

Abstract
The genetic basis of the large species differences in longevity and aging remains a mystery. Thanks to recent large-scale genome sequencing efforts, the genomes of multiple species have been sequenced and can be used for cross-species comparisons to study species divergence in longevity. By analyzing proteins under accelerated evolution in several mammalian lineages where maximum lifespan increased, we identified genes and processes that are candidate targets of selection when longevity evolves. We identified several proteins with longevity-specific selection patterns, including COL3A1 that has previously been related to aging and proteins related to DNA damage repair and response such as DDB1 and CAPNS1. Moreover, we found that processes such as lipid metabolism and cholesterol catabolism show such patterns of selection and suggest a link between the evolution of lipid metabolism, cholesterol catabolism, and the evolution of longevity. Lastly, we found evidence that the proteasome–ubiquitin system is under selection specific to lineages where longevity increased and suggest that its selection had a role in the evolution of longevity. These results provide evidence that natural selection acts on species when longevity evolves, give insights into adaptive genetic changes associated with the evolution of longevity in mammals, and provide evidence that at least some repair systems are selected for when longevity increases.
Electronic supplementary material
The online version of this article (doi:10.1007/s11357-011-9361-y) contains supplementary material, which is available to authorized users.
Keywords: Aging, Evolutionary genomics, Protein evolution, Mammals, Proteasome
Introduction
Although the remarkable variety of lifespans and rates of aging among mammals has been studied for decades, the mechanisms behind species differences in aging remain largely unknown (Finch 1990; Miller 2001; Austad 2005). One hypothesis is that long-lived species evolve better mechanisms to cope with different forms of molecular damage, including better repair mechanisms (Kirkwood and Austad 2000; Miller 2001). This view is supported by correlations between DNA repair and mammalian lifespan (Hart and Setlow 1974) and by evidence that cells from long-lived species are more resistant to some forms of stress (Harper et al. 2007). Similarly, two recent studies, one on the naked mole rat (Pérez et al. 2009), the longest living rodent known, and another on two long-lived bats (Salmon et al. 2009), suggest that an optimization of protein oxidation and turnover mechanisms may have been responsible for their exceptional longevity when compared to closely related species.
Evolutionary theory predicts that under low hazard conditions selection favors genes that confer a slow life history and specifically genes and pathways conferring longevity (Kirkwood and Austad 2000; de Magalhães and Church 2007). It has been argued that apolipoprotein E (APOE), involved in age-related diseases and associated with human longevity, is a meat-adaptive protein that contributed to the evolution of human lifespan (Finch and Stanford 2004). Nonetheless, the genetic basis for the evolution of longevity remains a major open question in spite of its widespread interest (Miller 2001).
Thanks to recent large-scale genome sequencing efforts, the genomes of multiple species have been sequenced and can be used for cross-species comparisons. This provides researchers the opportunity of performing genome-wide comparative studies across a large number of species to identify genes and processes associated with the evolution of longevity (Austad 2005; de Magalhães and Church 2007; Jobson et al. 2010). For example, some studies have employed mitochondrial genomic sequences from multiple species to associate features of mitochondrial proteins with longevity, such as methionine composition (Aledo et al. 2011). With the growing number of sequenced genomes, however, these studies have only scratched the surface. In fact, the studies published to date on large-scale analyses of genes involved in the evolution of longevity did not find evidence for selection in repair systems when longevity increases (de Magalhães and Church 2007; Jobson et al. 2010).
To identify candidate genes and processes underlying the evolution of longevity in mammals, we analyzed protein sequence evolution in multiple lineages with varying lifespans. More precisely, our approach involved 36 mammalian species and is based on detecting proteins with accelerated evolution in multiple lineages where longevity increased. Phylogenetic analysis and fossil evidence suggest that longevity generally increased during mammalian evolution, evolving particularly fast in the lineage leading to humans (Cutler 1979; de Magalhães and Toussaint 2002). Genetic alterations contributing to the evolution of longevity in mammals may have therefore common patterns—or “signatures”—that are detectable using cross-species genome comparisons. Indeed, we found evidence of such signatures and our results reveal genes and functional groups that are candidate targets of selection when longevity evolves. These include DNA repair genes and the ubiquitin pathway and thus provide evidence that at least some repair systems were selected for, and arguably optimized, in long-lived species.
