this structure was enriched in NPCs. The immunodetection of Olig2+ cells in the same brain sections demonstrated that Olig2 was expressed by a minority of nuclei within the SVZ (Figures 2B–2D). Confocal images of the SVZ confirmed that Olig2 was expressed by some cells within the SVZ, and that some co-expressed Sox2 (Figures 2E–2H, arrow in Figure 2G). However, most of the Sox2+ cells in the SVZ were Olig2−.
Similar to the results obtained from histochemical analysis, the NS cells in the NS culture system expressed Sox2 and Olig2. Furthermore, besides noticing that the expression of both these transcription factors was mutually exclusive in some cells, many others were clearly Sox2+/Olig2+ (Figures 2I–2L, arrow in Figure 2K). Dissociated NS cultures showed that 90% of the cells that make up an NS expressed the transcription factors Sox2 or Olig2, as a whole (Figures 2M–2P). Only a small fraction of the immunopositive cells expressed either one of the transcription factors, while most co-expressed them both (Figure 2P). These data confirmed there is a bias of SVZ NPCs in vitro towards glial lineages compared with what occurred in vivo, and that the large amounts of Sox2 and Olig2 double-labelled cells might correspond to NPCs that were transitioning towards a glial, or even OPC, phenotype.
Undifferentiated cells in NS
To further confirm that the cells in the SVZ-derived NS contained NPCs, we analysed SVZ-derived primary cultures with antibodies raised against acetylated α-tubulin. Although in vivo ependymal cells contain numerous cilia that are labelled for acetylated tubulin, a single immunopositive cilium is characteristic of the SVZ type B NPC that protrudes through the SVZ wall and contacts the CSF. A single acetylated tubulin+ cilium was detected in vitro on the cells within whole NS and in dissociated NS cultures (Figures 3A–3D, arrowheads in Figure 3B, arrows in Figure 3D).
The cell proliferation in whole NS was evaluated after a 4 h BrdU pulse. The NS cells incorporated BrdU and confirmed that cells within the NS were able to proliferate in vitro. A large proportion of cells within the NS expressed the Sox2 transcription factor, indicating their undifferentiated phenotype. Only a fraction of BrdU+ cells were Sox2+, demonstrating that NS contain proliferating cells other than NPC (Figures 3E–3H). The considerable amount of Sox2+/BrdU− cells might be a consequence of the relatively short BrdU pulse. NS were dissociated to a single cell suspension and exposed to a 24 h BrdU pulse to analyse the identity of proliferating cells. NS-derived cultures contained GFAP+ (glial fibrillary acidic protein) cells (20%), although only a small proportion of these cells were BrdU+ (Figures 3I–3L). In a similar manner, Nestin+ cells were also immunodetected, with small amounts of Nestin+/BrdU+ cells (Figures 3M–3P).
Glia and OPC-related markers in NS
A further characterization of the Olig2+ cells demonstrated that only 25% of them expressed the OPC marker NG2 (Figures 4A–4D), while PDGFRα (platelet-derived growth factor receptor α) was detected in 15% of the Olig2+ cells (Figures 4E–4H). Interestingly, some NG2+ cells lacked Olig2 expression (Figures 4A–4C, arrowhead). No Olig2−/PDGFRα+ cells were detected. The presence of large amounts of Olig2+/NG2− and Olig2+/PDGFRα− cells, together with the high proportion of cells co-expressing Olig2 and Sox2, suggests that most Olig2+ cells might be committed to the oligodendroglial lineage and have not yet activated the expression of the NG2 or PDGFRα genes. Alternatively, they might be committed to other neural lines, such as the astrocyte lineage, that do not express these surface markers.
Both NG2+ and PDGFRα+ cell populations grew in characteristic clusters, rather than spread out evenly among other cells. This might contribute to the knowledge that the SVZ-derived cultures contain distinct immature progenitors that are able to divide clonally, and give rise to groups of cells expressing the same cell surface markers as their progenitors. Notwithstanding, we decided to consider NG2+/PDGFRα+ cells as cells committed to the oligodendroglial lineage.
Effects of aTf on SVZ-derived cell proliferation
Free floating NS were cultured in the absence or presence of human aTf (100 μg/ml) in the culture media. The culture conditions were therefore termed CTL and aTf respectively. The size of NS was studied as a function of time, and the results indicated that NS were significantly larger in the aTf condition compared with CTL condition as from the sixth day in culture (Figure 5A). To confirm that the NS size increment observed in the aTf condition was a consequence of an increase in cell proliferation, the BrdU+ nuclei within the whole NS (non-dissociated) were quantified and normalized to the NS volume (Supplementary Figures S4A–S4F at http://www.asnneuro.org/an/005/an005e107add.htm). The data indicated that the cell proliferation rate was increased after the aTf exposure in vitro (Figure 5B). Furthermore, the use of a monoclonal antibody that blocked the TfRc1 in the culture media was able to significantly dampen the aTf effects on the NS size, and demonstrated that the TfRc1 was involved in the effects of aTf on cell proliferation in the NS cultures (Figure 5C).
Figure 5. Effects of aTf on the proliferation rate of SVZ-derived NS.

(A) Radius length quantification in free-floating NS in the absence (CTL) or presence of aTf (aTf) in the culture medium. (B) Quantification of the BrdU+ nuclei after normalizing values to the NS volume in 6-day-treated cultures. The NS volume was calculated from the radius length data, as indicated in Supplementary Figure S4. (C) Quantification of the effects of aTf and/or of the TfRc1 blocking antibody (5 μg/ml) on the NS radius length in 6-day-treated cultures. (D, E) Tf immunodetection (green) in CTL and aTf-treated cultures. (F) Quantitative analysis of Tf immunodetection by ICC in CTL and aTf-treated cultures. (G) Epi-fluorescence image showing the immunodetection of Tf (green) and of BrdU+ (red) cells in aTf-treated cultures. The BrdU+ nuclei are indicated with white asterisks. (H–J) Confocal microscopy images of aTf-treated cultures showing Tf (green) and TfRc1 (CD71, red) detection. The blue or magenta colour in images indicates the Hoechst nuclear dye. The bars in (A–C) and (F) represent means±S.E.M. The scale bar in (D) represents 250 μm in (D) and (E), the scale bar in (G) equals 50 μm and the scale bar in (J) equals 10 μm in (H–J). *P<0.05, **P<0.01, ***P<0.001.
