

Abstract
Over the past two decades, aberrant DNA methylation has emerged as a key player in the pathogenesis of chronic lymphocytic leukemia (CLL), and knowledge regarding its biological and clinical consequences in this disease has evolved rapidly. Since the initial studies relating DNA hypomethylation to genomic instability in CLL, a plethora of reports have followed showing the impact of DNA hypermethylation in silencing vital single gene promoters and the reversible nature of DNA methylation through inhibitor drugs. With the recognition that DNA hypermethylation events could potentially act as novel prognostic and treatment targets in CLL, the search for aberrantly methylated genes, gene families and pathways has ensued. Subsequently, the advent of microarray and next-generation sequencing technologies has supported the hunt for such targets, allowing exploration of the methylation landscape in CLL at an unprecedented scale. In light of these analyses, we now understand that different CLL prognostic subgroups are characterized by differential methylation profiles; we recognize DNA methylation of a number of signaling pathways genes to be altered in CLL, and acknowledge the role of DNA methylation outside of traditional CpG island promoters as fundamental players in the regulation of gene expression. Today, the significance and timing of altered DNA methylation within the complex epigenetic network of concomitant epigenetic messengers such as histones and miRNAs is an intensive area of research. In CLL, it appears that DNA methylation is a rather stable epigenetic mark occurring rather early in the disease pathogenesis. However, other consequences, such as how and why aberrant methylation marks occur, are less explored. In this review, we will not only provide a comprehensive summary of the current literature within the epigenetics field of CLL, but also highlight some of the novel findings relating to when, where, why and how altered DNA methylation materializes in CLL.
Keywords: chronic lymphocytic leukemia, hypomethylation, hypermethylation, CpG islands, gene silencing, microarrays, next-generation sequencing
Introduction
Indisputably, genetic alterations are key players in chronic lymphocytic leukemia (CLL) leukemogenesis, yet these lesions only partially explain the pathobiology of this disease.1 Today, growing evidence acknowledges the intricate interplay of genetic and epigenetic events shaping the complex molecular landscape in CLL. Unlike genomic lesions, epigenetic aberrations, such as anomalous histone and DNA methylation marks and dysregulated miRNAs, provoke changes to the chromatin/DNA without modifying the genomic sequence. DNA methylation is the most extensively studied epigenetic mark, involving the addition of a methyl group by DNA methyl-transferases (DNMTs) to the fifth position of the cytosine ring within CpG dinucleotides.2-4 Repetitive regions of chromosomes carry the highest density of CpG dinucleotides and in normal cells remain heavily methylated. In contrast, small concentrated regions of CpGs, referred to as CpG islands located primarily in gene promoters, are normally unmethylated, with the exception of imprinted and tissue-specific genes. In cancer, the opposite scenario ensues, where DNA methylation engages primarily, but not exclusively, within CpG island promoters, whereas repetitive elements become increasingly unmethylated.2,5 DNA methylation is also recognized as a relatively stable modification inducing transcriptional inactivation of both protein coding and non-coding regulatory miRNAs.2,6,7 For this reason, DNA methylation is now considered one of the hallmark mechanisms of aberrant gene silencing in cancer. In this review, we will not only survey the literature of the evolving field of CLL epigenetics during the last two decades, from the finding of general hypomethylation in CLL to studies of single genes promoters and, more lately, the application of whole-genome technologies, but will also emphasize important findings, providing hints to when and how the epigenetic landscape takes form in CLL.
Hypomethylation Contributes to Genomic Instability and Gene Activation in CLL
Early DNA methylation studies implicated the importance of hypomethylation as a key tumorigenic event promoting genomic instability and proto-oncogene activation in CLL and other cancers (Fig. 1).8-10 Over two decades ago, hypomethylation of ornithine decarboxylase, a vital downstream regulator of the MYC oncogene, was detected in CLL.11 However, it was not until four years later that high-pressure liquid chromatography (HPLC) analysis revealed the DNA of CLL to be globally hypomethylated relative to healthy controls. As mentioned, it is the repetitive sequences of the genome that mainly lend themselves to increased hypomethylation during tumorigenesis.12 Independent analysis in CLL corroborates this finding showing aberrant hypomethylation of repetitive sequences, such as ALU, LINE and SATα, to be particularly marked in aggressive CLL cases with TP53 aberrations.13 Interestingly, low SATα methylation levels have been further shown to be an independent predictor of time to first treatment in CLL.13 These latter findings are relevant since hypomethylation leading to genomic instability may be a contributing factor in the increased propensity of TP53-deleted/mutated cases to acquire genomic alterations. More recently, next-generation sequencing (NGS) of the DNA methylome has also noted gene body hypomethylation to be particularly widespread within enhancer regions in CLL patients.14

Figure 1. Illustration of epigenetic factors shaping the DNA methylome in CLL. This schematic details: (1) the possible timing and role of DNA methylation in CLL pathogenesis, (2) DNA hypermethylation silencing of vital tumor suppressor genes (TSGs), (3) DNA hypomethylation leading to genomic instability, (4) dysregulation of epigenetic regulators and machinery through aberrant methylation and (5) the interplay between DNA methylation and other epigenetic/microenvironmental factors.
After the CLL genome was discovered to be hypomethylated, the rising pursuit for aberrantly methylated oncogene targets revealed hypomethylation of BCL2, a key anti-apoptotic gene, to correlate with increased protein expression of BCL2 in CLL.15 Following this, MDR1, the multiple drug resistance gene,16 and TCL1, an activator of NF-κB, were subsequently found to be hypomethylated and upregulated in CLL.17 Through subsequent investigations, however, it became apparent that the activation of oncogenes through DNA hypomethylation was a rather infrequent lesion in CLL. Hence, with the landmark discovery of DNA hypermethylation silencing of tumor suppressor genes (TSGs) in cancer (Fig. 1),2 the search for aberrantly methylated genes serving as candidate prognostic and treatment targets soared. As a result, the hunt for such markers has led to the rapid evolution of DNA methylation technologies from the single gene approach to more global explorative, high-resolution methodologies.
Clinical and Biological Consequences of Hypermethylated Genes in CLL: Lessons Learned From the Study of Single Gene Promoters
In CLL, a myriad of semi/fully quantitative DNA methylation studies of single gene promoters have identified a plethora of targets of potential clinical and biological interest. One of the first frequently hypermethylated promoters observed in CLL was E-Cadherin (CHD1),18 a well-known suppressor of metastasis in solid tumors (as reviewed in ref. 19). Although a role for CHD1 methylation in leukemia remains elusive, studies have noted a reduced or absent expression of E-cadherin in hypermethylated CLL cases relative to normal B cell.18,20 Similarly, methylation of the telomerase enzyme hTERT promoter was found to be associated with low expression, low activity, shortened telomere length and poor overall survival in CLL (Table 1).21 Using one of the earliest genome-wide technologies, restriction landmark genome scanning (RLGS), TWIST2, a transcription factor and known silencer of p53, was shown be preferentially methylated in CLL.22 Through subsequent analysis, methylation of TWIST2 was demonstrated to be more frequent within favorable prognostic IGHV-mutated relative to poor-prognostic IGHV-unmutated CLL cases (Table 1).22
Table 1. Key genes inflicted with aberrant DNA methylation in CLL.