Materials and methods
Data retrieval and ancestral sequence prediction
Maximum lifespan was estimated from longevity records obtained from the AnAge database (de Magalhães and Costa 2009). Pairs of nine closely related species for which their maximal lifespans are significantly different were constructed as well as seven control pairs consisting of two species with similar maximal lifespans (Table 1). Since most of the species pairs represent species which have diverged during the Mesozoic era, our analysis represents the evolution of longevity over a long period of time and during the early mammalian radiation. Moreover, because the human and chimpanzee genomes are so closely related and our focus was on well conserved proteins, using humans and chimpanzees as one pair would be virtually useless in our method and therefore a human and orangutan pair was employed. These pairs of species were employed to find candidate genes and functional groups under higher selective pressure in phylogenetic branches where maximal lifespan increased (called MLI branches) compared to branches where maximal lifespan did not increase (MLS branches).
Table 1.
Experimental and control pairs of species
| Lineage where longevity evolved (i.e., long-lived lineage in pair) | MLSP | Lineage where longevity did not evolve | MLSP |
|---|---|---|---|
| Longevity divergent pairs | |||
| Choloepus hoffmanni | 37 | Dasypus novemcinctus | 22.3 |
| Equus caballus | 57 | Canis familiaris | 24 |
| Myotis lucifugus | 34 | Pteropus vampyrus | 20.9 |
| Loxodonta africana | 65 | Procavia capensis | 14.8 |
| Cavia porcellus | 12 | Ancestor of Mus musculus and Rattus norvegicus | 4 |
| Tursiops truncatus | 51.6 | Bos taurus | 20 |
| Homo sapiens | 122.5 | Pongo pygmaeus | 59 |
| Macaca mulatta | 40 | Callithrix jacchus | 16.5 |
| Erinaceus europaeus | 11.7 | Sorex araneus | 3.2 |
| Control pairs (longevity similar between both lineages) | |||
| Tarsius syrichta | 16 | Callithrix jacchus | 16.5 |
| Procavia capensis | 14.8 | Echinops telfairi | 19 |
| Vicugna pacos | 25.8 | Sus scrofa | 27 |
| Oryctolagus cuniculus | 9 | Ochotona princeps | 7 |
| Rattus norvegicus | 4 | Mus musculus | 4 |
| Spermophilus tridecemlineatus | 7.9 | Dipodomys ordii | 9.9 |
| Otolemur garnettii | 18.3 | Microcebus murinus | 18.2 |
Maximum lifespan potential (MLSP) estimated from the record longevity of the species obtained from the AnAge database
Ortholog mappings of proteins of 36 mammalian species to Homo sapiens were obtained from ENSEMBL (http://www.ensembl.org/) resulting in 15,350 proteins with at least one 1:1 ortholog (paralogs were excluded from our approach). Using these mappings and protein multiple sequence alignments along with a reference phylogenetic tree, ancestral protein sequences for the 15,350 proteins were predicted using Gapped Ancestral Sequence Prediction (Edwards and Shields 2004) for each node of the phylogenetic tree. Since any phylogenetic approach aiming to detect selection is highly sensitive to wrongly annotated splice variants or spurious alignments, protein orthologs with more than 10 substitutions out of a sliding window of 20 residues were removed. After this scan, 15,312 proteins had at least one other ortholog. Aging-related proteins based on findings from model organisms were obtained from the GenAge database of de Magalhães et al. (2009a).
