

Abstract
The electrophoretically-driven transport of double-stranded λ-phage DNA through focused ion beam (FIB) milled nanochannels is described. Nanochannels were fabricated having critical dimensions (width and depth) corresponding to 0.5×, 1×, and 2× the DNA persistence length – or 25 nm, 50 nm, and 100 nm, respectively. The threshold field strength required to drive transport, the threading mobility, and the transport mobility were measured as a function of nanochannel size. As the nanochannel dimensions decreased, the entropic barrier to translocation increased and transport became more constrained. Equilibrium models of confinement provide a framework in which to understand the observed trends, although the dynamic nature of the experiments resulted in significant deviations from theory. It was also demonstrated that the use of dynamic wall coatings for the purpose of electroosmotic flow suppression can have a significant impact on transport dynamics that may obfuscate entropic contributions. The non-intermittent DNA transport through the FIB milled nanochannels demonstrates that they are well suited for use in nanofluidic devices. We expect that an understanding of the dynamic transport properties reported here will facilitate the incorporation of FIB-milled nanochannels in devices for single molecule and ensemble analyses.
Keywords: DNA translocation, nanochannels, nanofluidics, entropic barrier, polyvinylpyrrolidone
The incorporation of nanofluidic components in lab-on-a-chip devices promises to enable analysis capabilities beyond those available on solely microfluidic platforms.1–4 One example that has attracted considerable attention is the use of nanofluidic channels to study the behavior of macromolecules under confinement.5–8 This is achieved by forcing macromolecules into nanochannels having widths and/or depths that are smaller than the molecules’ dimensions in free solution. Such studies are expected to advance the understanding of behavior in less stringently defined nanofluidic networks such as polymer melts, gels, and porous solids. In addition to insights gained about the ensemble behavior of macromolecules, opportunities exist for the precise characterization of single molecules. For example, single molecule analyses of DNA confined in nanochannels show promise in mapping, sizing, epigenetic analysis, and separations applications.9–20 Nanochannels have also been proposed as critical elements in single-molecule, direct-read sequencing devices.21,22 Such applications are enabled by the extension of a DNA molecule along the long axis of the nanochannel, which allows length determination and spatial mapping.
To date, the majority of these studies have focused on double-stranded DNA. It can be driven into nanochannels electrokinetically or using pressure driven flow, is readily stained with high-contrast intercalating dyes, and has a persistence length of ~50 nm in high ionic strength buffers.23,24 This length scale, which characterizes the bending stiffness of the chain, is therefore commensurate with the dimensions of nanochannels routinely fabricated using a variety of techniques.2,25–28 This fact has two important implications. First, it means that double-stranded DNA can be significantly extended in such nanochannels and thus mapped with high spatial resolution. Second, it enables experimental investigations of two distinct models of macromolecule confinement. If the nanochannel critical dimensions are significantly greater than the persistence length, then the molecule can fold over on itself and forms a string of blobs. If the nanochannel is significantly smaller than the persistence length, blob formation becomes energetically unfavorable because of the large bending strain and the molecule adopts a deflecting chain conformation. These two modes of polymer extension, under equilibrium conditions, were modeled by de Gennes and Odijk, respectively.29,30 More recently, theoretical and experimental efforts have begun to address the transition between the two regimes, with regard to the range of nanochannel sizes over which it occurs and the conformations assumed in the transition regime.31–33
The forces applied to DNA molecules required to load them into nanochannels can result in folded, compressed, or stretched conformations.11,12,17,34,35 In order to facilitate comparisons to equilibrium theories it is necessary to remove the forces driving transport and allow the molecules to relax to their lowest energy states. Many of these studies are performed in nanochannels having critical dimensions greater than 100 nm and show reasonable agreement with the predictions of de Gennes’ blob theory.14,36–38 However, measurements performed in a series of nanochannels of various sizes have shown equilibrium extension lengths, R, with a dependence on nanochannel dimensions, D, of R~D−0.8, deviating from the blob theory prediction of R~D−2/3.39,40 Data from nanochannels with dimensions smaller than the persistence length of DNA in standard electrophoresis buffers are limited. Reisner et al. observed extension consistent with Odijk’s deflecting chain model in 30 nm × 40 nm channels.39 The lack of data from nanochannels in this size range reflects the challenges of nanochannel fabrication below ~50 nm. An alternative approach to realizing deflecting chain conformations is to increase the DNA persistence length by lowering the ionic strength of the buffer, thereby reducing counter-ion condensation.11,41–46 Several groups have observed commensurately greater extension in nanoslits and nanochannels at ionic strengths below ~10 mM.11,41–43 It has been noted that the low ionic strength conditions employed in these experiments will also result in double layer overlap in nanochannels with charged surfaces, effectively depleting co-ion and enriching counter-ion concentrations.5 The effects of ionic strength on DNA and the nanochannel environment can be difficult to deconvolve.
This highlights the importance of considering how phenomena that are unique to nanofluidic channels may perturb behavior intrinsic to DNA’s molecular properties. For example, as the nanochannel size approaches molecular dimensions, hydrodynamic interactions are effectively screened, molecule-wall interactions become important, and electrical double-layer overlap can occur.47–49 Because of these phenomena, the nanochannel surface characteristics (i.e., surface chemistry, charge, and topography) may strongly affect transport dynamics. Indeed, in surveying studies that have explored the pressure or electrokinetically driven transport of DNA molecules through nanoslits and nanochannels, it becomes apparent that transport dynamics can be quite complex and sensitive to the nanochannels’ dimensions, materials, and method of fabrication.12,17,50–54 In some cases, the velocities of electrokinetically driven DNA molecules vary along the length of the nanochannels and may not be directly proportional to the applied voltage.51,52,54 Cross et al. observed size dependent DNA mobility in 19-nm deep but not in 70-nm deep nanoslits fabricated in fused silica.53 Campbell et al. observed an increase in DNA mobility with a decrease in the dimensions of FIB-milled nanochannels in a silicon device.51 This was attributed to the suppression of electroosmotic flow in smaller nanochannels due to double-layer overlap. In the studies cited above, the observed mobilities ranged from 6×10−5 to 2.5×10−3 cm2 V−1 s−1, depending on device and buffer conditions.