Tf was detected in dissociated NS cultures by ICC (Figures 5D and 5E). NS cultures grown in the CTL condition did not show any appreciable Tf immunodetection. The aTf-treated cultures showed an increase in Tf immunostaining (Figures 5E and 5F). Since aTf was shown to increase the cell proliferation rate in whole NS, we evaluated if there was a direct and proportional correlation between proliferation and Tf incorporation. We suspected cells undergoing the S phase of the cell cycle, and that were therefore BrdU+, would incorporate larger amounts of Tf compared with BrdU− cells. However, we were unable to notice significant changes in the Tf incorporation patterns of BrdU+ or BrdU− cells (Figures 5G). Nonetheless, these higher magnification images of aTf-treated cultures that showed small immunopositive puncta, suggested Tf was being incorporated into the cells via the canonical TfRc-mediated endocytic pathway (Figure 5G).
The Tf incorporation in aTf-treated cultures was analysed by confocal microscopy together with TfRc1 immunodetection (Figures 5H–5J). Tf was detected in all cells, and co-localized with TfRc1. The differences found in Tf immunoreactivity between Figures 5(G) and 5(H) could be attributable to the HCl treatment used for the BrdU antigen retrieval that could have partially damaged Tf epitopes. Although all cells expressed the TfRc1, this receptor was not evenly distributed within the cells. Some cells expressed the TfRc1 restricted to the soma, while others had TfRc1 expressed in both cell soma and its ramifications (Figures 5H–5J). Tf was also detected in cell ramifications where TfRc1 was not detected. This allowed us to speculate that the immunodetected Tf could have either been synthesized in those same cells, could have been incorporated via the TfRc2 or could have been incorporated by TfRc1 in the cell soma and then transported along the TfRc1− cell processes.
Tf incorporation in SVZ-derived primary cultures
We decided to use Tf-TR to identify the Tf being incorporated into the cells in culture, and simultaneously abrogate the detection of endogenously synthesized Tf. This method allowed us to confirm that not only was Tf being incorporated into the SVZ-derived cells in vitro but it was also specifically interacting with the ubiquitously expressed TfRc1 (CD71) (Supplementary Figures S5A–S5C at http://www.asnneuro.org/an/005/an005e107add.htm). The Tf-TR fluorescence was identified with the same punctate pattern as the immunodetected TfRc1. Tf-TR was also shown to be incorporated into BrdU+ cells (Supplementary Figures S5D–S5F), as well as into Sox2+ and Olig2+ cells (Supplementary Figures S5G–S5L). Since there were no appreciable differences in the quantities of Tf-TR incorporated into BrdU+ or BrdU− cells, we interpret that Tf-TR was internalized into the cells regardless of their engagement in the cell cycle S phase. Both NG2 and PDGFRα were able to incorporate Tf-TR (Supplementary Figures S5M–S5R). Some cells that did not express any of the cell makers previously mentioned incorporated great amounts of Tf-TR (Supplementary Figure S5L, arrow). These data confirmed that Tf uptake in SVZ-derived cells is detected with a punctate pattern, and that it is a widespread phenomenon that is not restricted to proliferating cells. Furthermore, it suggests Tf binding to cells must be following the canonical TfRc-mediated iron delivery pathway.
NG2 and PDGFRα expression in NS and N20.1 cell cultures
Since the SVZ in vivo and the cultured SVZ-derived cells contained Olig2+ cells, which are suggested to give rise to NG2+/PDGFRα+ OPC, we decided to evaluate if they could be a possible target of aTf in vivo. We injected Tf-TR into the right lateral ventricle of P2–4 rats and evaluated the red fluorescence in the cells surrounding the SVZ and neighbouring CC regions of the ipsilateral hemisphere. The rationale for the use of 2–4-day-old rat pups for the injections was based on the desire to evaluate the SVZ progenitors in vivo at the same age as those used to isolate the SVZ for the NS cultures. Furthermore, at this postnatal age the proliferating progenitors in the SVZ-niche and surrounding parenchyma are abundant and in a proliferative stage. Although there was a generalized and faint fluorescence dispersed throughout the tissue sections, some structures and cells incorporated large amounts of Tf-TR that were observed as distinct red-fluorescent puncta. Besides finding that the vasculature and some microglial cells incorporated large amounts of Tf-TR (data not shown), the NG2+ cells incorporated Tf-TR with a characteristic punctate pattern consistent with the Tf-TR endocytosis previously described in the in vitro experiments (Figures 6A–6C).
The analysis of BrdU+ nuclei revealed that almost 40% of all BrdU+ cells were positive for NG2, and to a lesser extent were PDGFR+ (Figures 6D, 6E and 6J). When analysing the proliferation rate of NG2+ and PDGFRα+ cell populations, 50% and 60% of them were BrdU+ respectively. Besides demonstrating that almost all NG2+ and PDGFRα+ cells belonged to the Olig2+ cell population (Figure 4), we evaluated the degree of co-localization of both these surface markers on dissociated NS cultures (Figures 6F and 6K). The results showed that the expression of these molecules overlapped in most of the immunopositive cells (Figure 6F, arrows), and we considered these double-labelled cells were OPC. We did notice that a small fraction of them expressed either one of the two markers (Figure 6F, whole and empty arrowheads).
On the basis of these results, we questioned whether the NG2/PDGFRα cell population could be responsible for the increased proliferative response of NS-derived cells to the Tf treatment. In order to test this hypothesis, we decided to use N20.1 immortalized oligodendroglial cell line grown in culture conditions in which cells are maintained in proliferation as undifferentiated progenitors. The N20.1 cells were enriched as a whole in NG2+ and PDGFRα+ cells when compared with the NS primary cultures, and were considered an appropriate system to evaluate Tf effects on the OPC population (Figures 6F, 6I and 6K). Interestingly, when PDGFRα was only partially expressed in some cells, this expression was concentrated in areas where cell membranes contacted each other (Figure 6I, arrowheads). We noticed that the polarized PDGFRα expression took place when at least one of the contact partner membranes expressed NG2, in agreement with data suggesting PDGFRα interacts with NG2 on surface membranes of cells.
The proliferation capacity of both NG2+ and PDGFRα+ cells in the N20.1 cell line was evaluated as well, since approximately half the NG2+ cells lacked PDGFRα expression. Besides confirming their high proliferative rate, there were no distinct differences among them according to this parameter (Figures 6G, 6H and 6L).
Effects of aTf on N20.1 cell cultures
We added 2% FCS in the culture media so that the addition of exogenous aTf (100 μg/ml) would be appreciably different from the untreated culture condition. The total proteins present in the DMEM/F12 culture media were analysed by SDS/PAGE at different percentages of FCS supplementation, and in the presence or absence of added aTf. The optimal FCS supplementation percentage that allowed us to identify a significant increase in Tf content in the culture medium proteins compared with the control condition was 2% (Supplementary Figures S6A and S6B, available at http://www.asnneuro.org/an/005/an005e107add.htm). We made sure the N20.1 cells in the 2% FCS-supplemented culture medium were proliferating.