| Gene/pathway | Functional role | References |
|---|---|---|
|
CLL prognostic genes | ||
|
ZAP70 |
Involved in B cell/T cell signaling. Methylation related with decreased expression and IGHV-mutated CLL. |
23
,
42
|
|
LPL |
Known CLL prognosticator. Methylation associated with decreased expression and IGHV-mutated CLL. |
42
,
81
|
|
CLLU1 |
Prognostic marker in CLL. Methylation correlates with decreased expression and IGHV-mutated CLL. |
42
,
83
|
|
NOTCH1 |
Mutations of NOTCH1 are associated progressive CLL disease and are a marker of poor prognosis. |
42
,
84
|
|
Tumor suppressors | ||
|
WISP3 |
Involved in WNT signaling. Implicated in breast cancer. Preferentially methylated in IGHV-mutated CLL. |
42
,
85
|
|
VHL |
Associated with von Hippel-Lindau syndrome, a hereditary cancer syndrome predisposing to cancer. Differentially methylated between IGHV-mutated and unmutated CLL. Preferentially methylated in IGHV-unmutated CLL |
44
,
86
|
|
ABI3 |
ABI3 protein inhibits metastasis and tumor migration. ABI3 was found to be differentially methylated between IGHV-mutated and unmutated CLL. Frequently methylated in IGHV-unmutated CLL |
42
,
44
,
87
|
|
DAPK1 |
Positive mediator of apoptosis. Potential tumor suppressor. Commonly methylated in CLL, indicated in familial CLL. |
31
|
|
Survival/proliferation related genes | ||
|
MYB |
Proto-oncogene regulates miRNA155 in CLL. Methylated in IGHV-mutated CLL |
42
,
88
|
|
LEF1 |
Involved in WNT signaling. Pro-survival factor in CLL, methylated in good-prognostic CLL. |
42
,
89
|
|
SNRP2 and SNRP4 |
WNT signaling inhibitors commonly methylated in CLL. |
20
|
|
Transcription factor genes | ||
|
TWIST2 |
Known silencer of p53 in hematological malignancies. Hypermethylation common in IGHV-mutated CLL. |
22
|
|
HOXA4 |
Transcription factor important for cell development. Commonly methylated in poor-prognostic CLL. |
26
|
|
miRNAs* | ||
|
miR-34a, miR-129, miR-708,miR-124–1, miR-203, miR-9 |
Regulatory sequences thought to regulate gene expression of key genes involved in CLL pathogenesis such as p53, XPO1 and NOTCH1 |
49
-
51
,
58
|
|
Epigenetic regulator genes |
|
|
| HDAC4/HDAC9 | Histone deacetylases, differentitally methylated between prognostic CLL subgroups. | 42 , 90 |
Anticipated targets of the bold marked miRNAs are correspondingly highlighted in bold.
Around the same time, ZAP70, a known prognosticator in CLL and an intracellular tyrosine kinase involved in B cell (and T cell) signaling, was shown to be differentially methylated in CLL (Fig. 2 and Table 1).23 Accordingly, several studies have found good-prognostic IGHV-mutated CLL to have low ZAP70 expression associated with DNA methylation silencing, whereas poor-prognostic, high ZAP70-expressing cases demonstrate less methylated promoters.23-25 More specifically, Corcoran et al. described the methylation status of C-334, a CpG site 334 bp away from the transcription start site of ZAP70, to be predictive of prognosis and associated with expression and IGHV gene mutational status.23 Similarly, Chantepie et al. have determined the methylation status of 4 CpG pairs in the first intron (C-223, C-243, C-254 and C-267) of ZAP70 to correlate to clinical outcome and expression.24 More recently, Claus and colleagues identified loss of methylation at a single CpG site within the 5′ regulatory region to correlate not only to mRNA expression and prognosis but also to protein expression and ZAP70 activity.25 Since the ZAP70 expression level in a particular sample, as measured by flow cytometry or real-time quantitative PCR, may be influenced by other immune cells expressing ZAP70, these latter studies endorse the use of quantitative ZAP70 methylation measurement in clinical routine.24,25
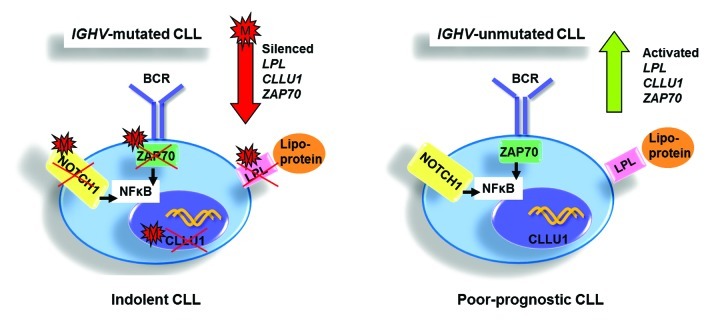
Figure 2. Illustration summarizing the differential methylation status of key prognostic genes, i.e., LPL, CLLU1 and ZAP70, and their biological/clinical effects in favorable-prognostic IGHV-mutated vs. poor-prognostic IGHV-unmutated CLL. M: methylated.
Another recurrently methylated target, the HOXA4 promoter, inversely correlates to HOXA4 gene expression in CLL (Table 1).26 HOXA4 is part of the HOXA gene cluster, a family of transcription factors important for cell development whose expression is commonly altered in lymphoid malignancies.27,28 Interestingly, hypermethylation of HOXA4 is more commonly detected among poor-prognostic IGHV-unmutated CLL cases.26 More recently, HOXA4 was included in a panel of methylation markers that was proposed to improve the risk stratification in CLL. Relative to the individual loci, combining the methylation status of HOXA4 along with BTG4 and CD38 to produce an overall methylation score was indicated to be a strong predictor of time to first treatment, independent of IGHV mutational and CD38 expression status. Notably, this panel could identify a subset of IGHV-mutated patients who had a greater possibility of progressive disease.29
DNA methylation changes have also been associated with CLL transformation and familial CLL.30,31 In 2007, a major breakthrough highlighting the importance of DAPK1 DNA methylation in CLL came to light. Here, Raval and colleagues demonstrated DNA methylation silencing of DAPK1, a pro-apoptotic gene, to occur in almost all sporadic cases of CLL. Remarkably, they further showed DAPK1 downregulation through promoter methylation to contribute to a heritable predisposition to CLL (Table 1).30,31 More recent discoveries include hypermethylation of the CRY1 gene in high risk CLL32 and frequent promoter methylation of the SLIT2 gene, a candidate tumor suppressor frequently inactivated in lung and breast cancer.33 Since CRY1 is a circadian gene involved in the expression of cell cycle and DNA damage response genes, and given the putative role of SLIT2 in other cancers, it is interesting to speculate that deregulation of these genes is involved in CLL leukemogenesis.
As the single gene approach gave little insight into aberrantly methylated pathways at play in CLL, DNA methylation studies evolved to include more comprehensive investigations of signaling cascades and gene families. One of the most extensively studied is the WNT pathway, a key pathway in B cell development, constitutively activated in CLL.34-36 Chim et al. found WNT signaling activation to be related to hypermethylation of WNT inhibitor genes.35 In this study, a large proportion of the cohort showed methylation of all 7 WNT inhibitor genes studied, i.e., WIF1, DKK3, APC, SFRP1, SFRP2, SFRP4 and SFRP5 (Table 1). Interestingly, over half of CLL cases showed methylation of at least one inhibitor.35 Later studies by Seeliger and Liu further corroborated the particularly frequent hypermethylation of the SFRP1, SFRP2 and SFRP4 genes in CLL and suggesting these as key players in leukemogenesis (Table 1).20,34 More recently, another group of genes involved in the Salvador-Warts-Hippo (SWH) pathway, the RASF family gene members 1–10 and 2 upstream members of the SHW circuit, KIBRA and CRB3, have been studied at the DNA methylation level.37 Here, the most frequently methylated genes were RASF10, followed by RASF6. The upstream regulator KIBRA was also found to be recurrently methylated and to be associated with poor-prognostic factors, such as unmutated IGHV genes and CD38 expression.37
Global Patterns of Aberrant DNA Methylation in CLL: The Genome-Wide Perspective
The characterization of methylation patterns of single genes in CLL, although limited, highlighted the ability of DNA methylation to influence a range of functionally diverse genes of clinical and biological importance in CLL. These initial findings have instigated a wave of genome-wide investigations searching to map the global DNA methylation landscape and determine the role of DNA methylation outside of traditional CpG island promoters. In the beginning, through capillary electrophoresis-laser induced detection, the overall DNA methylation level was found to be rather heterogeneous between CLL patients. Nonetheless, a high genomic methylation level was shown to be associated with poor-prognostic IGHV-unmutated CLL.38 More recently, Yu and colleagues, through HPLC analysis, indicated that CLL patients with a higher methylation index (MI) relative to age-matched controls have an increased likelihood of requiring treatment, whereas a lower MI was observed in CLL without need of therapy.39 In light of the fact that these latter methods lacked the capacity to identify target sequences influenced by DNA methylation, attention turned to the development of global methods facilitating the analysis of specific targets.