The dynamic properties of single DNA molecules have also been monitored as the molecules relax to or fluctuate around their equilibrium conformations and diffuse in the nanofluidic structures.11,14,17,34,35,37–39,50,55–59 This behavior can be understood in the context of the conformational models described above. Adoption of a particular conformation affects not only static extension but also dynamic properties since the coefficient of drag is dependent upon the molecular configuration.5,39 Diffusion and relaxation are found to be slower in nanochannels and nanoslits compared to bulk solution, consistent with confinement scaling theories.5,38,57,58 These studies have primarily imaged single molecules at equilibrium or under modest applied forces. In the case of molecules being rapidly driven through nanochannels, configurations and dynamics become more complex.12,60
We recently reported the fabrication of nanochannels in quartz substrates with critical dimensions as small as 5 nm using focused ion beam (FIB) milling.61 Here we describe the transport of double-stranded λ-phage DNA (48.5 kbp) through unity aspect ratio nanochannels thus fabricated with dimensions 0.5×, 1×, and 2× the biopolymer’s persistence length (i.e., 25 nm, 50 nm and 100 nm, respectively). We focus on dynamic transport properties that are relevant to “on-the-fly” single-molecule characterizations, in contrast to measurements on pre-loaded and equilibrated molecules. Specifically, we determined the voltage necessary to initiate transport, the velocity of DNA threading into the nanochannels, and the electrophoretic mobility of DNA in the various nanochannels. A comparison of transport in electrophoresis buffers lacking or including polyvinylpyrrolidone (PVP), an additive to suppress electroosmotic flow, offers some additional insights on the impact of this routinely used surface coating.
EXPERIMENTAL SECTION
Device Fabrication
A series of experimental devices, one for each nanochannel dimension, were fabricated in quartz substrates. Each consisted of an array of ten identical 50-μm long nanochannels interfaced to microfluidic channels. The microfluidic channels were patterned using standard photolithography and wet etching techniques and were typically 20-μm wide and 5-μm deep. Access vias were drilled from the substrate backside using abrasive powder blasting. The nanochannels were patterned by FIB milling through a chromium film, a process described in detail elsewhere,61 using a Helios NanoLab 600 DualBeam instrument (FEI Company). After milling, the Cr was removed from the substrate using a Chromium Mask Etchant (Transene Company, Inc.) and a thin AuPd film was sputtered for imaging with SEM using the DualBeam instrument. The AuPd film was removed with an aqua regia etch. A substrate and a 0.13-mm thick quartz coverslip (Esco Products, Inc.) were cleaned using Nanostrip 2X (Cyantek Corporation), brought into contact, and heated in a furnace to 900°C to achieve fusion bonding. Devices that are subjected to an identical thermal treatment without being sealed with a coverslip were imaged using SEM to ensure that nanochannel collapse did not occur under these conditions. Reservoirs were affixed over the vias on the backside with UV-cured epoxy.62 Microfluidic devices were also fabricated without FIB-milled nanochannels in which 20-μm wide × 5-μm deep microchannels were patterned in a simple cross geometry. These devices were used to determine the electroosmotic mobility in the buffers used in the DNA transport experiments by measuring the mobility of a neutral fluorescent dye, Rhodamine B, that was introduced to the analysis channel using a pinched injection.63,64
Sample Preparation and Transport Measurements
DNA transport experiments were conducted in 2× TBE electrophoresis buffer [180 mM Tris base (Fisher Scientific), 180 mM boric acid (Mallinckrodt Chemicals), and 4 mM EDTA (Sigma-Aldrich)]. In some experiments, polyvinylpyrrolidone (PVP, 10 kDa molecular weight, Sigma-Aldrich) was added to the buffer (2% by mass) as a dynamic coating to suppress electroosmotic flow (EOF). Additionally, β-mercaptoethanol (Fisher Scientific) was added to all solutions (4% by volume) to reduce photoinduced DNA cleavage.34 Solutions were initially introduced to the nanofluidic devices by adding filtered methanol to all reservoirs and pulling vacuum on one of the reservoirs of each pair addressing a microfluidic channel (~1 bar) for 10 min. The reservoirs were then emptied and this was repeated twice with filtered distilled deionized water, then twice with the electrophoresis buffer. After filling the micro and nanochannels with buffer, the ionic conductance was measured between all reservoir pair combinations in order to determine the fraction of the applied voltage dropped across the nanochannel arrays. The percentage of the applied voltage dropped across the nanochannel arrays was 51%, 73%, and 81% for the 100-nm, 50-nm, and 25-nm arrays, respectively, with the remainder of the voltage dropped across the microfluidic channels that accessed the nanochannel arrays. The electric fields reported in this Article were calculated from the voltage dropped across the nanochannel arrays and the measured length of the nanochannels. After the transport experiments, these ionic conductance measurements were repeated to verify that device performance remained constant during the experiments (see Supporting Information for additional details).
λ-phage DNA (48.5 kbp, Promega Corporation) was used as received and stained with YOYO-1 intercalating dye (Invitrogen) at a base-pair:dye ratio of 5:1. Stock solutions were allowed to equilibrate at room temperature overnight prior to dilution for experiments. Experiments were run using solutions having a DNA concentration of 0.5 ng/μL (16 pM). DNA molecules were electrokinetically driven through the nanochannels by immersing platinum electrodes in the reservoirs on either side of the nanochannel array and applying a bias using a variable voltage DC power supply (Agilent). Being negatively charged, DNA was driven towards the anode. Fluorescence microscopy was performed on an Eclipse TE2000-U inverted microscope (Nikon) using a 100×/1.4 NA oil immersion objective (Nikon). Fluorescence was excited and observed using a 100-W mercury arc lamp and a GFP-3035B filter set (Semrock). Images were acquired using a Cascade II EM-CCD camera (Photometrics) collecting at 350–400 frames s−1. Images were recorded and analyzed using NIS Elements Advanced Research software (Nikon). A binary threshold was manually defined to delimit the intensity of a fluorescent DNA molecule from the background. The automated measurement of this binary layer tabulated the length of the molecule and the coordinates of the leading end, trailing end, and intensity-weighted center of mass in each frame. These parameters, together with the recorded time for each frame and the electric field strength driving transport were used to generate the results presented in this Article.