To analyse the effects of aTf on the OL cell line in a similar assay as the one used with the SVZ primary cultures, we developed a method to grow N20.1 cells as free-floating spheres. In this case, since the cells used to form the spheres belonged to an OL-enriched culture, we decided to term the spherical structures formed from N20.1 cells as OS (Figure 7A). A 4 h BrdU pulse allowed us to observe a large number of proliferating cells within the OS (Figure 7B). We detected a significant increase in the mean size of the OS when aTf was added to the culture medium compared with the CTL condition (Figure 7C).
Figure 7. Tf effects on the N20.1 cell line.

(A) Bright field image of free-floating OS in 2% FCS-supplemented DMEM/F12. (B) Representative image of BrdU+ nuclei (red) in CTL-treated OS after a 4 h BrdU pulse. (C) Quantitative analysis of the OS radius in the absence (CTL) or the presence (aTf) of aTf in the culture medium at 5 and 7 days after treatment initiation. (D) Evaluation of BrdU incorporation by ICC in dissociated N20.1 cells after exposing them to BrdU pulses of different durations. (E) Quantification of BrdU+ cell proportions in CTL and aTf-treated cultures after a 48 h treatment and a 4 h BrdU pulse. (F) Fluorometric quantification of DNA in cell cultures treated for 6 days with different concentrations of aTf. (G) Quantitative analysis of caspase III+ cells in 48 h treated CTL and aTf cultures. The blue colour in microscopy images corresponds to the Hoechst nuclear dye. The bars in (C–G) represent means±S.E.M. The scale bar in (A) represents 250 μm in (A, B). *P<0.05, **P<0.01.
Due to the amount of proliferating cells within whole OS, their quantification in this three-dimensional structure was cumbersome. Therefore the proliferation rate of the N20.1 cell line was analysed by BrdU incorporation in dissociated monolayer cultures. We first evaluated the increase in the number of BrdU+ cells after exposing the cells to different BrdU time pulses (Figure 7D). We decided to use a 4 h BrdU pulse and confirmed by ICC that the proliferation of the N20.1 cell line was increased when exposed to aTf for 48 h in the culture medium compared with the CTL condition (Figure 7E). A fluorometric assay was used to quantify DNA. This method was considered an indirect approach to measuring the cumulative effects of an increased proliferation rate, and could serve to complement the BrdU incorporation experiments. The results showed that not only did the 6-day aTf-treatment increase the DNA amounts but also indicated that this response to aTf was dose dependent (Figure 7F).
Since the cell viability could be an additional variable responsible for the increase in DNA amounts and in the BrdU+ cell numbers in the aTf-treated cultures, we decided to evaluate if cell death by apoptosis was decreased in these cultures compared with controls. The immunodetection of activated caspase III+ cells was used to quantify the number of apoptotic cells in either CTL or in aTf-treated cultures. There was no difference in the amount of Caspase III+ cells among the different treatments, which allowed us to confirm that aTf increased the cell proliferation rather than protected cells from cell death by apoptosis (Figure 7G).
Tf incorporation in the N20.1 cell line
Similar to the rat NS primary cultures, we decided to make sure that the added aTf in the culture medium was internalized into the cells by using the ICC technique. Simultaneously, we evaluated the Tf-TR fluorescent pattern in the same cultures to discriminate the FCS-derived, or the endogenously synthesized Tf, from the human aTf we added to the culture media. When no aTf was added to the culture medium (CTL), there was a faint red fluorescence background and significant punctate immunodetection for Tf (Figures 8A and 8B). The data in Figure 8(A) indicate that the Tf detected could belong to that synthesized by the N20.1 cells or to the Tf of bovine origin contained in the FCS. As expected, when aTf was added to the culture medium (aTf condition), although there still was no significant red fluorescence detection, there was a noticeable increase in the amount and intensity of Tf+ immunoreactive puncta (Figures 8C and 8D). This result is in agreement with the SDS/PAGE analysis of the culture medium proteins belonging to CTL and aTf-supplemented culture medium (Supplementary Figure S6), where the overall Tf content is appreciably increased in the aTf supplemented condition compared with the control condition.
Figure 8. Tf incorporation in the N20.1 cell line.

Tf immunodetection is labelled in green, and Tf-TR fluorescence is labelled in red. (A, B) Images of cell cultures grown for 2 days in the absence of added aTf and Tf-TR. (C, D) Images corresponding to cell cultures grown in the presence of aTf (100 μg/ml) after a 2-day treatment. (E–G) Cell cultures treated for 4 h with Tf-TR. (G′) Co-localization plots of the merged image (G), where the green fluorescence intensity of pixels is plotted as a function of their red intensity. (G″) The co-localization mask determined according to the selected area in (G′), was used on (G). The white plotted area in (G″) corresponds to the double-labelled (green-red) pixels in (G). (H) Confocal image of the Tf immunodetection (green) in aTf-treated cells. (I, J) BrdU incorporation (red) and Tf immunodetection (green) in the absence (CTL) or the presence (aTf) of aTf in the culture medium. Inset in (J) is shown in (J′) at a higher magnification. The blue colour in the images corresponds to the Hoechst nuclear dye. Scale bar in (A) represents 100 μm for (A–F), scale bar in (H) represents 10 μm, and scale bar in (I) represents 50 μm in (I) and (J).
When Tf-TR was added to the culture medium, the TR fluorescence was detected at significantly higher levels than in the previous conditions (Figure 8F compared with Figures 8B or 8D). Furthermore, the red fluorescence had a punctate pattern that coincided with that of the immunodetected Tf (Figures 8E–8G). We performed a qualitative analysis to confirm that Tf-TR was co-localized in the same puncta as the immunodetected Tf by plotting the intensity of the green fluorescent pixels of two-dimensional epi-fluorescence images against the intensity of the red fluorescent pixels (Figure 8G′). The region of the plot where most pixels contained red and green fluorescence was selected and used to apply a mask on the merged images of either experimental condition. The results indicated that the pixels with the highest green and red fluorescence had a punctate pattern, as expected for the Tf internalization (Figure 8G″). Therefore these data reinforce the fact that the significant increase in the Tf immunodetected in the aTf-treated cultures (Figure 8C compared with Figure 8A) corresponds to the extracellular aTf that was added to the culture medium.
Further image analysis revealed that some cells in the Tf-TR treated cultures had a large round-shaped structure adjacent to the nuclei that seemed to deform or indent the normal nuclei ovoid shape (Figure 8G, arrowheads). The use of confocal imaging allowed us to confirm that the Tf+ puncta resembled the endosomal-mediated entry of Tf into the cells, and that the Tf puncta was located within the cell cytoplasm (Figure 8H).