The advent of such technologies gave rise to RLGS, a 2D electrophoresis method interrogating ~3,000 CpG sites globally. By applying RLGS to 10 CLL cases, Rush et al. demonstrated between 2.5–8.1% of CpG islands to be aberrantly methylated relative to healthy donors.40 Of the 193 methylated sequences noted, 93% maintained CpG island characteristics and 90% had homology to expressed genes, such as known transcription factors (e.g., FOXE1 and TBX3).40 Since then, a number of microarray-based studies with an ever increasing resolution and rising number of CpG targets have come to light in CLL. Using two different microarrays, Rahmatpanah et al. identified over 100 genes to be hypermethylated in CLL relative to normal B cells.41 Although the majority of genes maintained the same DNA methylation status across CLL samples with different CD38 expression levels, a panel of genes was shown to segregate according to high or low CD38 expression. For example, NRP2, SFRP2 and ADAM12 were preferentially methylated in “CD38 high” cases (poor-prognostic), whereas methylation of DLEU7 was found in “CD38 low” cases (good-prognostic). Notably, an overrepresented number of WNT signaling genes, particularly the WNT inhibitory genes, were affected by methylation, a finding reported earlier using single gene promoter applications (see above).41
Other microarray studies have successfully identified aberrant methylation patterns in certain prognostic subgroups of CLL.42-44 For instance, Tong et al. have revealed 280 aberrantly methylated targets in the poor-prognostic 17p-deleted CLL subgroup. These targets were shown to cover numerous functional networks and were more frequently found within chromosomes 11, 17 and 19. Interestingly, four aberrantly methylated genes identified on chromosome 17 where known to interact with p53.43 At the global level, our research group has identified a differential methylation pattern distinguishing poor-prognostic IGHV-unmutated from favorable-prognostic IGHV-mutated CLL patients (Fig. 3).42,44 In our first 27K microarray study, a number of TSGs, such as ABI and VHL, were observed to be preferentially methylated in IGHV-unmutated relative to IGHV-mutated CLL (Table 1). Furthermore, genes occupying MAPK and NF-κB pathways, involved in cell proliferation and progression, were found to be unmethylated in IGHV-unmutated cases compared with IGHV-mutated patients. Additionally, we also observed distinct methylation patterns deciphering poor-prognostic IGHV3–21 CLL from IGHV-mutated and unmutated cases.44 Using the same type of array, we also compared the methylation profiles in three major and paradigmatic CLL subsets with stereotyped B-cell receptors: the poor-prognostic subsets #1 (IGHV1/5/7/IGKV1–39) and #2 (IGHV3–21/IGVL3–21) and the favorable-prognostic subset #4 (IGHV4–34/IGKV2–30), which revealed distinct methylation profiles for each subset. Interestingly, gene ontology analysis of the differentially methylated genes showed a striking enrichment of genes involved in immune response, such as B-cell activation, which were generally methylated in subset #1 vs. subset #2 and, in particular, subset #4. As a prime example, the co-stimulatory molecules CD80 and CD86 were methylated and not expressed in subset #1, while these remained unmethylated and were expressed at high levels in subset #4, pointing to a key role for these molecules in the cross-talk with the microenvironment in subset #4 CLL cells.45
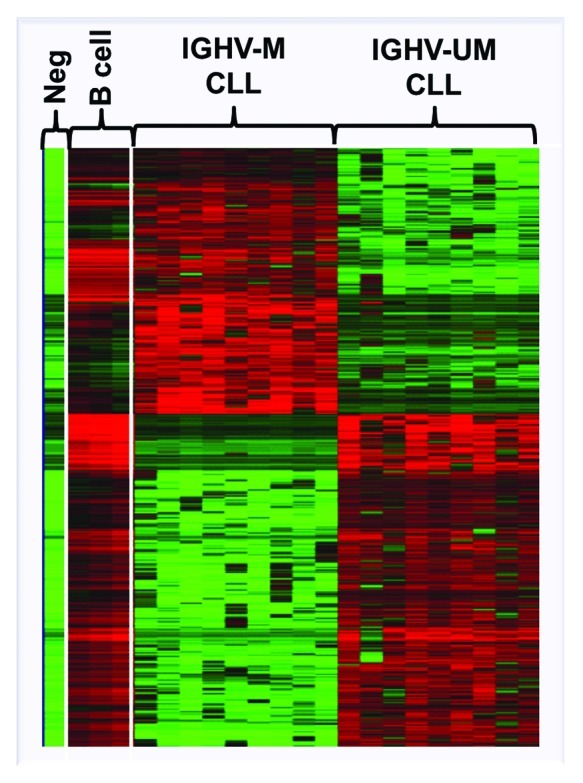
Figure 3. Heat-map showing the global differential DNA methylation profile distinguishing IGHV-mutated (IGHV-M) from IGHV-unmutated (IGHV-UM) CLL. Adapted from Cahill et al. 2012. Neg: whole genome amplified negative control, B-cell: aged-matched normal B cells.42
More recently, using 450K-array analysis, interrogating over 485,000 CpG sites, we revealed a set of CLL prognostic genes, i.e., CLLU1, LPL, ZAP70 and NOTCH1 (Fig. 2 and Table 1), as well as epigenetic regulators (i.e., HDAC9, HDAC4), the B-cell signaling IBTK gene and numerous signaling targets involved in TGF-β and NF-κB/TNF pathways to be alternatively methylated between IGHV-mutated and unmutated CLL.42 Furthermore, using these arrays, we for the first time noted DNA methylation in CLL to be relatively stable over time and similar in CLL cells derived from proliferative (lymph node) and resting (peripheral blood) microenvironments.42 Unlike other array studies in CLL, we could characterize CpG sites outside of CpG islands and found a large proportion of the differentially methylated sites identified between IGHV-mutated and unmutated CLL to reside in CpG shores, regions positioned up to 2 kb away from the promoter (Fig. 4). Interestingly, Irizarry and colleagues have shown that the methylation status of these shore regions strongly correlates with gene expression.46 In addition, on investigating the position of these CpG sites in relation to the gene orientation, we noted a large proportion of aberrantly methylated sites to occupy gene-bodies, a finding which has been also evidenced more recently using next-generation bisulfite sequencing in CLL.14 As a proof of principle, we and others have also demonstrated the biological relevance and reversible nature of DNA methylation through reactivation of select genes using methyl and HDAC inhibitors.43,44,47 For instance, by treating primary CLL B cells with concomitant methyl and deacetyl inhibitors, we could induce a reduction in DNA methylation and reinstate the expression of TSGs ABI3 and VHL in CLL.44
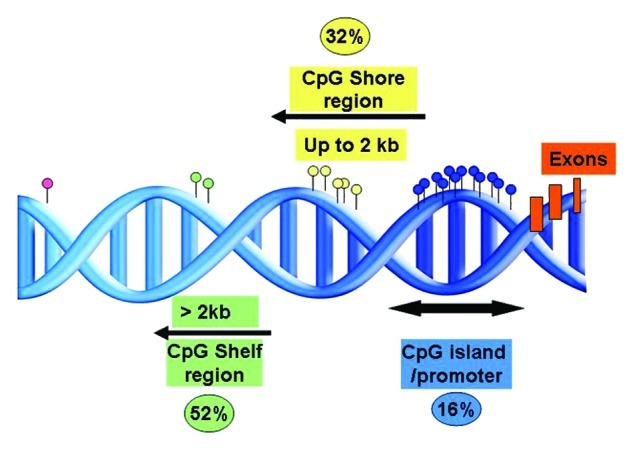
Figure 4. Representation of the proportion and positions of the annotated differentially methylated sites between IGHV-mutated and unmutated CLL in relation to the CpG island.42
Now, with the advent of NGS technologies, the limitations of microarray, such as restricted genome coverage, can be overcome. To date, only two NGS studies have been conducted in CLL, one employing the reduced representation bisulfite sequencing (RRBS) technique48 and, the second, whole-genome bisulfite sequencing (WGBS).14 Using RRBS, interrogation of 1.8–2.3 million CpGs were determined revealing ~45% of sites to be positioned in more than 23,000 CpG islands. However, global CpG methylation was determined to be rather similar between CLL and normal controls. That being said, 1,764 gene promoters were shown to be differential methylated in at least one CLL case relative to normal control. Almost 20% of the differentially methylated genes were implicated in transcription regulation. Of interest, aberrant methylation was found to be enriched in WNT signaling genes and all HOX gene clusters were subject to anomalous methylation. Using this technology, NFATc1 hypomethylation was identified and was further shown to be associated with increased mRNA and protein expression suggesting hypomethylation as a mechanism of constitutive activation of NFATc1 in CLL.48 RRBS offers a rather high coverage of the genome; however, it is still limited by the fact that this technology is based on prior selection of the regions of interest. With sequencing costs becoming more affordable, WGBS, a complete genome-wide method, is set to revolutionize the DNA methylome mapping, providing unbiased coverage at single base resolution. Most recently, the validity of WGBS as a reliable method to characterize the CLL DNA methylome has been described. Using this technology, albeit on a rather small sample set, the methylation values retrieved using WGBS were found to be concordant to those given by the 450K microarray.14 This conjoint sequencing/microarray study again revealed IGHV-mutated and unmutated CLL to differ at the global methylation level. Furthermore, they noted that extensive gene body DNA hypomethylation targeting mainly enhancer sites could distinguish these two molecular CLL subtypes relative to normal B cells and differentiate naive from memory B cells. Additionally, a relationship between gene body hypomethylation and gene expression was established.14
Undoubtedly, the aforementioned studies have broadened our knowledge of the DNA methylome in CLL providing us with numerous candidate aberrantly methylated targets and sites which maybe of biological and clinical significance. Nevertheless, further investigation, verification and careful interpretation of the above findings is needed, especially when considering whether the functional consequences are directly the result of aberrant methylation or due to other epigenetic regulatory marks. Additionally, it must be kept in mind that a number of biological and technical factors can influence the results. For instance, the type of technology used, batch bias, the cut-offs used to call the level of methylation, the type of cohort studied, the number of patients included and sample purity among others must be considered. Now with the abundance of aberrantly methylated marks identified, future challenges such as deciphering passenger from driver lesions, establishing the function of methylation outside of CpG islands and unraveling the role of 5-hydroxymethyl-cytosine in CLL awaits us.