RESULTS AND DISCUSSION
The FIB milled nanochannels used here have approximately square cross-sections with nearly vertical sidewalls. Figure 1 shows a scanning electron microscope (SEM) image of one of the nanochannel arrays, along with the cross-section images used to determine the actual nanochannel dimensions (width × depth) of 25 nm × 29 nm, 57 nm × 49 nm, and 98 nm × 103 nm. The contiguousness and low surface roughness of the nanochannels were also verified for all nanochannels during SEM imaging and prior to device bonding. Atomic force microscopy (AFM) scans of FIB-milled features similar to the nanochannels in these arrays confirm the low roughness of the bottom surface (Supporting Information). For nanochannels of this size, analysis of both the SEM and AFM images indicates standard deviations in nanochannel width and depth of ~0.5 nm.61 DNA transport through nanochannels that satisfied these quality requirements occurred with constant velocity (i.e., sticking or trapping in the nanochannels was rare). A representative series of frames from each of the nanochannel devices is presented in Figure 2.
Figure 1.

Scanning electron micrographs of nanochannels interfaced to a microfluidic device showing (a) a top view of an array of 50-μm long nanochannels and (b–d) tilted views (52°) of nanochannel cross-sections having critical dimensions of (b) 100 nm, (c) 50 nm, and (d) 25 nm. The roughness observed in the cross-section micrographs is due to a 2-nm AuPd film sputtered on the surfaces for imaging. A schematic (e) showing the experimental setup where stained DNA solutions are added to the device reservoirs, DNA is electrokinetically driven through the nanochannels, and transport events are observed using fluorescence microscopy. The inset shows a magnified view of the nanochannel array.
Figure 2.

Representative series of frames showing the transport of fluorescently-stained λ-DNA molecules through a single (a) 100-nm nanochannel, (b) 50-nm nanochannel, and (c) 25-nm nanochannel. Scale bars are 20 μm. The time between frames in each of these series is approximately 2.8 ms.
The introduction of a DNA molecule into a nanochannel smaller than the molecule’s hydrodynamic radius requires a force sufficient to exceed the entropic barrier. This requisite force has not been well characterized in nanochannel experiments since many such experiments measure DNA molecules that have been introduced into the nanochannels and then allowed to equilibrate after the transport driving force (i.e., pressure or voltage) has been removed. However, when on-the-fly characterizations of DNA molecules during transport are desired, the entropic barrier establishes a lower limit to the driving force, and consequently to the molecules’ transport velocity. Large entropic barriers, and fast transport velocities, may exceed the temporal resolution of some detection methods, such as the fluorescence imaging used in these studies. We therefore characterized the threshold electric field strengths required to drive transport through the various nanochannels.
This was accomplished by decreasing the driving voltage until the frequency of translocation events dropped to ~0.002 s−1, or ~1 event/10 min. This event frequency was comparable to near threshold capture rates measured using solid-state nanopores.65 This approach overestimates the value of the true entropic barrier, since translocations are, in fact, observed. However, it provides an estimate of an experimentally practical threshold that is consistent across devices. The threshold field strengths thus measured are plotted in Figure 3 as a function of nanochannel dimensions in both 2X TBE and 2X TBE containing 2% PVP. Initially, experiments were attempted using 1X TBE electrophoresis buffer solutions. However, we found that DNA molecules could not be electrophoretically driven through the nanochannels in the absence of the EOF suppressor. While the net DNA mobility remained towards the positively biased electrode, the EOF through the nanochannels appeared to produce a counterflow sufficient to inhibit DNA threading into the nanochannels. At high field strengths (>5 kV/cm) DNA migrated toward the nanochannel array but was excluded from the regions near the nanochannel entrances, with the depletion regions increasing in volume as the applied voltage was increased. These effects are similar to behavior reported by Cross et al. for 19-nm and 70-nm deep nanoslits in fused silica using 5X TBE buffers.53 While this counterflow can be effectively reduced by the addition of EOF suppressors such as PVP, it may also be desirable to monitor DNA behavior in additive-free buffers. A change in electrolyte concentration was therefore explored as an alternative means to decrease the electroosmotic flow. In nanochannels, EOF can be reduced by decreasing the buffer’s ionic strength and inducing electrical double-layer overlap.49,66 Alternatively, increasing the buffer concentration can also reduce EOF by shielding surface charge on the channel walls and consequently decreasing their zeta potential.67–69 Because the EOF at low ionic strengths would be suppressed to significantly different degrees for various nanochannel sizes, we applied the second strategy to ensure more similar electroosmotic mobility in the three nanochannel sizes investigated here. In our devices, merely doubling the ionic strength was sufficient for electrophoretically-driven transport to dominate. For 2X TBE, the Debye length is estimated to be ~1 nm and the consequent suppression in the electroosmotic flow in the smallest nanochannels is expected to be ~10% of flow in the microfluidic channels.49,69,70 This corresponds to a small error in the DNA mobilities due to slightly different electroosmotic mobilities in each device. In nanofluidic devices where the wall zeta potential differs due to differences in materials, fabrication methods, or device conditioning protocols, other buffer conditions may be optimal.
Figure 3.

Dependence of the threshold field strength on the nanochannel dimensions, measured in 2X TBE and 2X TBE with 2% PVP added to suppress electroosmotic flow. The scalings shown are empirical fits to the data and deviate from theoretical predictions, most significantly upon addition of PVP to the buffer. The error bars represent the standard deviations of triplicate measurements for each device and buffer.