The detection of Tf by ICC was hampered by the HCl treatment used during the BrdU antigen retrieval protocol (Figures 8I and 8J) compared with cells that had not been exposed to the HCl treatment (Figures 8C and 8E). However, Tf was still clearly detected in the aTf-treated cultures. The Tf incorporation pattern was not homogenous between treated cells, since some cells incorporated more Tf than others (Figure 8J). The BrdU incorporation did not strictly correlate with the amount of Tf detected in the cells, since not all the BrdU+ nuclei incorporated large amounts of Tf, nor were all the strong Tf-labelled cells positive for BrdU.
Effects of Tf overexpression on N20.1 cell proliferation rates
To evaluate Tf effects on cell proliferation in an alternative system, we decided to overexpress rat Tf in the murine N20.1 cell line. For BrdU analysis, the cells were transfected and cultured for 48 h until treatment termination. The Tf overexpression was analysed by immunodetecting the Tf content in whole-cell homogenates and in culture medium supernatants, and indicated that Tf was not only synthesized by the cells but also followed the canonical secretion pathway (Figures 9A and 9B). As time progressed after the transfection protocol, Tf increased in the cultures overexpressing Tf (pExpTf) compared with the control condition (CTL). The amount of Tf synthesized in the pExpTf cultures was semi-quantified by WB where known amounts of human aTf were used as standards. The amount of Tf in the culture medium was similar to the aTf concentration used to treat the cell cultures in the previously described experiments. The quantitative analysis of strongly labelled Tf+ cells by ICC 48 h after transfection revealed that the transfection efficiency with the pExpTf plasmid ranged between 15 and 18% (Figures 9C and 9D). The BrdU incorporation analysis showed that both Tf+ and Tf− cells in Tf-overexpressing cultures (48 h after transfection) incorporated BrdU in similar proportions. As a whole, Tf-overexpressing cultures had a higher number of BrdU+ nuclei compared with controls (Figure 9F). This suggested that Tf was increasing the cell proliferation rate in Tf overexpressing cells, and paracrinally acting on the non-transfected neighbouring cells as well.
Figure 9. Effects of Tf overexpression on N20.1 cell proliferation rates.

(A) Tf detection by WB in whole cell homogenates belonging to CTL and pExpTf-transfected cells at different time points after transfection. (B) Semi-quantitative analysis of Tf detection by WB in culture medium samples belonging to CTL and pExpTf-transfected cultures at different time points after transfection. The vertical line drawn across the WB membrane indicates the image was built from two non-contiguous regions of the same membrane. The bar colour reference is equal to the one shown in A. (C, D) Immunocytochemical analysis of Tf (green) in CTL (C) and pExpTf-transfected (D) cells 48 h after transfection. (E) Immunodetection of BrdU+ cells (red nuclei labelled with asterisks) and Tf+ cells (green) in a representative pExpTf-transfected cell culture. (F) Quantitative analysis of BrdU+ nuclei proportions 48 h after transfection. (G) DNA quantitative analysis in cells transfected with Pts1-digested pExpTf vector (pExp, interrupted Tf coding sequence) and with the BamH1-digested pExpTf vector (pExpTf, containing the full length Tf sequence) 6 days after transfection. Scale bars in graphs (A and B) represent mean values of representative experiments, and bars in (F and G) represent means±S.E.M. The blue colour in the microscopy images indicates Hoechst nuclear dye. *P<0.05.
We further decided to evaluate if the overall cell density would eventually increase over time in response to the increased cell proliferation. We therefore decided to transfect the N20.1 cells with linear versions of the CTL and the pExpTf vectors and culture the transfected cells for 6 days. After the 6th day, the total DNA content of the cultures was quantified using a fluorometric assay. The DNA fluorometric assay indicated that the cultures overexpressing Tf had increased DNA amounts compared with the control cultures (Figure 9G).
Effect of iron addition to CTL and aTf-treated cultures
The aTf treatment of NS and of the N20.1 cell cultures indicated that Tf was able to increase cell proliferation in a dose-dependent manner, and that at least TfRc1 was involved in this process. However, little is known about the mechanisms involved. We considered that iron was a plausible candidate responsible for these effects. We used the quantitative DNA assay to study the effects aTf had on cell cultures when increasing amounts of ferrous or ferric iron were added to the N20.1 cells for 6 days. We used a multifactorial designed experiment to analyse the influence of iron on the DNA content of the cells in this culture system. The two-way ANOVA allowed us not only to analyse the independent effects of aTf or iron, but also analyse if the different iron sources had differential effects on Tf function according to their concentration in the culture media.
Ferrous addition to cultures in the absence of aTf had a significant and negative effect on DNA amounts, and was considered the major source of variation (Figure 10A). The aTf supplementation was considered a significant source of variation as well, while there was no evidence to sustain a possible interaction among the factors (ferrous iron and aTf). The Bonferroni post-test indicated that the difference between the CTL and aTf treatment was only significant at the lowest ferrous iron concentration (asterisk in Figure 10A). We suppose the ferrous iron addition to the culture media might be generating an oxidizing environment due to the free radicals generated from Fenton's reaction that overrides the effects of aTf when added at the 2.5 nM and 2.5 μM concentrations.
Figure 10. Effect of iron addition to CTL and aTf-treated cultures.

(A, B) Ferrous sulfate and ferric iron citrate were used to quantify DNA in the N20.1 cell line after 6 days of treatment, where culture medium was changed every 48 h. Both iron forms were added at three different concentrations: 0, 2.5 nM and 2.5 μM. Datasets represented with white bars correspond to the control (CTL) treatment, and the datasets in black belong to the aTf treated condition. DNA fluorescence is expressed as a percentage compared with the CTL condition with no iron supplementation. The error bars represent the S.E.M. for each point. The data were analysed using a two-way ANOVA and a Bonferroni post-test. *P<0.05, **P<0.01. The asterisks indicate at which iron concentration aTf has a significant effect on the culture DNA content.
In the case of the ferric citrate treatment, both aTf and the ferric iron were a significant source of variation (Figure 10B). Although the effect aTf had on the DNA content was previously considered significant when adding ferrous iron, or when analysed by a one-way ANOVA (Figure 7F), the multifactorial design with a Bonferroni post-test indicated that the difference between the CTL and aTf treatment was significant at the 2.5 nM ferric iron concentration (asterisks in Figure 10B). However, similar to the highest ferrous iron treatment, high ferric citrate concentrations decreased the DNA content regardless of aTf supplementation.
DISCUSSION
We decided to evaluate in further depth the effects of Tf on NS proliferation by using the SVZ tissue belonging to neonatal rat pups. We first made sure the amount of aTf added to the culture medium (100 μg/ml) was significantly different from the endogenous Tf present in either DMEM/F12-B27 or 2% FCS-DMEM/F12. We also determined that the aTf concentration used for in vitro treatments closely resembled the physiological Tf levels found in the brain.