Aberrant DNA Methylation Leads to Dysregulation of Non-Coding miRNAs in CLL
Most noteworthy, recent studies have found aberrant DNA methylation to be a key mechanism in the deregulation of miRNAs in CLL (Fig. 1 and Table 1).49-51 These single stranded non-coding miRNAs, through their downregulation of target proteins, act either in an oncogenic or tumor suppressive manner. For this reason, aberrant miRNA expression is now accepted as one of the main signatures in CLL pathogenesis.52-57 The mechanisms underlying altered miRNA expression are poorly known, however it is increasingly apparent that genetic and/or epigenetic manipulation may play a role at least in some CLL cases. As a prime example, the common 13q deletion, which includes miR15a and miR16–1, leads to upregulation of BCL2 and deregulated apoptosis, a well-described mechanism in human and murine leukemogenesis.52,56
Until recently, the role of aberrant methylation in tumor suppressor miRNAs was rather undefined in CLL. Pallasch and colleagues noted a number of deregulated miRNA promoters associated with decreased miRNAs expression, to have gain of methylation in many CLL cases compared with normal B cells.59 In particular, promoters of miR-139 and miR-582 showed a significant gain of methylation in CLL.59 Furthermore, through combining DNA methylation and miRNAs promoter profiles, Baer et al. have defined a panel of 128 recurrent novel and known miRNAs targets subject to altered promoter DNA methylation in CLL.51 For instance, hypomethylation of miR-21, miR-29a, miR-34a, miR-155, miR-574 and miR1204 was shown to correlate with an upregulated expression of these respective regulatory miRNAs. Conversely, hypermethylation of miR-124–2, miR-129–2, miR-9–2, miR-551 and miR-708 correlated with a reduced expression of these miRNAs. Interestingly, increased expression of XPO1, a predicted target of miR-129, POT1 an inferred target of miR-9, and, NOTCH1, an anticipated target of miR-708, correlated with a reduction in expression of their respective regulatory miRNAs (Table 1).51 Nevertheless, this is probably just the tip of the iceberg and further studies will hopefully clarify which of these aberrantly methylated miRNAs that are key to the pathobiology of CLL.
Complex Regulatory Mechanisms Shaping the Aberrant DNA Methylation Landscape in CLL
DNA methylation partakes in a hierarchal order of epigenetic events that concomitantly work together to regulate nuclear structure and gene activity. For instance, gene inactivation is preceded by repressive histone alterations and DNA methylation forming a condensed genomic architecture impeding the binding of transcriptional machinery and thus gene expression. Conversely, gene activation is permitted through the lack of repressive histone and DNA methylation marks. Through these finely tuned events, normal processes, such as cell differentiation, tissue specific expression, genomic imprinting and transposon silencing, are coordinated.3,60 The ability of epigenetic marks to manipulate the DNA and chromatin relies on the stringent regulation of the epigenetic machinery such as the DNMTs, histone modification enzymes, methyl-binding proteins, polycomb complexes and miRNAs, among others.60 In normal cells, the order and timing in which these regulators cross talk to synchronize epigenetic marks at specific targets is not fully elucidated. Nonetheless, evidence of when aberrant DNA methylation takes place, how altered DNA methylation occurs and where DNA methylation partakes in the sequence of epigenetic events at specific gene targets in CLL is slowly emerging. In the following sections, we will discuss some of these issues governing when and how altered DNA methylation takes place in CLL. A fully comprehensive discussion of this topic is not provided since it is beyond the scope of this review.
When Does Aberrant DNA Methylation Take Place in CLL Pathogenesis?
The Eμ-TCL1 transgenic mouse model, referred to as the TCL1 model, has been instrumental in deciphering the timing of epigenetic events, particularly DNA methylation, in CLL leukemogenesis.61,62 This model overexpresses TCL1, a known oncogene, leading to a CLL disease phenotype similar to that of human poor-prognostic IGHV-unmutated CLL. Normally, these mice develop a CLL-like disease at around 11 mo; however, these mice demonstrated aberrant methylation as early as 3 mo before disease indications appeared.61 A genome-wide scan for promoter methylation during the course of disease observed methylation to steadily increase from 0.4% at 3 mo to 0.6%, 1.2% and 1.9% at 5, 7 and 9 mo, respectively. Finally, during advanced disease, a markedly increased methylation level of 3.9% was detected. Intriguingly, the majority of early silenced genes were found to be subsequently methylated in human CLL. Furthermore, hypermethylated genomic repeat sequences found in wild type mice were also found to be already hypomethylated in 7-mo-old TCL1 mice. Similarly, in human CLL, hypomethylation of LINE repeat sequences were found to be more pronounced in late stage patients relative to early stage cases.61,62 As previously mentioned, our 450K-array analysis, encompassing patient-paired diagnostic and follow-up samples, noted DNA methylation to relatively stable over time.42 More specifically, we found no recurrent differences to occur in IGHV-mutated CLL and few recurrent changes in IGHV-unmutated over the course of disease.42 That said, in light of the heterogeneous nature of CLL, we determined a variable number of non-recurrent changes in both prognostic subgroups of CLL and found the amount of changes to be more pronounced in IGHV-unmutated CLL.42 As IGHV-unmutated cases are more prone to acquire genomic lesions over time,63,64 perhaps the aggressive character of IGHV-unmutated CLL allow the acquisition of a higher number of “passenger” epimutations during disease evolution. All together, these studies indicate aberrant DNA methylation to be early initiating lesions in CLL leukemogenesis.42
How Does Altered DNA Methylation Occur in CLL?
There is growing evidence that the epigenetic machinery falls victim to deregulation. Genomic alteration of DNMT enzymes and histone alteration enzymes that catalyze the addition or removal of epigenetic marks, as well as altered expression and epigenetic modification of the epigenetic machinery have all contributed to aberrant epigenetic lesions observed in different cancers.65-68 Additionally, even so called epi-miRNAs, in which the miRNA components of the epigenetic machinery themselves target further epigenetic modifiers have been described in lymphoma.65
In CLL, no difference in gene expression of the DNA methylation maintenance enzyme DNMT1 has been noted relative to normal B cells.69 However, downregulated expression of the de novo methylating DNMT3B gene has been evidenced in CLL.69 Interestingly, expression of the histone methyltransferase 1 enzyme HMT1 has also been associated with more advanced disease stages of CLL.69 Given the functional redundancy of these enzymes and the fact that their relative expression maybe related to the proliferative rate of cells, expression analysis must be interpreted carefully.