In 2X TBE the threshold field strength is inversely proportional to the nanochannel critical dimension, D, the geometric average of the width and depth for nanochannels with approximately square cross sections. This behavior is intermediate between the expectations predicted by the de Gennes and Odijk models for which the free energy of confinement scales with D−5/3 and D−2/3, respectively.29–31,71 As noted above, recent theoretical work has begun to examine the gradual transition between these two regimes.31–33 A comparison between experiment and these emerging theories will require investigations with a large number of nanochannel dimensions over a wider size range. Nonetheless, we can conclude that the observed results are in qualitative agreement with theory and that the increase of this entropic barrier with decreasing nanochannel dimensions has practical implications for studies monitoring DNA molecules during transport. By reducing the EOF, which runs counter to DNA migration, we expect to see a reduction in the field strengths required to drive translocation. Upon addition of 2% PVP to the buffer, a different trend in the threshold field strength also emerges, in which it increases linearly with a decrease in nanochannel size. This change in behavior suggests contributions that are not considered in the confinement models. Such effects may include hydrodynamic forces at the nanochannel entrances or enthalpic contributions due to short-range attractive interactions between DNA and the wall coating.72 While the exact mechanism is unclear, the inclusion of PVP in the system results in a significant decrease in the voltages required to drive transport in the smallest nanochannels.
As mentioned above, one motivation for reducing the field strength is to lessen the detection requirements vis-à-vis sampling frequency and sensitivity. A second consideration is the minimization of molecular deformations resulting from large strain rates. It has been previously reported that the significant stress experienced by DNA molecules as they are threaded into nanochannels results in non-equilibrium conformations that take several seconds to relax.11,12,17,34,35 In the present case, DNA stretching occurs during threading due to resistance by the entropic force and the drag experienced by the portion of the molecule in the microchannel. Once the DNA molecule is pulled fully into the nanochannel, a degree of relaxation occurs, as is apparent in the frame series in Figure 2. Due to the relatively short length of these nanochannels, however, the maximum translocation time is ~100 ms and the molecule is not expected to be fully equilibrated to its confinement-induced conformation. The extension lengths of λ-phage DNA, measured both immediately after full insertion into the nanochannels and near the exit of the nanochannels, are shown in Figure 4 and compared to the values predicted by the de Gennes and Odijk models:
| (1) |
| (2) |
where L is the DNA contour length, P is its persistence length, and w its effective diameter.5 We use values of L=20 μm (accounting for the effect of staining), P=50 nm, and w=3 nm (accounting for electrostatic contributions).12,56 Although the dynamic extension lengths cannot be quantitatively compared to the equilibrium models, the theoretical values provide a useful frame of reference. The extension lengths increase with confinement, as expected, and are found to be greater than those predicted by the de Gennes model, as previously reported.12 We also note that there is no discernible effect of PVP addition on the extension lengths. For applications such as DNA sizing and mapping, the perceived advantages of operating a nanofluidic device in a constant driving mode are simplicity and throughput. The stretching of DNA molecules during their introduction into the nanochannels may provide additional spatial resolution beyond that expected from confinement-induced extension. It remains to be seen if such gains would be offset by uncertainties from the greater distribution of pre-equilibrated conformations.60
Figure 4.

Extension lengths as determined from the fluorescence images. The initial lengths describe the average lengths immediately after the DNA molecules have been pulled completely into the nanochannels. The final lengths are measured near the end of the nanochannels, before ejection into the microchannel. These dynamic values are compared to the equilibrium values predicted by the de Gennes (dashed line) and Odijk (dashed-dotted line) theories. N=10–20 molecules analyzed for each device and buffer.
We consider now the effects of confinement on the electrophoretic mobility of λ-phage DNA. The translocation process consists of three distinct stages: threading or injection into the nanochannel, transport within the nanochannel, and ejection from the nanochannel exit. The threading process is expected to be the slowest because it is resisted by entropic and frictional forces.34,73 Conversely, the ejection process is expected to be the fastest as the entropic force adds to the electrostatic force driving transport in the nanochannel.34,74 With the present data, it was possible to quantify the dynamics of the threading and transport processes; the effect of the entropic force on ejection was not discernible in these experiments.
An inspection of the frame series in Figure 2 reveals that DNA is transported through each nanochannel with a constant velocity. This is in contrast to earlier reports that indicated the presence of steric or dielectrophoretic trapping sites in some devices that resulted in intermittent stationary or slowed behavior.51,52,54,75 The behavior observed in our FIB-milled nanofluidic devices suggests the absence of such trapping sites, confirming the low roughness of the nanochannels apparent in electron micrographs. We note that intermittent transport can also result from enthalpic interactions between DNA molecules and the nanochannel surface. In such cases, stick-slip motion would be observed for an attractive DNA/wall interaction if the driving force was less than the interfacial forces.76 We do not observe such effects, however, indicating that the electrostatic forces in our experiments overwhelm surface interactions and/or that the dynamics occur so quickly that they are not measurable on our experimental time scale.
The dependence of velocity on field strength also conforms to expectations, appearing linear in the voltage range investigated. Figure 5 shows this linear dependence for DNA molecules being transported through the various nanochannels in 2X TBE with 2% PVP. The DNA mobilities measured in the various nanochannels are shown in Figure 6. These values were determined from center of mass measurements performed on images in which a DNA molecule was fully contained within the nanochannel (i.e., no portion of the molecule extended into either microchannel), thus avoiding contributions from the threading or ejection processes. Furthermore, in order to facilitate the comparison of transport behavior in the two electrolyte solutions, the values were corrected for the contribution of EOF by adding the electroosmotic mobilities measured in microfluidic devices to the DNA mobilities. The electroosmotic mobilities were found to be 1.58(±0.01) ×10−4 and 0.79(±0.01) ×10−4 cm2 V−1 s−1 in 2X TBE and 2X TBE with 2% PVP, respectively.
Figure 5.