We detected Tf mRNA synthesis in SVZ tissue in vivo as well as in NS cultures by RT–PCR, suggesting an additional source for Tf in the brain besides the previously reported biosynthesis in OL and the choroid plexus (Bloch et al., 1985). Furthermore, mRNA belonging to TfRc 1 and 2 were detected as well. In the case of TfRc2, a higher molecular mass PCR product, other than the expected classic isoforms α and β, was detected in all tissue samples of neonatal rats, while a single PCR product was detected in mRNA samples obtained from in vitro cultures. Since the initial aim behind our experiments was to study the effects of Tf on SVZ-derived cells in vitro, the detection of the TfRc in the cell cultures provided compelling evidence that there was an underlying mechanism through which Tf could exert its effects. Falcão et al. (2012) have already demonstrated a close relationship between the secretion of choroid plexus soluble factors to the CSF and their influence on SVZ cells, including Tf.
The large amounts of Sox2+ nuclei in the SVZ in vivo indicated the NPC identity of the cells in this niche. We found that the expression of this transcription factor is also maintained in vitro. The acetylated α-tubulin immunodetection, together with GFAP and Nestin, provide further evidence that cells contained within neonatal brain-derived NS are NPCs, expressing the same markers as adult SVZ type B NPCs (Mirzadeh et al., 2008).
Cells expressing Olig2 were detected within the SVZ and in the NS. Although immunopositive cells for Sox2 or Olig2 were not quantified in vivo, there was a noticeable difference in the overlapping degree of both transcription factors between the in vivo and the in vitro cell samples. The higher proportions of Olig2+ cells in the NS suggest a considerable bias of the SVZ-derived NPC towards glial lineages in vitro compared with the default neuronal phenotype that predominates in vivo. This is in agreement with data published by Brazel et al. (2005), who described phenotypically distinct mouse brain-derived NS able to generate different proportion of neurons and glia.
The addition of aTf to the culture medium, while NS were under formation, demonstrated that the size of the NS increased as a function of time compared with untreated cultures. The BrdU incorporation analysis, after normalizing the data to the NS size, proved that the increase in the sphere size after aTf treatment was a direct consequence of an increment in cell proliferation. Besides the fact that Tf seems to be sufficient to increment NS proliferation, Erickson et al. (2008) provide additional results supporting Tf as a necessary factor for NS formation. The use of the anti-TfRc1 antibody with blocking activity in the NS culture medium demonstrated that the aTf effects on cell proliferation in this culture system were mediated, at least in part, by TfRc1. This finding is in agreement with Chirasani et al. (2009) who described a correlation in glioma cells between TfRc expression and tumour proliferation.
Although we do not demonstrate TfRc1 is expressed at the protein level in our cultures by WB, the RT–PCR and ICC results we show are supported by that of Cao et al. (2012) who used the NPC line C17.2 and stable isotope-labelled amino acids to quantify membrane proteins under different experimental conditions. They observed that one of the various membrane proteins that significantly vary between undifferentiated and differentiated cell conditions is TfRc1, where the protein expression of this particular receptor is highest in the cells in their undifferentiated state. According to Sergent-Tanguy et al. (2006), 50% of the NPCs that were obtained from whole brains of 18-day-old rat embryos, and that were cultured in the form of NS, showed TfRc1 expression.
Since GFAP+ and Nestin+ cells were demonstrated to have negligible proliferation rates in vitro, we decided to evaluate if there were other cell types in the SVZ-derived cultures with higher proliferation rates. Olig2+ nuclei were numerous in this system and are capable of giving rise to glial restricted progenitors, among which are OPCs. Surprisingly, besides finding Olig2+ nuclei co-expressing NG2 and PDGFRα, many other Olig2+ nuclei did not express either cell surface marker, which suggests that Olig2+ cells might have differentiated to the oligodendroglial lineage and silenced the expression of NG2 and/or PDGFRα. Alternatively, these cells might be considered progenitors that have not entirely committed to the oligodendroglial lineage and have not yet activated the expression of NG2 and PDGFRα. Dimou et al. (2008) have demonstrated that the Olig2+ cells in vivo at this postnatal stage can give rise both to OL or astrocytes. The high degree of co-localization among NG2+ and PDGFRα+ cells is in agreement with data published by Rivers et al. (2008), although there were a small proportion of cells that only expressed either of the two surface markers. These data allow for speculation regarding the chronological order of appearance of these two surface markers on OPCs. Furthermore, it highlights the heterogeneity of this particular cell population in terms of the degree of specification and commitment towards the oligodendroglial lineage. As a whole, immunodetection of different markers demonstrate that the SVZ-derived NS assay is a highly heterogeneous system containing different cell types, and contains cells at different stages along the differentiation pathway of a given neural lineage.
The in vivo analysis in the brain demonstrated that NG2+ cells were one of the main cell types to incorporate Tf-TR, and made them interesting putative targets to continue studying in vitro. The BrdU analysis in dissociated NS cultures reinforced our interest in NG2+ and PDGFRα+ cells since they represented a significant proportion of the proliferating cells. The use of the N20.1 cell line was decided because of the need for a cell culture system enriched in NG2 and/or PDGFRα expressing cells. The immature OL cell line N20.1 was shown to respond to the added aTf in the culture medium by increasing the proliferation rate. Moreover, the BrdU incorporation rate was proven to respond to aTf in a dose-dependent manner. We further included a quantitative DNA assay that could detect the cumulative effects of the increased cell proliferation rate in response to aTf, and demonstrated that there was a discrete increase in DNA amounts after the treatment compared with the controls.
Besides analysing the expression levels of the TfRc by RT–PCR in the N20.1 cell line, the use of Tf-TR confirmed that Tf was readily incorporated into the cells, and was further confirmed by the use of confocal microscopy. When Tf was overexpressed in the N20.1 cell line, Tf was secreted to the culture medium and continued to have a positive effect on cell proliferation of neighbouring cells, similar to that exerted by human aTf. These data imply that Tf has to be secreted to the culture medium and bind to its receptors on the extracellular side of the plasma membrane, rather than acting directly within the cell cytoplasm of Tf-overexpressing cells.