As mentioned earlier, the TCL1 mouse model, which resembles human IGHV-unmutated CLL, has shown increased methylation in CLL with disease progression and this has subsequently been shown to follow the pattern of de novo methylating DNMT activity.61,62 Here, protein levels of DNMT3A/3B were found to be absent during transformation, yet levels were shown to rise at later stages of disease.61,62 Reduced expression of miRNA29a and miRNA29c was also noted in the TCL1 mouse at 5 and 7 mo. This reduction correlated with increasing DNMT3A and 3B protein expression at a later age, suggesting miRNA29s to be a direct regulator of DNMT3A/3B in CLL, an interaction also apparent in lung cancer.61,62,70 Hence, increased activity of DNMT3A and 3B was proposed as a possible factor triggering increased GpG island hypermethylation at distinct promoters in CLL.61,62
In support of the above findings, and also illustrating the pivotal role of miRNA29 in CLL leukemogenesis, miRNA29 transgenic mice display expanding CD5+ B cell populations with CLL characteristics. This miRNA was also found to be preferentially expressed in indolent CLL relative to aggressive CLL cases.71 Moreover, histone deacetylase (HDAC)-mediated elimination of acetyl groups, triggering compact chromatin formation, has been evidenced to mediate silencing of miRNA29b and other miRNAs in CLL.72 All together, these studies highlight the important role aberrant regulatory miRNAs at play in CLL pathogenesis.
More recent studies of the TCL1 transgenic mouse have found TCL1 to physically interact and inhibit de novo DNMT3A activity.73 On analyzing the B-cells from 4–6 week old TCL1 transgenic mice, who characteristically display no signs of disease, a significant decrease in DNA methylation was noted compared with wild type controls.73 Similarly, human CLL cells with high TCL1 expression had a lower DNA methylation level relative to patients with low expression.73 In a somewhat contradictory manner to the aforementioned study by Chen et al.,61 which indicated increased DNA methylation on CLL progression, Palamarchuk et al. suggest that in these disease-free mice, inhibition of de novo DNA methylation maybe a common mechanism leading to DNA hypomethylation of distinctive CpG sites perhaps during early pathogenesis.73 Nonetheless, these studies clearly implicate altered regulation of de novo methylation enzymes to play a key role in leukemogenesis.
Lessons from normal cell development may provide vital clues as to how DNA methylation may be elicited and where in the hierarchal order of coordinated epigenetic marks DNA methylation is placed. One scenario proposes that histone marks direct DNA methylation events by acting as platforms permitting the recruitment or inhibition of the DNA methylation machinery.4,74 The opposing notion suggests DNA methylation to serve as messenger directing the assembly of histone alterations.4,75 In CLL, the importance of cross-talk between DNA methylation and other chromatin modifications in defining the aberrant epigenetic landscape is slowly emerging. For instance, the TCL1 mouse model shows that 70% of the early methylated promoters identified in preclinical disease were targets of the FOXD3 transcription factor, a frequently methylated/silenced gene in the TCL1 mice and human CLL with high TCL1 expression.61,62 Importantly, FOXD3 expression was shown to be repressed in these mice prior to methylation through an NF-κB p50/p50:HDAC1 repressor complex, eventually leading to methylation of downstream targets.61 In human CLL, DNA methylation and histone modification cross-talk has been described for ZAP7075 and Aiolos,76 a transcription factor regulating the BCL2 family members. Additionally, the ID4 transcription factor is known to have a relatively high but rather variable level of DNA methylation at its promoter.77 Nevertheless, ID4 mRNA and protein levels are shown to be universally silenced in CLL, implying that other silencing components are initially responsible for gene deregulation, with DNA methylation occurring at later stages.77 Similarly, PTPROt, a protein tyrosine phosphatase primarily involved in lymphocyte survival, was initially demonstrated to be frequently methylated in CLL.78 However, lessons from the TCL1 mouse now describe that up to 60% of PTPROt gene inactivation is the consequence of other inactivating complexes working independently of DNA methylation.79 Overall, these latter studies highlight the importance of coordinating regulatory modifications in the control of gene transcription. For the most part, these studies portray DNA methylation as a stabilizing secondary modification in a hierarchal order of epigenetic events.
Finally, even the cell microenvironment in which the cells reside is suggested to prompt epigenetic mechanisms in CLL (Fig. 1). Intriguingly, recent in vitro studies describe a demethylation process triggered by a set of stimulators typically found within the CLL microenvironment.80 Here, Morena and colleagues observed that induction of the leukemic clone with CD40L/IL4 and anti-IgM resulted in demethylation of LPL,80 a prominent prognostic gene also known to be differentially methylated in good and poor CLL subgroups.42,81 On the global level, we ourselves have searched for DNA methylation differences in patient-matched samples derived from alternative microenvironments. However, we observed the global methylation profiles to be rather similar in CLL cells derived from the proliferative lymph node and resting peripheral blood compartments.42,81
Concluding Remarks
Compared with other epigenetic marks, DNA methylation is deemed to be a rather stable event, a property that has enabled this modification to be extensively studied for more than two decades. Today, we recognize DNA methylation to influence a number of signaling pathway genes and key tumor suppressors of biological and potential clinical importance to CLL. Furthermore, we are now beginning to understand the extent of global aberrant DNA methylation in different prognostic subsets, the fundamental role of DNA methylation in sites positioned outside of CpG islands and the collaboration of DNA methylation with other regulatory messengers in shaping the CLL methylome. In the wake of the increasing knowledge provided by genome-wide studies, it has also become apparent that similar to the genomic scenario, an abundance of ‘passenger’ DNA methylation events likely accompany this disease. In the next coming years, we should focus our efforts to identifying key epigenetic lesions that act as ‘driver’ epimutations early during CLL pathogenesis, probably by investigating DNA methylation (1) in relation to potential CLL precursors, such as the novel CD5+/CD27+ post-germinal center B-cell subset, recently identified by Seifert et al.,82 and (2) among clinically relevant and more homogenous subsets of CLL patients. Furthermore, we should also harmonize methodology to measure DNA methylation and investigate larger cohorts of samples in order to fully address the clinical impact of DNA methylation of certain key genes or pathways. Finally, deciphering the functional consequences of these lesions as well as determining the functional relationships between DNA methylation and other epigenetic messengers will be crucial in order to better understand the complex regulation of epigenetics in CLL, which ultimately has the potential to lead to tailored epigenetic-based therapies.