Linear relationship between λ-phage DNA velocity and field strength as measured in 2X TBE with 2% PVP added to suppress electroosmotic flow. N=10–20 molecules analyzed for each device and field strength.
Figure 6.
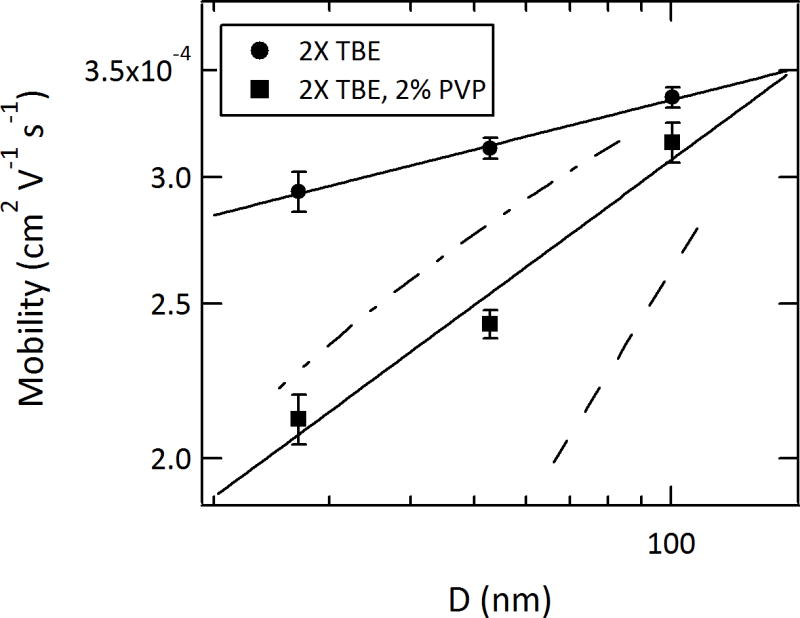
Effect of increased confinement on the electrophoretic mobility of λ-phage DNA. The decrease in mobility with nanochannel dimensions is weaker than predicted by blob (de Gennes) scaling. See text for details. N=20–40 molecules analyzed for each device and buffer.
In both buffers, the DNA electrophoretic mobility was found to decrease with decreasing nanochannel size. In an earlier study of DNA mobility in a series of larger nanochannels (having dimensions of 150 nm × 180 nm, 170 nm × 340 nm, and 240 nm × 400 nm (width × depth)) Campbell et al. observed the opposite trend, and EOF reduction due to electrical double-layer overlap was postulated as an explanation.51 In the present study, it should be noted that the electrophoretic mobilities measured in 100-nm nanochannels show little significant decrease from that observed in bulk solution. This suggests that confinement-induced perturbations to electrophoretic mobility do not begin to emerge until the critical nanochannel dimensions approach 100 nm. In larger nanochannels, particularly in the lower ionic strength buffer used by Campbell et al., EOF effects are likely more important. We note that the trends observed in the two buffers, represented by the fits in Figure 6, converge at a value approximately equal to the bulk mobility and at a nanochannel dimension of ~150 nm, which corresponds to the diameter of a Pincus thermal blob.77 In nanochannels larger than the Pincus blob diameter but smaller than the molecule’s hydrodynamic radius, molecular rotation and flexion are inhibited but the blob size and number of blobs are not determined by the nanochannel dimensions. Consequently, the hydrodynamic drag opposing migration is not strongly impacted by the nanochannel. This differs from diffusion, which remains significantly hindered in channels larger than the molecule’s hydrodynamic radius.5
Also presented in Figure 6 is the scaling expected for the de Gennes and Odijk models of confinement. The electrophoretic mobility, μep, is related to the charge per unit length, q; the length of the molecule, L; and the drag coefficient of the molecule, ξ, by the following equation:
| (3) |
In the de Gennes model,
| (4) |
where η is the solution viscosity.5 Therefore,
| (5) |
Similarly, the drag coefficient in the Odijk model is5
| (6) |
And consequently,
| (7) |
These models consider the hydrodynamic drag due to the molecular conformation and the screening of hydrodynamic interactions. They do not take into account any additional friction terms from the interaction with the nanochannel walls. Despite this omission, the experimental mobilities exhibit a weaker dependence on nanochannel dimensions than theoretically predicted. The mobilities in 2X TBE and 2X TBE with 2% PVP are found to scale with D0.10±0.01 and D0.30±0.06, respectively. We compare these results to previous studies investigating the effect of nanoslit depth on the diffusivity of double-stranded DNA molecules.56,58 Since diffusivity also scales inversely with the drag coefficient, a notable finding in those studies was that the confinement dependence of diffusivity was weaker than the expected blob theory scaling. The deviation from theory was less dramatic than our results, however, with exponents in the range of 0.45–0.55.56,58 This difference likely originates in the non-equilibrium conformations of DNA molecules during driven transport. The fact that the molecular conformation appears to be strongly influenced by the force applied during threading and transport implies a lesser contribution from confinement. In other words, the molecule does not experience the full extent of its confinement, in contrast to diffusivity measurements made on molecules equilibrated to their boundary conditions. One implication of this argument is that the velocity of molecules may decrease during transport in sufficiently long nanochannels as they relax to conformations defined by confinement.
With the addition of PVP, the electrophoretic mobility of DNA in the nanochannels decreased and became more strongly dependent upon the degree of confinement. Given the lesser force required to drive transport (Fig. 3) and the lower velocity of fluid flowing against the DNA molecules, this might result from the molecules assuming conformations having greater drag coefficients. This explanation seems unlikely, however, given the similarity in DNA extension lengths in the two buffers (Fig. 4) and the invariance in electrophoretic mobility with field strength. Alternatively, the lower mobility might be due to an increased affinity between DNA molecules and the PVP-coated nanochannel walls, or a higher viscosity in the nanochannels due to the presence of PVP.78 Such effects are expected to increase as the nanochannel dimensions decrease and the surface area per unit volume increases. An enthalpic contribution resulting from DNA-PVP affinity is consistent with the decreased voltage threshold shown in Figure 3. We suspect that the similarity in the scaling observed in 2X TBE with PVP and that predicted by the Odijk model is coincidental, considering the fact that the trends in the threshold voltage and molecular extension described above did not conform to the deflecting chain theory.