The role played by iron and its relationship with the aTf used to supplement the culture medium was analysed by iron supplementation experiments and evaluating the DNA content in CTL and aTf-treated N20.1 cells. Previous reports have demonstrated the negative effect of iron depletion on cell proliferation in vitro (Renton and Jeitner, 1996; Fu and Richardson, 2007; Nurtjahja-Tjendraputra et al., 2007) and in vivo (Richardson, 2005; Steegmann-Olmedillas, 2011). Iron must be in its ferric form to bind to the Tf protein. Furthermore, Tf in its diferric form (holoTransferrin) has an average affinity for its receptor at least 50 times higher than aTf at a physiological pH. When ferric iron was added to the culture medium at low doses together with aTf, the DNA content increased compared with the same treatment lacking aTf, and suggests that ferric iron was able to (i) bind to Tf and increase its saturation and (ii) promote the binding to its receptors. However, the addition of higher concentrations of ferric iron to the culture medium did not have the same effect, and may be indicative of cell damage due to iron overload.
The possible negative effects of ferrous iron addition to the culture medium might be related to an oxidative stress burden mediated by free radical production through the Fenton reaction. The fact that aTf addition to the culture medium did not reverse the negative effects of the ferrous iron might be highlighting the inability of iron in its ferrous state to bind to aTf. It is possible that the cell line might lack an effective ferroxidase system that oxidizes the ferrous iron to its ferric form, such as that provided by astroglial ceruloplasmin.
However, further determinations will be needed in the future to confirm if the effects of Tf on cell proliferation are mediated in these culture systems by supplying the cells with the much-required iron for cell cycle progression and DNA replication (Yu et al., 2007). Other mechanisms might be involved as well in the proliferation process, such as the activation of the MAPK (mitogen-activated protein kinase)-ERK1/2 (extracellular-signal-regulated kinase 1/2) transduction signalling pathways downstream of the Tf–TfRc2 complex formation (Calzolari et al., 2006). Besides, it would be interesting to neatly discriminate the weight each of the TfRc have on proliferation after Tf treatment in culture. A possible answer might be reached with antibodies with blocking activity that recognize TfRc2, or by using interference RNA to silence either TfRc. In addition, the different TfRc2 isoforms might have differential roles in SVZ-derived cells in response to Tf.
Overall, the data presented in this article provide evidence for the use of Tf in the culture media to improve cell amplification of SVZ-derived cells, considering their implications in the upcoming field of NSC (neural stem cell) replacement therapies for CNS diseases (Low et al., 2008; Kim and de Vellis, 2009; Buchet et al., 2011; Feng and Gao, 2012; Gögel et al., 2011; Potter et al., 2011). Some treatments have even been suggested for clinical trials (Trounson et al., 2011). Furthermore, in the industrial biotechnology field there is great interest in finding alternate components that can replace animal-derived supplements in culture media, and Tf is one of them. Human Tf has proven to have a larger growth-promoting effect than the bovine analogue on human cells (Brandsma et al., 2011).
The aim of this work is to provide a contribution to the culturing of neural cells and data that could be considered when formulating the culture medium for NPC amplification in vitro.
Online data
ACKNOWLEDGEMENTS
We thank Dr M. Pandolfo and Dr C. Caravia (Secc. de Hematología, Dpto. De Análisis Clínicos, Hospital de Clínicas José de San Martin, CABA) for the help with the iron determinations in FCS. We also thank Dr A. Ferrari (Laboratorio de Immunología Básica Aplicada y Patológica, IDEHU, UBA-CONICET) for his dedication and help with the DNA fluorimetric determinations, and Julia Perez (IQUIFIB, UBA-CONICET) for the constructive critiques provided during the preparation of the paper. We thank Dr A. Paganelli (IBCN, UBA-CONICET) for sharing the anti-acetylated α-tubulin antibody.
FUNDING
This work was supported by the CONICET [grant number PIP 112-200801-01767], ANPCyT [grant number BID-PICT 0791], UBACyT [grant number 20020090200481] and ANPCyT [grant number BID-PICT 2040].
References
- Anderson G, Vulpe C. Mammalian iron transport. Cell Mol Life Sci. 2009;66:3241–3261. doi: 10.1007/s00018-009-0051-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloch B, Popovici T, Levin M, Tuil D, Kahn A. Transferrin gene expression visualized in oligodendrocytes of the rat brain by using in situ hybridization and immunohistochemistry. Proc Natl Acad Sci USA. 1985;82:6706–6710. doi: 10.1073/pnas.82.19.6706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandsma M, Jevnikar A, Ma S. Recombinant human transferrin: beyond iron binding and transport. Biotechnol Adv. 2011;29:230–238. doi: 10.1016/j.biotechadv.2010.11.007. [DOI] [PubMed] [Google Scholar]
- Brazel C, Limke T, Osborne J, Miura I, Cai J, Pevny L, Rao M. Sox expression defines a heterogeneous population of neurosphere-forming cells in the adult murine brain. Aging Cell. 2005;4:197–207. doi: 10.1111/j.1474-9726.2005.00158.x. [DOI] [PubMed] [Google Scholar]
- Brewer G, Torricelli J, Evege E, Price P. Optimized survival of hippocampal neurons in B27-supplemented neurobasal™, a new serum-free medium combination. J Neurosci Res. 1993;35:567–576. doi: 10.1002/jnr.490350513. [DOI] [PubMed] [Google Scholar]
- Broadwell R, Baker-Cairns B, Friden P, Oliver C, Villegas J. Transcytosis of protein through the mammalian cerebral epithelium and endothelium. Exp Neurol. 1996;142:47–65. doi: 10.1006/exnr.1996.0178. [DOI] [PubMed] [Google Scholar]
- Buchet D, Garcia C, Deboux C, Nait-Oumesmar B, Baron-Van Evercooren A. Human neural progenitors from different foetal forebrain regions remyelinate the adult mouse spinal cord. Brain. 2011;134:1168–1183. doi: 10.1093/brain/awr030. [DOI] [PubMed] [Google Scholar]