Acknowledgments
This research was supported by the Nordic Cancer Union, the Swedish Cancer Society, the Swedish Research Council, and the Lion’s Cancer Research Foundation, Uppsala.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/epigenetics/article/23439
References
- 1.Zenz T, Mertens D, Küppers R, Döhner H, Stilgenbauer S. From pathogenesis to treatment of chronic lymphocytic leukaemia. Nat Rev Cancer. 2010;10:37–50. doi: 10.1038/nrc2764. [DOI] [PubMed] [Google Scholar]
- 2.Baylin SB, Herman JG. DNA hypermethylation in tumorigenesis: epigenetics joins genetics. Trends Genet. 2000;16:168–74. doi: 10.1016/S0168-9525(99)01971-X. [DOI] [PubMed] [Google Scholar]
- 3.Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- 4.Cedar H, Bergman Y. Linking DNA methylation and histone modification: patterns and paradigms. Nat Rev Genet. 2009;10:295–304. doi: 10.1038/nrg2540. [DOI] [PubMed] [Google Scholar]
- 5.Takai D, Jones PA. Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proc Natl Acad Sci U S A. 2002;99:3740–5. doi: 10.1073/pnas.052410099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Esteller M. Epigenetic gene silencing in cancer: the DNA hypermethylome. Hum Mol Genet. 2007;16(Spec No 1):R50–9. doi: 10.1093/hmg/ddm018. [DOI] [PubMed] [Google Scholar]
- 7.Weber B, Stresemann C, Brueckner B, Lyko F. Methylation of human microRNA genes in normal and neoplastic cells. Cell Cycle. 2007;6:1001–5. doi: 10.4161/cc.6.9.4209. [DOI] [PubMed] [Google Scholar]
- 8.Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002;3:415–28. doi: 10.1038/nrg816. [DOI] [PubMed] [Google Scholar]
- 9.Eden A, Gaudet F, Waghmare A, Jaenisch R. Chromosomal instability and tumors promoted by DNA hypomethylation. Science. 2003;300:455. doi: 10.1126/science.1083557. [DOI] [PubMed] [Google Scholar]
- 10.Rodriguez J, Frigola J, Vendrell E, Risques RA, Fraga MF, Morales C, et al. Chromosomal instability correlates with genome-wide DNA demethylation in human primary colorectal cancers. Cancer Res. 2006;66:8462–9468. doi: 10.1158/0008-5472.CAN-06-0293. [DOI] [PubMed] [Google Scholar]
- 11.Lipsanen V, Leinonen P, Alhonen L, Jänne J. Hypomethylation of ornithine decarboxylase gene and erb-A1 oncogene in human chronic lymphatic leukemia. Blood. 1988;72:2042–4. [PubMed] [Google Scholar]
- 12.Wahlfors J, Hiltunen H, Heinonen K, Hämäläinen E, Alhonen L, Jänne J. Genomic hypomethylation in human chronic lymphocytic leukemia. Blood. 1992;80:2074–80. [PubMed] [Google Scholar]
- 13.Fabris S, Bollati V, Agnelli L, Morabito F, Motta V, Cutrona G, et al. Biological and clinical relevance of quantitative global methylation of repetitive DNA sequences in chronic lymphocytic leukemia. Epigenetics. 2011;6:188–94. doi: 10.4161/epi.6.2.13528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kulis M, Heath S, Bibikova M, Queirós AC, Navarro A, Clot G, et al. Epigenomic analysis detects widespread gene-body DNA hypomethylation in chronic lymphocytic leukemia. Nat Genet. 2012;44:1236–42. doi: 10.1038/ng.2443. [DOI] [PubMed] [Google Scholar]
- 15.Hanada M, Delia D, Aiello A, Stadtmauer E, Reed JC. bcl-2 gene hypomethylation and high-level expression in B-cell chronic lymphocytic leukemia. Blood. 1993;82:1820–8. [PubMed] [Google Scholar]
- 16.Kantharidis P, El-Osta A, deSilva M, Wall DM, Hu XF, Slater A, et al. Altered methylation of the human MDR1 promoter is associated with acquired multidrug resistance. Clin Cancer Res. 1997;3:2025–32. [PubMed] [Google Scholar]
- 17.Yuille MR, Condie A, Stone EM, Wilsher J, Bradshaw PS, Brooks L, et al. TCL1 is activated by chromosomal rearrangement or by hypomethylation. Genes Chromosomes Cancer. 2001;30:336–41. doi: 10.1002/gcc.1099. [DOI] [PubMed] [Google Scholar]
- 18.Melki JR, Vincent PC, Brown RD, Clark SJ. Hypermethylation of E-cadherin in leukemia. Blood. 2000;95:3208–13. [PubMed] [Google Scholar]
- 19.Berx G, Cleton-Jansen AM, Nollet F, de Leeuw WJ, van de Vijver M, Cornelisse C, et al. E-cadherin is a tumour/invasion suppressor gene mutated in human lobular breast cancers. EMBO J. 1995;14:6107–15. doi: 10.1002/j.1460-2075.1995.tb00301.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Seeliger B, Wilop S, Osieka R, Galm O, Jost E. CpG island methylation patterns in chronic lymphocytic leukemia. Leuk Lymphoma. 2009;50:419–26. doi: 10.1080/10428190902756594. [DOI] [PubMed] [Google Scholar]
- 21.Bechter OE, Eisterer W, Dlaska M, Kühr T, Thaler J. CpG island methylation of the hTERT promoter is associated with lower telomerase activity in B-cell lymphocytic leukemia. Exp Hematol. 2002;30:26–33. doi: 10.1016/S0301-472X(01)00760-3. [DOI] [PubMed] [Google Scholar]
- 22.Raval A, Lucas DM, Matkovic JJ, Bennett KL, Liyanarachchi S, Young DC, et al. TWIST2 demonstrates differential methylation in immunoglobulin variable heavy chain mutated and unmutated chronic lymphocytic leukemia. J Clin Oncol. 2005;23:3877–85. doi: 10.1200/JCO.2005.02.196. [DOI] [PubMed] [Google Scholar]
- 23.Corcoran M, Parker A, Orchard J, Davis Z, Wirtz M, Schmitz OJ, et al. ZAP-70 methylation status is associated with ZAP-70 expression status in chronic lymphocytic leukemia. Haematologica. 2005;90:1078–88. [PubMed] [Google Scholar]
- 24.Chantepie SP, Vaur D, Grunau C, Salaün V, Briand M, Parienti JJ, et al. ZAP-70 intron1 DNA methylation status: determination by pyrosequencing in B chronic lymphocytic leukemia. Leuk Res. 2010;34:800–8. doi: 10.1016/j.leukres.2009.10.018. [DOI] [PubMed] [Google Scholar]
- 25.Claus R, Lucas DM, Stilgenbauer S, Ruppert AS, Yu LB, Zucknick M, et al. Quantitative DNA methylation analysis identifies a single CpG dinucleotide important for ZAP-70 expression and predictive of prognosis in chronic lymphocytic leukemia. J Clin Oncol. 2012;30:2483–91. doi: 10.1200/JCO.2011.39.3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Strathdee G, Sim A, Parker A, Oscier D, Brown R. Promoter hypermethylation silences expression of the HoxA4 gene and correlates with IgVh mutational status in CLL. Leukemia. 2006;20:1326–9. doi: 10.1038/sj.leu.2404254. [DOI] [PubMed] [Google Scholar]
- 27.Cillo C, Cantile M, Faiella A, Boncinelli E. Homeobox genes in normal and malignant cells. J Cell Physiol. 2001;188:161–9. doi: 10.1002/jcp.1115. [DOI] [PubMed] [Google Scholar]
- 28.Strathdee G, Holyoake TL, Sim A, Parker A, Oscier DG, Melo JV, et al. Inactivation of HOXA genes by hypermethylation in myeloid and lymphoid malignancy is frequent and associated with poor prognosis. Clin Cancer Res. 2007;13:5048–55. doi: 10.1158/1078-0432.CCR-07-0919. [DOI] [PubMed] [Google Scholar]
- 29.Irving L, Mainou-Fowler T, Parker A, Ibbotson RE, Oscier DG, Strathdee G. Methylation markers identify high risk patients in IGHV mutated chronic lymphocytic leukemia. Epigenetics. 2011;6:300–6. doi: 10.4161/epi.6.3.14038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fülöp Z, Csernus B, Tímár B, Szepesi A, Matolcsy A. Microsatellite instability and hMLH1 promoter hypermethylation in Richter’s transformation of chronic lymphocytic leukemia. Leukemia. 2003;17:411–5. doi: 10.1038/sj.leu.2402792. [DOI] [PubMed] [Google Scholar]
- 31.Raval A, Tanner SM, Byrd JC, Angerman EB, Perko JD, Chen SS, et al. Downregulation of death-associated protein kinase 1 (DAPK1) in chronic lymphocytic leukemia. Cell. 2007;129:879–90. doi: 10.1016/j.cell.2007.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hanoun M, Eisele L, Suzuki M, Greally JM, Hüttmann A, Aydin S, et al. Epigenetic silencing of the circadian clock gene CRY1 is associated with an indolent clinical course in chronic lymphocytic leukemia. PLoS One. 2012;7:e34347. doi: 10.1371/journal.pone.0034347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dunwell TL, Dickinson RE, Stankovic T, Dallol A, Weston V, Austen B, et al. Frequent epigenetic inactivation of the SLIT2 gene in chronic and acute lymphocytic leukemia. Epigenetics. 2009;4:265–9. doi: 10.4161/epi.9137. [DOI] [PubMed] [Google Scholar]