The mobilities are also given in Table 1 for the purpose of comparing them to the threading mobilities. The threading mobilities are calculated from the velocities of the leading ends of DNA molecules as they thread into the nanochannels. In practice, this consisted of analyzing the first three frames (~8 ms) of each event, providing a somewhat gross estimate of the initial threading velocity. It was expected that the threading mobilities would be slower than the transport mobilities due to the entropic and drag forces that resist DNA migration and this was found to be the case. As the threading process proceeds, however, the volume of the DNA molecule in the microchannel decreases, as does the consequent drag. The mobility is therefore expected to increase gradually from an initial minimum value to the transport value. More precisely characterizing this transition would require greater temporal resolution than was available in the present study. The differences between the threading and transport rates indicated in Table 1 for the various nanochannels provide a qualitative measure of the stretching force applied along a DNA molecule’s length. These results therefore reinforce the finding of highly strained, non-equilibrated molecules observed more directly in the fluorescence images (Figs. 2,4).
Table 1.
Summary of the Transport and Threading Mobilitiesa
| 2× TBE
|
2× TBE, 2% PVP
|
|||
|---|---|---|---|---|
| channel size (nm) | μep, transport × 104 (cm2 V−1 s−1) | μep, threading × 104 (cm2 V−1 s−1) | μep, transport × 104 (cm2 V−1 s−1) | μep, threading × 104 (cm2 V−1 s−1) |
| 100 | 3.37±0.05 | 3.2±0.1 (5%) | 3.17±0.08 | 2.8±0.1 (10%) |
| 50 | 3.13±0.05 | 2.7±0.3 (14%) | 2.43±0.05 | 1.9±0.2 (22%) |
| 25 | 2.94±0.09 | 2.2±0.1 (25%) | 2.12±0.08 | 1.2±0.1 (43%) |
In parenthesis is given the percent decrease in mobility observed for the threading process into each nanochannel, relative to the transport mobility through the same nanochannel.
CONCLUSIONS
In summary, the characteristics of DNA transport through a series of FIB milled nanochannels are described. As the nanochannel size decreases, the field strength required to drive transport increases. This results not only in faster transport velocities but also in significant extensional forces applied to DNA molecules. The increase in transport velocity is offset somewhat by lower electrophoretic mobilities in smaller nanochannels. This dependence is weaker than theoretically predicted, however, likely due to the non-equilibrium conditions of the experiments. The lack of intermittent transport dynamics indicates the suitability of the smooth-walled FIB milled nanochannels for use in nanofluidic devices. Finally, the addition of the EOF suppressor, PVP, was found to modify the behavior of DNA in significant ways. These perturbations should be considered when attempting to reconcile experimental results and theoretical predictions.
Supplementary Material
Acknowledgments
This work was sponsored in part by a grant from the National Human Genome Research Institute, National Institutes of Health (R01HG02647-05).
References
- 1.Eijkel JCT, van den Berg A. Microfluid Nanofluid. 2005;1:249–267. [Google Scholar]
- 2.Abgrall P, Nguyen NT. Anal Chem. 2008;80:2326–2341. doi: 10.1021/ac702296u. [DOI] [PubMed] [Google Scholar]
- 3.Kovarik ML, Jacobson SC. Anal Chem. 2009;81:7133–7140. doi: 10.1021/ac900614k. [DOI] [PubMed] [Google Scholar]
- 4.Piruska A, Gong M, Sweedler JV, Bohn PW. Chem Soc Rev. 2010;39:1060–1072. doi: 10.1039/b900409m. [DOI] [PubMed] [Google Scholar]
- 5.Hsieh C-C, Doyle PS. Korea-Aust Rheol J. 2008;20:127–142. [Google Scholar]
- 6.Mannion JT, Craighead HG. Biopolymers. 2007;85:131–143. doi: 10.1002/bip.20629. [DOI] [PubMed] [Google Scholar]
- 7.Levy SL, Craighead HG. Chem Soc Rev. 2010;39:1133–1152. doi: 10.1039/b820266b. [DOI] [PubMed] [Google Scholar]
- 8.Persson F, Tegenfeldt JO. Chem Soc Rev. 2010;39:985–999. doi: 10.1039/b912918a. [DOI] [PubMed] [Google Scholar]
- 9.Riehn R, Lu M, Wang Y-M, Lim SF, Cox EC, Austin RH. Proc Natl Acad Sci USA. 2005;102:10012–10016. doi: 10.1073/pnas.0503809102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reisner W, Larsen NB, Silahtaroglu A, Kristensen A, Tommerup N, Tegenfeldt JO, Flyvbjerg H. Proc Natl Acad Sci USA. 2010;107:13294–13299. doi: 10.1073/pnas.1007081107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jo K, Dhingra DM, Odijk T, de Pablo JJ, Graham MD, Runnheim R, Forrest D, Schwartz DC. Proc Natl Acad Sci USA. 2007;104:2673–2678. doi: 10.1073/pnas.0611151104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reccius CH, Stavis SM, Mannion JT, Walker LP, Craighead HG. Biophys J. 2008;95:273–286. doi: 10.1529/biophysj.107.121020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Foquet M, Korlach J, Zipfel W, Webb WW, Craighead HG. Anal Chem. 2002;74:1415–1422. doi: 10.1021/ac011076w. [DOI] [PubMed] [Google Scholar]