- Calzolari A, Raggi C, Deaglio S, Sposi N, Stafsnes M, Fecchi K, Parolini I, Malavasi F, Peschle C, Sargiacomo M, Testa U. TfR2 localizes in lipid raft domains and is released in exosomes to activate signal transduction along the MAPK pathway. J Cell Sci. 2006;119:4486–4498. doi: 10.1242/jcs.03228. [DOI] [PubMed] [Google Scholar]
- Cao R, Chen K, Song Q, Zang Y, Li J, Wang X, Chen P, Liang S. Quantitative proteomic analysis of membrane proteins involved in astroglial differentiation of neural stem cells by SILAC labeling coupled with LC-MS/MS. J Proteome Res. 2012;11:829–838. doi: 10.1021/pr200677z. [DOI] [PubMed] [Google Scholar]
- Connor J, Menzies S. Cellular management of iron in the brain. J Neurol Sci. 1995;134:33–44. doi: 10.1016/0022-510x(95)00206-h. [DOI] [PubMed] [Google Scholar]
- Chirasani S, Markovic D, Synowitz M, Eichler S, Wisniewski P, Kaminska B, Otto A, Wanker E, Schäfer M, Chiarugi P, Meier J, Kettenmann H, Glass R. Transferrin-receptor-mediated iron accumulation controls proliferation and glutamate release in glioma cells. J Mol Med. 2009;87:153–167. doi: 10.1007/s00109-008-0414-3. [DOI] [PubMed] [Google Scholar]
- Dautry-Varsat A. Receptor-mediated endocytosis: the intracellular journey of transferrin and its receptor. Biochimie. 1986;68:375–381. doi: 10.1016/s0300-9084(86)80004-9. [DOI] [PubMed] [Google Scholar]
- Dimou L, Simon C, Kirchoff F, Takebayashi H, Götz M. Progeny of Olig2-expressing progenitors in the gray and white Matter of the adult mouse cerebral cortex. J Neurosci. 2008;28:10434–10442. doi: 10.1523/JNEUROSCI.2831-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson R, Paucar A, Jackson R, Visnyei K, Kornblum H. Roles of insulin and transferrin in neural progenitor survival and proliferation. J Neurosci Res. 2008;86:1884–1894. doi: 10.1002/jnr.21631. [DOI] [PubMed] [Google Scholar]
- Eriksson P, Perfilieva E, Björk-Eriksson T, Alborn A, Nordborg C, Peterson D, Gage F. Neurogenesis in the adult human hippocampus. Nat Med. 1998;4:1313–1317. doi: 10.1038/3305. [DOI] [PubMed] [Google Scholar]
- Escobar Cabrera O, Bongarzone E, Soto E, Pasquini J. Single intracerebral injection of apotransferrin in young rats induces increased myelination. Dev Neurosci. 1994;16:248–254. doi: 10.1159/000112116. [DOI] [PubMed] [Google Scholar]
- Escobar Cabrera O, Zakin M, Soto E, Pasquini J. Single intracranial injection of apotransferrin in young rats increases the expression of specific myelin protein mRNA. J Neurosci Res. 1997;47:603–608. [PubMed] [Google Scholar]
- Espinosa de los Monteros A, Chiapelli F, Fisher R, de Vellis J. Transferrin: an early marker of oligodendrocytes in culture. Int J Dev Neurosci. 1988;6:167–175. doi: 10.1016/0736-5748(88)90042-1. [DOI] [PubMed] [Google Scholar]
- Falcão A, Marques F, Novais A, Sousa N, Palha J, Sousa J. The path from the choroid plexus to the subventricular zone: go with the flow! Front Cell Neurosci. 2012;6:1–8. doi: 10.3389/fncel.2012.00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Z, Gao F. Stem cell challenges in the treatment of neurodegenerative disease. CNS Neurosci Ther. 2012;18:142–148. doi: 10.1111/j.1755-5949.2011.00239.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster L, Phan T, Verity A, Bredesen D, Campagnoni A. Generation and analysis of normal and shiverer temperature-sensitive immortalized cell lines exhibiting phenotypic characteristics of OLs at several stages of differentiation. Dev Neurosci. 1993;15:100–109. doi: 10.1159/000111322. [DOI] [PubMed] [Google Scholar]
- Fu D, Richardson D. Iron chelation and regulation of the cell cycle: 2 mechanisms of posttranscriptional regulation of the universal cyclin-dependent kinase inhibitor p21CIP1/WAF1 by iron depletion. Blood. 2007;110:752–761. doi: 10.1182/blood-2007-03-076737. [DOI] [PubMed] [Google Scholar]
- Gallagher S. Quantification of DNA and RNA with absorption and fluorescence spectroscopy. Curr Protoc Cell Biol. 2000:A.3D.1–A.3D.8. doi: 10.1002/0471143030.cba03ds07. [DOI] [PubMed] [Google Scholar]
- Gögel S, Gubernator M, Minger S. Progress and prospects: stem cells and neurological diseases. Gene Therapy. 2011;18:1–6. doi: 10.1038/gt.2010.130. [DOI] [PubMed] [Google Scholar]
- Gould E. How widespread is adult neurogenesis in mammals? Nat Rev. 2007;3:481–488. doi: 10.1038/nrn2147. [DOI] [PubMed] [Google Scholar]
- Guardia Clausi M, Pasquini L, Soto E, Pasquini J. Apotransferrin-induced recovery after hypoxia/ischemic injury on myelination. ASN Neuro. 2010;2:e00048. doi: 10.1042/AN20100020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jablonska B, Aguirre A, Raymond M, Szabo G, Kitabatake Y, Sailor K, Ming G, Song H, Gallo V. Chordin-induced lineage plasticity of adult SVZ neuroblasts after demyelination. Nat Neurosci. 2010;13:541–550. doi: 10.1038/nn.2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawabata H, Yang R, Hirama T, Vuong P, Kawano S, Gombart A, Koeffler H. Molecular cloning of transferrin receptor 2. A new member of the transferrin receptor-like family. J Biol Chem. 1999;274:20826–20832. doi: 10.1074/jbc.274.30.20826. [DOI] [PubMed] [Google Scholar]
- Kim S, de Vellis J. Stem cell-based cell therapy in neurological diseases: a review. J Neurosci Res. 2009;87:2183–2200. doi: 10.1002/jnr.22054. [DOI] [PubMed] [Google Scholar]
- Kokovay E, Shen Q, Temple S. The incredible elastic brain: how neural stem cells expand our minds. Neuron. 2009;60:420–429. doi: 10.1016/j.neuron.2008.10.025. [DOI] [PubMed] [Google Scholar]
- Kriegstein A, Alvarez-Buylla A. The glial nature of embryonic and adult neural stem cells. Annu Rev Neurosci. 2009;32:149–184. doi: 10.1146/annurev.neuro.051508.135600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitner D, Connor J. Functional roles of transferin in the brain. Biochim Biophys Acta. 2012;1820:393–402. doi: 10.1016/j.bbagen.2011.10.016. [DOI] [PubMed] [Google Scholar]
- Lois C, Alvarez-Buylla A. Proliferating subventricular zone cells in the adult mammalian forebrain can differentiate into neurons and glia. Proc Natl Acad Sci USA. 1993;90:2074–2077. doi: 10.1073/pnas.90.5.2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low C, Liou Y, Tang B. Neural differentiation and potential use of stem cells from the human umbilical cord for central nervous system transplantation therapy. J Neurosci Res. 2008;86:1670–1679. doi: 10.1002/jnr.21624. [DOI] [PubMed] [Google Scholar]