- 34.Liu TH, Raval A, Chen SS, Matkovic JJ, Byrd JC, Plass C. CpG island methylation and expression of the secreted frizzled-related protein gene family in chronic lymphocytic leukemia. Cancer Res. 2006;66:653–8. doi: 10.1158/0008-5472.CAN-05-3712. [DOI] [PubMed] [Google Scholar]
- 35.Chim CS, Pang R, Liang R. Epigenetic dysregulation of the Wnt signalling pathway in chronic lymphocytic leukaemia. J Clin Pathol. 2008;61:1214–9. doi: 10.1136/jcp.2008.060152. [DOI] [PubMed] [Google Scholar]
- 36.Moskalev EA, Luckert K, Vorobjev IA, Mastitsky SE, Gladkikh AA, Stephan A, et al. Concurrent epigenetic silencing of wnt/β-catenin pathway inhibitor genes in B cell chronic lymphocytic leukaemia. BMC Cancer. 2012;12:213. doi: 10.1186/1471-2407-12-213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shinawi T, Hill V, Dagklis A, Baliakas P, Stamatopoulos K, Agathanggelou A, et al. KIBRA gene methylation is associated with unfavorable biological prognostic parameters in chronic lymphocytic leukemia. Epigenetics. 2012;7:211–5. doi: 10.4161/epi.7.3.19222. [DOI] [PubMed] [Google Scholar]
- 38.Lyko F, Stach D, Brenner A, Stilgenbauer S, Döhner H, Wirtz M, et al. Quantitative analysis of DNA methylation in chronic lymphocytic leukemia patients. Electrophoresis. 2004;25:1530–5. doi: 10.1002/elps.200305830. [DOI] [PubMed] [Google Scholar]
- 39.Yu MK, Bergonia H, Szabo A, Phillips JD. Progressive disease in chronic lymphocytic leukemia is correlated with the DNA methylation index. Leuk Res. 2007;31:773–7. doi: 10.1016/j.leukres.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 40.Rush LJ, Raval A, Funchain P, Johnson AJ, Smith L, Lucas DM, et al. Epigenetic profiling in chronic lymphocytic leukemia reveals novel methylation targets. Cancer Res. 2004;64:2424–33. doi: 10.1158/0008-5472.CAN-03-2870. [DOI] [PubMed] [Google Scholar]
- 41.Rahmatpanah FB, Carstens S, Hooshmand SI, Welsh EC, Sjahputera O, Taylor KH, et al. Large-scale analysis of DNA methylation in chronic lymphocytic leukemia. Epigenomics. 2009;1:39–61. doi: 10.2217/epi.09.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cahill N, Bergh AC, Kanduri M, Goransson-Kultima H, Mansouri L, Isaksson A, et al. 450K-array analysis of chronic lymphocytic leukemia cells reveals global DNA methylation to be relatively stable over time and similar in resting and proliferative compartments. Leukemia. 2013;27:150–8. doi: 10.1038/leu.2012.245. [DOI] [PubMed] [Google Scholar]
- 43.Tong WG, Wierda WG, Lin E, Kuang SQ, Bekele BN, Estrov Z, et al. Genome-wide DNA methylation profiling of chronic lymphocytic leukemia allows identification of epigenetically repressed molecular pathways with clinical impact. Epigenetics. 2010;5:499–508. doi: 10.4161/epi.5.6.12179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kanduri M, Cahill N, Göransson H, Enström C, Ryan F, Isaksson A, et al. Differential genome-wide array-based methylation profiles in prognostic subsets of chronic lymphocytic leukemia. Blood. 2010;115:296–305. doi: 10.1182/blood-2009-07-232868. [DOI] [PubMed] [Google Scholar]
- 45.Kanduri M, Marincevic M, Halldórsdóttir AM, Mansouri L, Junevik K, Ntoufa S, et al. Distinct transcriptional control in major immunogenetic subsets of chronic lymphocytic leukemia exhibiting subset-biased global DNA methylation profiles. Epigenetics. 2012;7:1435–42. doi: 10.4161/epi.22901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet. 2009;41:178–86. doi: 10.1038/ng.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Halldórsdóttir AM, Sander B, Göransson H, Isaksson A, Kimby E, Mansouri M, et al. High-resolution genomic screening in mantle cell lymphoma--specific changes correlate with genomic complexity, the proliferation signature and survival. Genes Chromosomes Cancer. 2011;50:113–21. doi: 10.1002/gcc.20836. [DOI] [PubMed] [Google Scholar]
- 48.Pei LR, Choi JH, Liu JM, Lee EJ, McCarthy B, Wilson JM, et al. Genome-wide DNA methylation analysis reveals novel epigenetic changes in chronic lymphocytic leukemia. Epigenetics. 2012;7:567–78. doi: 10.4161/epi.20237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chim CS, Wong KY, Qi Y, Loong F, Lam WL, Wong LG, et al. Epigenetic inactivation of the miR-34a in hematological malignancies. Carcinogenesis. 2010;31:745–50. doi: 10.1093/carcin/bgq033. [DOI] [PubMed] [Google Scholar]
- 50.Wong KY, So CC, Loong F, Chung LP, Lam WW, Liang R, et al. Epigenetic inactivation of the miR-124-1 in haematological malignancies. PLoS One. 2011;6:e19027. doi: 10.1371/journal.pone.0019027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Baer C, Claus R, Frenzel LP, Zucknick M, Park YJ, Gu L, et al. Extensive promoter DNA hypermethylation and hypomethylation is associated with aberrant microRNA expression in chronic lymphocytic leukemia. Cancer Res. 2012;72:3775–85. doi: 10.1158/0008-5472.CAN-12-0803. [DOI] [PubMed] [Google Scholar]
- 52.Calin GA, Dumitru CD, Shimizu M, Bichi R, Zupo S, Noch E, et al. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A. 2002;99:15524–9. doi: 10.1073/pnas.242606799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Calin GA, Pekarsky Y, Croce CM. The role of microRNA and other non-coding RNA in the pathogenesis of chronic lymphocytic leukemia. Best Pract Res Clin Haematol. 2007;20:425–37. doi: 10.1016/j.beha.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 54.Nicoloso MS, Kipps TJ, Croce CM, Calin GA. MicroRNAs in the pathogeny of chronic lymphocytic leukaemia. Br J Haematol. 2007;139:709–16. doi: 10.1111/j.1365-2141.2007.06868.x. [DOI] [PubMed] [Google Scholar]
- 55.Visone R, Rassenti LZ, Veronese A, Taccioli C, Costinean S, Aguda BD, et al. Karyotype-specific microRNA signature in chronic lymphocytic leukemia. Blood. 2009;114:3872–9. doi: 10.1182/blood-2009-06-229211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Klein U, Lia M, Crespo M, Siegel R, Shen Q, Mo T, et al. The DLEU2/miR-15a/16-1 cluster controls B cell proliferation and its deletion leads to chronic lymphocytic leukemia. Cancer Cell. 2010;17:28–40. doi: 10.1016/j.ccr.2009.11.019. [DOI] [PubMed] [Google Scholar]
- 57.Zenz T, Häbe S, Denzel T, Mohr J, Winkler D, Bühler A, et al. Detailed analysis of p53 pathway defects in fludarabine-refractory chronic lymphocytic leukemia (CLL): dissecting the contribution of 17p deletion, TP53 mutation, p53-p21 dysfunction, and miR34a in a prospective clinical trial. Blood. 2009;114:2589–97. doi: 10.1182/blood-2009-05-224071. [DOI] [PubMed] [Google Scholar]
- 58.Chim CS, Wong KY, Leung CY, Chung LP, Hui PK, Chan SY, et al. Epigenetic inactivation of the hsa-miR-203 in haematological malignancies. J Cell Mol Med. 2011;15:2760–7. doi: 10.1111/j.1582-4934.2011.01274.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pallasch CP, Patz M, Park YJ, Hagist S, Eggle D, Claus R, et al. miRNA deregulation by epigenetic silencing disrupts suppression of the oncogene PLAG1 in chronic lymphocytic leukemia. Blood. 2009;114:3255–64. doi: 10.1182/blood-2009-06-229898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis. 2010;31:27–36. doi: 10.1093/carcin/bgp220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen SS, Raval A, Johnson AJ, Hertlein E, Liu TH, Jin VX, et al. Epigenetic changes during disease progression in a murine model of human chronic lymphocytic leukemia. Proc Natl Acad Sci U S A. 2009;106:13433–8. doi: 10.1073/pnas.0906455106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chen SS, Sherman MH, Hertlein E, Johnson AJ, Teitell MA, Byrd JC, et al. Epigenetic alterations in a murine model for chronic lymphocytic leukemia. Cell Cycle. 2009;8:3663–7. doi: 10.4161/cc.8.22.9957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Stilgenbauer S, Sander S, Bullinger L, Benner A, Leupolt E, Winkler D, et al. Clonal evolution in chronic lymphocytic leukemia: acquisition of high-risk genomic aberrations associated with unmutated VH, resistance to therapy, and short survival. Haematologica. 2007;92:1242–5. doi: 10.3324/haematol.10720. [DOI] [PubMed] [Google Scholar]