- 14.Tegenfeldt JO, Prinz C, Cao H, Chou S, Reisner WW, Riehn R, Wang YM, Cox EC, Sturm JC, Silberzan P, Austin RH. Proc Natl Acad Sci USA. 2004;101:10979–10983. doi: 10.1073/pnas.0403849101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cipriany BR, Zhao RQ, Murphy PJ, Levy SL, Tan CP, Craighead HG, Soloway PD. Anal Chem. 2010;82:2480–2487. doi: 10.1021/ac9028642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Han J, Craighead HG. Science. 2000;288:1026–1029. doi: 10.1126/science.288.5468.1026. [DOI] [PubMed] [Google Scholar]
- 17.Yasui T, Kaji N, Ogawa R, Hashioka S, Tokeshi M, Horiike Y, Baba Y. Anal Chem. 2011;83:6635–6640. doi: 10.1021/ac201184t. [DOI] [PubMed] [Google Scholar]
- 18.Strychalski EA, Lau HW, Archer LA. J Appl Phys. 2009;106:024915. doi: 10.1063/1.3183953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Das SK, Austin MD, Akana MC, Deshpande P, Cao H, Xiao M. Nucl Acids Res. 2010;38:e177. doi: 10.1093/nar/gkq673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lam ET, Hastie A, Lin C, Ehrlich D, Das SK, Austin MD, Deshpande P, Cao H, Nagarajan N, Xiao M, Kwok P-Y. Nat Biotech. 2012;30:771–776. doi: 10.1038/nbt.2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liang X, Chou SY. Nano Lett. 2008;8:1472–1476. doi: 10.1021/nl080473k. [DOI] [PubMed] [Google Scholar]
- 22.Min SK, Kim WY, Cho Y, Kim KS. Nat Nanotechnol. 2011;6:162–165. doi: 10.1038/nnano.2010.283. [DOI] [PubMed] [Google Scholar]
- 23.Hagerman PJ. Ann Rev Biophys Biophys Chem. 1988;17:265–286. doi: 10.1146/annurev.bb.17.060188.001405. [DOI] [PubMed] [Google Scholar]
- 24.Rye HS, Yue S, Wemmer DE, Quesada MA, Haugland RP, Mathies RA, Glazer AN. Nucleic Acids Res. 1992;20:2803–2812. doi: 10.1093/nar/20.11.2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Salieb-Beugelaar GB, Dorfman KD, van den Berg A, Eijkel JCT. Lab Chip. 2009;9:2508–2523. doi: 10.1039/b905448k. [DOI] [PubMed] [Google Scholar]
- 26.Mijatovic D, Eijkel JCT, van den Berg A. Lab Chip. 2005;5:492–500. doi: 10.1039/b416951d. [DOI] [PubMed] [Google Scholar]
- 27.Perry JL, Kandlikar SG. Microfluid Nanofluid. 2006;2:185–193. [Google Scholar]
- 28.Douville N, Huh D, Takayama S. Anal Bioanal Chem. 2008;391:2395–2409. doi: 10.1007/s00216-008-1995-y. [DOI] [PubMed] [Google Scholar]
- 29.Daoud M, de Gennes PG. J Phys (Paris) 1977;38:85–93. [Google Scholar]
- 30.Odijk T. Macromolecules. 1983;16:1340–1344. [Google Scholar]
- 31.Chen JZY, Sullivan DE. Macromolecules. 2006;39:7769–7773. [Google Scholar]
- 32.Odijk T. Phys Rev E. 2008;77:060901. doi: 10.1103/PhysRevE.77.060901. [DOI] [PubMed] [Google Scholar]
- 33.Wang Y, Tree DR, Dorfman KD. Macromolecules. 2011;44:6594–6604. doi: 10.1021/ma201277e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mannion JT, Reccius CH, Cross JD, Craighead HG. Biophys J. 2006;90:4538–4545. doi: 10.1529/biophysj.105.074732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Levy SL, Mannion JT, Cheng J, Reccius CH, Craighead HG. Nano Lett. 2008;8:3839–3844. doi: 10.1021/nl802256s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abad E, Juarros A, Retolaza A, Merino S, Marie R, Kristensen A. Microelectron Eng. 2011;88:300–304. [Google Scholar]
- 37.Thamdrup LH, Klukowska A, Kristensen A. Nanotechnology. 2008;19:125301. doi: 10.1088/0957-4484/19/12/125301. [DOI] [PubMed] [Google Scholar]
- 38.Reccius CH, Mannion JT, Cross JD, Craighead HG. Phys Rev Lett. 2005;95:268101. doi: 10.1103/PhysRevLett.95.268101. [DOI] [PubMed] [Google Scholar]
- 39.Reisner W, Morton KJ, Riehn R, Wang YM, Yu ZN, Rosen M, Sturm JC, Chou SY, Frey E, Austin RH. Phys Rev Lett. 2005;94:196101. doi: 10.1103/PhysRevLett.94.196101. [DOI] [PubMed] [Google Scholar]
- 40.Persson F, Utko P, Reisner W, Larsen NB, Kristensen A. Nano Lett. 2009;9:1382–1385. doi: 10.1021/nl803030e. [DOI] [PubMed] [Google Scholar]
- 41.Kim Y, Kim KS, Kounovsky KL, Chang R, Jung GY, de Pablo JJ, Jo K, Schwartz DC. Lab Chip. 2011;11:1721–1729. doi: 10.1039/c0lc00680g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reisner W, Beech JP, Larsen NB, Flyvbjerg H, Kristensen A, Tegenfeldt JO. Phys Rev Lett. 2007;99:058302. doi: 10.1103/PhysRevLett.99.058302. [DOI] [PubMed] [Google Scholar]