- Marta C, Escobar Cabrera O, Garcia C, Villar M, Pasquini J, Soto E. Oligodendroglial cell differentiation in rat brain is accelerated by the intracranial injection of apotransferrin. Cell Mol Biol. 2000;46:529–539. [PubMed] [Google Scholar]
- Merkle F, Alvarez-Buylla A. Neural stem cells in mammalian development. Curr Opin Cell Biol. 2006;18:704–709. doi: 10.1016/j.ceb.2006.09.008. [DOI] [PubMed] [Google Scholar]
- Miller F, Gauthier-Fisher A. Home at last: neural stem cell niches defined. Cell Stem Cell. 2009;4:507–510. doi: 10.1016/j.stem.2009.05.008. [DOI] [PubMed] [Google Scholar]
- Mirzadeh Z, Merkle F, Soriano-Navarro M, Garcia-Verdugo J, Alvarez-Buylla A. Neural stem cells confer unique pinwheel architecture to the ventricular surface in neurogenic regions of the adult brain. Cell Stem Cell. 2008;3:265–278. doi: 10.1016/j.stem.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moos T, Rosengren Nielsen T, Skjorringe T, Morgan E. Iron trafficking inside the brain. J Neurochem. 2007;103:1730–1740. doi: 10.1111/j.1471-4159.2007.04976.x. [DOI] [PubMed] [Google Scholar]
- Morris C, Candy J, Keith A, Oakley A, Taylor G, Pullen R, Bloxham C, Gocht A, Edwardson J. Brain iron homeostasis. J Inorg Biochem. 1992;41:257–265. doi: 10.1016/0162-0134(92)84071-t. [DOI] [PubMed] [Google Scholar]
- Nait-Oumesmar B, Picard-Riera N, Kerninon C, Decker L, Seilhean D, Höglinger G, Hirsch E, Reynolds R, Baron-Van Evercooren A. Activation of the subventricular zone in multiple sclerosis: Evidence for early glial progenitors. Proc Natl Acad Sci USA. 2007;104:4694–4699. doi: 10.1073/pnas.0606835104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurtjahja-Tjendraputra E, Fu D, Phang J, Richardson D. Iron chelation regulates cyclin D1 expression via the proteasome: a link to iron deficiency-mediated growth suppression. Blood. 2007;190:4045–4054. doi: 10.1182/blood-2006-10-047753. [DOI] [PubMed] [Google Scholar]
- Picard-Riéra N, Decker L, Delarasse C, Goude K, Nait-Oumesmar B, Liblau R, Pham-Dinh D, Evercooren A. Experimental autoimmune encephalomyelitis mobilizes neural progenitors from the subventricular zone to undergo oligodendrogenesis in adult mice. Proc Natl Acad Sci USA. 2002;99:13211–13216. doi: 10.1073/pnas.192314199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter G, Rowich D, Petryniak M. Myelin restoration: progress and prospects for human cell replacement therapies. Arch lmmunol Ther Exp. 2011;59:179–193. doi: 10.1007/s00005-011-0120-7. [DOI] [PubMed] [Google Scholar]
- Renton F, Jeitner T. Cell cycle-dependent inhibition of the proliferation of human neural tumor cell lines by iron chelators. Biochem Pharmacol. 1996;51:1553–1561. doi: 10.1016/0006-2952(96)00099-8. [DOI] [PubMed] [Google Scholar]
- Richardson D. Molecular mechanisms of iron uptake by cells and the use of iron chelators for the treatment of cancer. Curr Med Chem. 2005;12:2711–2729. doi: 10.2174/092986705774462996. [DOI] [PubMed] [Google Scholar]
- Richardson W, Young K, Tripathi R, McKenzie I. Ng2-glia as multipotent neural stem cells–fact or fantasy? Neuron. 2011;70:661–673. doi: 10.1016/j.neuron.2011.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivers L, Young K, Rizzi M, Jamen F, Psachoulia K, Wade A, Kessaris N, Richardson W. PDGFRA/NG2-positive glia generate myelinating oligodendrocytes and cortical projection neurons in the adult mouse CNS. Nat Neurosci. 2008;11:1392–1401. doi: 10.1038/nn.2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonberg D, McTigue D. Iron is essential for oligodendrocyte genesis following intraspinal macrophage activation. Exp Neurol. 2009;218:64–74. doi: 10.1016/j.expneurol.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sergent-Tanguy S, Véziers J, Bonnamain V, Boudin H, Neveu I, Naveilhan P. Cell surface antigens on rat neural progenitors and characterization of the CD3 (+)/CD3 (−) cell populations. Differentiation. 2006;74:530–541. doi: 10.1111/j.1432-0436.2006.00098.x. [DOI] [PubMed] [Google Scholar]
- Silvestroff L, Franco P, Pasquini J. Apotransferrin: dual role on adult subventricular zone-derived neurospheres. PLoS ONE. 2012;7:e33937. doi: 10.1371/journal.pone.0033937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steegmann-Olmedillas J. The role of iron in tumour cell proliferation. Clin Transl Oncol. 2011;13:71–76. doi: 10.1007/s12094-011-0621-1. [DOI] [PubMed] [Google Scholar]
- Tripathi R, Rivers L, Young K, Jamen F, Richardson W. NG2 glia generate new oligodendrocytes but few astrocytes in a murine experimental autoimmune encephalomyelitis model of demyelinating disease. J Neurosci. 2010;30:16383–16390. doi: 10.1523/JNEUROSCI.3411-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trounson A, Thakar R, Lomax G, Gibbons D. Clinical trials for stem cell therapies. BMC Med. 2011;9:52. doi: 10.1186/1741-7015-9-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trowbridge I, Lopez F. Monoclonal antibody to transferrin receptor blocks transferrin binding and inhibits human tumor cell growth in vitro. Proc Natl Acad Sci USA. 1982;79:1175–1179. doi: 10.1073/pnas.79.4.1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verity A, Bredesen D, Vonderscher C, Handley V, Campagnoni A. Expression of myelin protein genes and other myelin components in an oligodendrocyte cell line conditionally immortalized with a temperature-sensitive retrovirus. J Neurochem. 1993;60:577–587. doi: 10.1111/j.1471-4159.1993.tb03188.x. [DOI] [PubMed] [Google Scholar]
- Wang J, Pantopoulos K. Regulation of cellular iron metabolism. Biochem J. 2011;434:365–381. doi: 10.1042/BJ20101825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Kovacevic Z, Richardson D. Turning cell cycle regulation with an iron key. Cell Cycle. 2007;6:1982–1994. doi: 10.4161/cc.6.16.4603. [DOI] [PubMed] [Google Scholar]
- Zhao C, Zawadzka M, Roulois A, Bruce C, Franklin R. Promoting remyelination in multiple sclerosis by endogenous adult neural stem/precursor cells: defining cellular targets. J Neurol Sci. 2008;265:12–16. doi: 10.1016/j.jns.2007.05.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.