- 64.Gunnarsson R, Mansouri L, Isaksson A, Göransson H, Cahill N, Jansson M, et al. Array-based genomic screening at diagnosis and during follow-up in chronic lymphocytic leukemia. Haematologica. 2011;96:1161–9. doi: 10.3324/haematol.2010.039768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sander S, Bullinger L, Klapproth K, Fiedler K, Kestler HA, Barth TF, et al. MYC stimulates EZH2 expression by repression of its negative regulator miR-26a. Blood. 2008;112:4202–12. doi: 10.1182/blood-2008-03-147645. [DOI] [PubMed] [Google Scholar]
- 66.Chen S, Bohrer LR, Rai AN, Pan Y, Gan L, Zhou X, et al. Cyclin-dependent kinases regulate epigenetic gene silencing through phosphorylation of EZH2. Nat Cell Biol. 2010;12:1108–14. doi: 10.1038/ncb2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ernst T, Chase AJ, Score J, Hidalgo-Curtis CE, Bryant C, Jones AV, et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat Genet. 2010;42:722–6. doi: 10.1038/ng.621. [DOI] [PubMed] [Google Scholar]
- 68.Yan XJ, Xu J, Gu ZH, Pan CM, Lu G, Shen Y, et al. Exome sequencing identifies somatic mutations of DNA methyltransferase gene DNMT3A in acute monocytic leukemia. Nat Genet. 2011;43:309–15. doi: 10.1038/ng.788. [DOI] [PubMed] [Google Scholar]
- 69.Kn H, Bassal S, Tikellis C, El-Osta A. Expression analysis of the epigenetic methyltransferases and methyl-CpG binding protein families in the normal B-cell and B-cell chronic lymphocytic leukemia (CLL) Cancer Biol Ther. 2004;3:989–94. doi: 10.4161/cbt.3.10.1137. [DOI] [PubMed] [Google Scholar]
- 70.Fabbri M, Garzon R, Cimmino A, Liu Z, Zanesi N, Callegari E, et al. MicroRNA-29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3A and 3B. Proc Natl Acad Sci U S A. 2007;104:15805–10. doi: 10.1073/pnas.0707628104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Santanam U, Zanesi N, Efanov A, Costinean S, Palamarchuk A, Hagan JP, et al. Chronic lymphocytic leukemia modeled in mouse by targeted miR-29 expression. Proc Natl Acad Sci U S A. 2010;107:12210–5. doi: 10.1073/pnas.1007186107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sampath D, Liu C, Vasan K, Sulda M, Puduvalli VK, Wierda WG, et al. Histone deacetylases mediate the silencing of miR-15a, miR-16, and miR-29b in chronic lymphocytic leukemia. Blood. 2012;119:1162–72. doi: 10.1182/blood-2011-05-351510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Palamarchuk A, Yan PS, Zanesi N, Wang L, Rodrigues B, Murphy M, et al. Tcl1 protein functions as an inhibitor of de novo DNA methylation in B-cell chronic lymphocytic leukemia (CLL) Proc Natl Acad Sci U S A. 2012;109:2555–60. doi: 10.1073/pnas.1200003109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ooi SK, Qiu C, Bernstein E, Li K, Jia D, Yang Z, et al. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature. 2007;448:714–7. doi: 10.1038/nature05987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Amin S, Walsh M, Wilson C, Parker AE, Oscier D, Willmore E, et al. Cross-talk between DNA methylation and active histone modifications regulates aberrant expression of ZAP70 in CLL. J Cell Mol Med. 2012;16:2074–84. doi: 10.1111/j.1582-4934.2011.01503.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Billot K, Soeur J, Chereau F, Arrouss I, Merle-Béral H, Huang ME, et al. Deregulation of Aiolos expression in chronic lymphocytic leukemia is associated with epigenetic modifications. Blood. 2011;117:1917–27. doi: 10.1182/blood-2010-09-307140. [DOI] [PubMed] [Google Scholar]
- 77.Chen SS, Claus R, Lucas DM, Yu L, Qian J, Ruppert AS, et al. Silencing of the inhibitor of DNA binding protein 4 (ID4) contributes to the pathogenesis of mouse and human CLL. Blood. 2011;117:862–71. doi: 10.1182/blood-2010-05-284638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Motiwala T, Majumder S, Kutay H, Smith DS, Neuberg DS, Lucas DM, et al. Methylation and silencing of protein tyrosine phosphatase receptor type O in chronic lymphocytic leukemia. Clin Cancer Res. 2007;13:3174–81. doi: 10.1158/1078-0432.CCR-06-1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Motiwala T, Zanesi N, Datta J, Roy S, Kutay H, Checovich AM, et al. AP-1 elements and TCL1 protein regulate expression of the gene encoding protein tyrosine phosphatase PTPROt in leukemia. Blood. 2011;118:6132–40. doi: 10.1182/blood-2011-01-323147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Moreno P, Abreu C, Borge M, Morande P, Pegazzano M, Palacios F, et al. Lipoprotein lipase expression in unmutated CLL patients is the consequence of a demethylation process induced by the microenvironment. Leukemia. 2012 doi: 10.1038/leu.2012.212. In Press. [DOI] [PubMed] [Google Scholar]
- 81.Kaderi MA, Kanduri M, Buhl AM, Sevov M, Cahill N, Gunnarsson R, et al. LPL is the strongest prognostic factor in a comparative analysis of RNA-based markers in early chronic lymphocytic leukemia. Haematologica. 2011;96:1153–60. doi: 10.3324/haematol.2010.039396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Seifert M, Sellmann L, Bloehdorn J, Wein F, Stilgenbauer S, Dürig J, et al. Cellular origin and pathophysiology of chronic lymphocytic leukemia. J Exp Med. 2012;209:2183–98. doi: 10.1084/jem.20120833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Buhl AM, Jurlander J, Geisler CH, Pedersen LB, Andersen MK, Josefsson P, et al. CLLU1 expression levels predict time to initiation of therapy and overall survival in chronic lymphocytic leukemia. Eur J Haematol. 2006;76:455–64. doi: 10.1111/j.0902-4441.2005.t01-1-EJH2530.x. [DOI] [PubMed] [Google Scholar]
- 84.Fabbri G, Rasi S, Rossi D, Trifonov V, Khiabanian H, Ma J, et al. Analysis of the chronic lymphocytic leukemia coding genome: role of NOTCH1 mutational activation. J Exp Med. 2011;208:1389–401. doi: 10.1084/jem.20110921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kleer CG, Zhang Y, Pan Q, van Golen KL, Wu ZF, Livant D, et al. WISP3 is a novel tumor suppressor gene of inflammatory breast cancer. Oncogene. 2002;21:3172–80. doi: 10.1038/sj.onc.1205462. [DOI] [PubMed] [Google Scholar]
- 86.Latif F, Tory K, Gnarra J, Yao M, Duh FM, Orcutt ML, et al. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science. 1993;260:1317–20. doi: 10.1126/science.8493574. [DOI] [PubMed] [Google Scholar]
- 87.Matsuda S, Yokozaki S, Yoshida H, Kitagishi Y, Shirafuji N, Okumura N. Insulin receptor substrate protein 53 (IRSp53) as a binding partner of antimetastasis molecule NESH, a member of Abelson interactor protein family. Ann Oncol. 2008;19:1356–7. doi: 10.1093/annonc/mdn293. [DOI] [PubMed] [Google Scholar]
- 88.Vargova K, Curik N, Burda P, Basova P, Kulvait V, Pospisil V, et al. MYB transcriptionally regulates the miR-155 host gene in chronic lymphocytic leukemia. Blood. 2011;117:3816–25. doi: 10.1182/blood-2010-05-285064. [DOI] [PubMed] [Google Scholar]
- 89.Gutierrez A, Jr., Tschumper RC, Wu X, Shanafelt TD, Eckel-Passow J, Huddleston PM, 3rd, et al. LEF-1 is a prosurvival factor in chronic lymphocytic leukemia and is expressed in the preleukemic state of monoclonal B-cell lymphocytosis. Blood. 2010;116:2975–83. doi: 10.1182/blood-2010-02-269878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Moreno DA, Scrideli CA, Cortez MA, de Paula Queiroz R, Valera ET, da Silva Silveira V, et al. Differential expression of HDAC3, HDAC7 and HDAC9 is associated with prognosis and survival in childhood acute lymphoblastic leukaemia. Br J Haematol. 2010;150:665–73. doi: 10.1111/j.1365-2141.2010.08301.x. [DOI] [PubMed] [Google Scholar]