- 43.Zhang C, Zhang F, van Kan Ja, van der Maarel JRC. J Chem Phys. 2008;128:225109. doi: 10.1063/1.2937441. [DOI] [PubMed] [Google Scholar]
- 44.Odijk T. J Polym Sci. 1977;15:477–483. [Google Scholar]
- 45.Skolnick J, Fixman M. Macromolecules. 1977;10:944–948. [Google Scholar]
- 46.Dobrynin AV. Macromolecules. 2006;39:9519–9527. [Google Scholar]
- 47.Bakajin OB, Duke TAJ, Chou CF, Chan SS, Austin RH, Cox EC. Phys Rev Lett. 1998;80:2737–2740. [Google Scholar]
- 48.Das S, Chakraborty S. Electrophoresis. 2008;29:1115–1124. doi: 10.1002/elps.200700265. [DOI] [PubMed] [Google Scholar]
- 49.Rice CL, Whitehead R. J Phys Chem. 1965;69:4017–4024. [Google Scholar]
- 50.Stein D, van der Heyden FHJ, Koopmans WJA, Dekker C. Proc Natl Acad Sci USA. 2006;103:15853–15858. doi: 10.1073/pnas.0605900103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Campbell LC, Wilkinson MJ, Manz A, Camilleri P, Humphreys CJ. Lab Chip. 2004;4:225–229. doi: 10.1039/b312592k. [DOI] [PubMed] [Google Scholar]
- 52.Chantiwas R, Hupert ML, Pullagurla SR, Balamurugan S, Tamarit-López J, Park S, Datta P, Goettert J, Cho Y-K, Soper SA. Lab Chip. 2010;10:3255–3264. doi: 10.1039/c0lc00096e. [DOI] [PubMed] [Google Scholar]
- 53.Cross JD, Strychalski EA, Craighead HG. J Appl Phys. 2007;102:024701. [Google Scholar]
- 54.Salieb-Beugelaar GB, Teapal J, Nieuwkasteele JV, Wijnperlé D, Tegenfeldt JO, Lisdat F, van den Berg A, Eijkel JCT. Nano Lett. 2008;8:1785–1790. doi: 10.1021/nl080300v. [DOI] [PubMed] [Google Scholar]
- 55.Tang J, Trahan DW, Doyle PS. Macromolecules. 2010;43:3081–3089. doi: 10.1021/ma902689c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Balducci A, Mao P, Han JY, Doyle PS. Macromolecules. 2006;39:6273–6281. [Google Scholar]
- 57.Balducci A, Hsieh CC, Doyle PS. Phys Rev Lett. 2007;99:238102. doi: 10.1103/PhysRevLett.99.238102. [DOI] [PubMed] [Google Scholar]
- 58.Strychalski EA, Levy SL, Craighead HG. Macromolecules. 2008;41:7716–7721. [Google Scholar]
- 59.Stavis SM, Geist J, Gaitan M, Locascio LE, Strychalski EA. Lab Chip. 2012;12:1174–1182. doi: 10.1039/c2lc21152a. [DOI] [PubMed] [Google Scholar]
- 60.Menard LD, Mair CE, Woodson ME, Alarie JP, Ramsey JM. ACS Nano. 2012;6:9087–9094. doi: 10.1021/nn303322r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Menard LD, Ramsey JM. Nano Lett. 2011;11:512–517. doi: 10.1021/nl103369g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jacobson SC, Moore AW, Ramsey JM. Anal Chem. 1995;67:2059–2063. [Google Scholar]
- 63.Jacobson SC, Hergenröder R, Koutny LB, Warmack RJ, Ramsey JM. Anal Chem. 1994;66:1107–1113. [Google Scholar]
- 64.Wang W, Zhou F, Zhao L, Zhang J-R, Zhu J-J. J Chromatogr A. 2007;1170:1–8. doi: 10.1016/j.chroma.2007.08.083. [DOI] [PubMed] [Google Scholar]
- 65.Wanunu M, Morrison W, Rabin Y, Grosberg AY, Meller A. Nat Nanotechnol. 2010;5:160–165. doi: 10.1038/nnano.2009.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Levine S, Marriott JR, Robinson K. J Chem Soc, Faraday Trans 2. 1975;71:1–11. [Google Scholar]
- 67.Sadr R, Yoda M, Zheng Z, Conlisk AT. J Fluid Mech. 2004;506:357–367. [Google Scholar]
- 68.VanOrman BB, Liversidge GG. J Microcol Sep. 1990;2:176–180. [Google Scholar]
- 69.Kirby BJ, Hasselbrink EF. Electrophoresis. 2004;25:187–202. doi: 10.1002/elps.200305754. [DOI] [PubMed] [Google Scholar]
- 70.Hsieh CC, Balducci A, Doyle PS. Nano Lett. 2008;8:1683–1688. doi: 10.1021/nl080605+. [DOI] [PubMed] [Google Scholar]
- 71.Burkhardt TW. J Phys A. 1997;30:L167–172. [Google Scholar]
- 72.Kumar KK, Sebastian KL. Phys Rev E. 2000;62:7536–7539. doi: 10.1103/physreve.62.7536. [DOI] [PubMed] [Google Scholar]
- 73.Storm AJ, Storm C, Chen JH, Zandbergen H, Joanny JF, Dekker C. Nano Lett. 2005;5:1193–1197. doi: 10.1021/nl048030d. [DOI] [PubMed] [Google Scholar]
- 74.Turner SWP, Cabodi M, Craighead HG. Phys Rev Lett. 2002;88:128103. doi: 10.1103/PhysRevLett.88.128103. [DOI] [PubMed] [Google Scholar]
- 75.Castillo-Fernandez O, Salieb-Beugelaar GB, Nieuwkasteele JWv, Bomer JG, Arundell M, Samitier J, Berg Avd, Eijkel JCT. Electrophoresis. 2011;32:2402–2409. doi: 10.1002/elps.201100278. [DOI] [PubMed] [Google Scholar]
- 76.Luan B, Afzali A, Harrer S, Peng H, Waggoner P, Polonsky S, Stolovitzky G, Martyna G. J Phys Chem B. 2010;114:17172–17176. doi: 10.1021/jp108865q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chen YL, Graham M, de Pablo JJ, Randall G, Gupta M, Doyle PS. Phys Rev E. 2004;70:060901. doi: 10.1103/PhysRevE.70.060901. [DOI] [PubMed] [Google Scholar]
- 78.Kaneta T, Ueda T, Hata K, Imasaka T. J Chromatogr A. 2006;1106:52–55. doi: 10.1016/j.chroma.2005.08.062. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.