Scoring evolutionary selection
Because under low hazard conditions genes conferring longevity are expected to be under selection (de Magalhães and Church 2007), proteins involved in the evolution of longevity are expected to undergo more changes in MLI branches than in MLS branches, and our algorithm was specifically devised to detect such proteins. For each of the 15,312 proteins, substitution scores based on the predicted ancestral protein sequence and on the physiochemical properties of the residue substitutions (Grantham 1974) were computed (see Supplementary material) in each branch as a proxy for selective pressure akin to Zhang et al. (2002), where the number of residue substitutions was used as a measure for evolutionary pressure. It should be noted here that the use of similar matrices, such as the BLOSUM matrix, did not alter the results obtained. For each protein and branch, the expected value of the number of residue substitutions was computed according to the empirical distribution of the residue substitutions in the branch in all proteins by comparing predicted ancestral protein sequences at each node of the phylogenetic tree. This expected value was used to normalize the protein score for each branch in order to minimize the effects of different branch lengths, amino acid compositions, and protein lengths across different lineages. Although our normalization strategy is simplistic, it is able to capture differences in generation time, rate of mutation, biases in amino acid substitutions, and biases in protein lengths, unlike Zhang et al. (2002) who used divergence time as proxy for rates of evolution. Our normalization strategy does not take into consideration the variation of substitution rate across amino acid sites, and multiple substitution events, but our focus was on well-conserved proteins which obviate the need for a more complex model. Although the expected value is an under-estimate of the true number of substitutions due to the possibility of multiple substitutions at a single residue site, this effect is minimal as the lineages used in our study are short and the proteins are chosen to be well conserved. After all scores have been normalized, for each of the pairs of species with divergent lifespan (for which an ortholog exists in both branches) and each of the control pairs, an approximate selection score was calculated to measure the relative selective pressure in one lineage versus the other by the binomial score:
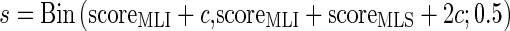 |
Where scoreMLI is the substitution score for the protein in the MLI branch in the pair, scoreMLS is the substitution score for the same orthologous protein in the MLS branch of the pair, and c is a pseudo-count (c = 3 in this study) so that s is stable for conserved proteins. The parameters are then truncated for the binomial test. For the control pairs where both branches (A and B) are MLS branches, only the smallest selection score is kept:
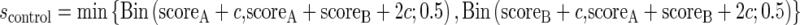 |
An assumption of our method is that selection for longevity only occurs in branches where longevity increased. The evolutionary theory of aging predicts that aging per se and the evolution of short lifespans are not under direct selection, while in species under low hazard conditions, selection will favor genes that confer longevity (de Magalhães and Church 2007). Moreover, longevity is expected to have increased in most mammalian species (Cutler 1979; de Magalhães and Toussaint 2002). Therefore, for a given pair of species with divergent lifespans, it is likely that longevity evolution contributed and was even the most prevalent force in the observed longevity differences. It is possible that for some species pairs used in this work, in particular pairs less closely related, that longevity decreased in the MLS branch, but while this may increase the noise of that particular branch, it will not introduce any systematic bias since protein alterations from species with the smaller MLSP are not incorporated into the results.
Detecting longevity-specific selectivity in proteins
The fact that different proteins are under different evolutionary pressure and have different lengths was taken into consideration by considering three thresholds for significance (t = 0.05 (stringent), 0.1 (moderate), 0.2 (relaxed)). To understand the advantage of having three different selection score thresholds, consider a well-conserved protein with a weak signature of selectivity specific to MLI branches. Under the stringent selectivity criteria, this protein will show no signature of selectivity in any branch at all, but under the relaxed selectivity criteria, it will obtain a high longevity-specific selectivity (LSS) score. Conversely, consider a protein that exhibits a signature of selectivity in many different branches but much stronger in MLI branches. The variability of the strength of selective pressure is big and so many branches will be considered to be under selection with respect to a relaxed selectivity criteria, hence yielding a low score because of the lack of specificity to MLI branches. Therefore, employing different selectivity criteria allows coverage of proteins with varying molecular evolution rates and lengths.
To assess whether a particular protein is associated with increased longevity, the number of pairs, NMLI, where the proteins has a selection score such that s < t is recorded. The number of pairs, NMLS, such that 1-s < t is also recorded as well as the number of control pairs, Ncontrol, such that scontrol < t. With these numbers, a normalized “longevity-specific selectivity” score for each protein can be computed as follows:
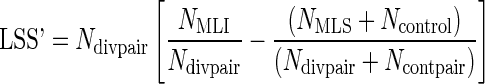 |
Where Ndivpair is the number of pairs with divergent lifespans for which a protein ortholog exists in both branches, and Ncontpair is the number of control pairs for which a protein ortholog exists in both branches. Therefore NMLI/Ndivpair is the percentage of MLI branches in which the protein is selected and 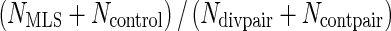 is the percentage of MLS branches in which the protein is selected. It can be seen that
is the percentage of MLS branches in which the protein is selected. It can be seen that 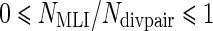 and
and 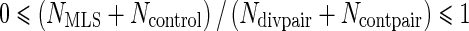 so that
so that 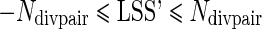 also holds with equality when either (1) the protein is selected in all MLI branches and no MLS branch or (2) the protein is selected in all MLS branches in pairs with divergent lifespans and in one branch in all control pairs. Moreover, assuming a trivial threshold of t = 1, both
also holds with equality when either (1) the protein is selected in all MLI branches and no MLS branch or (2) the protein is selected in all MLS branches in pairs with divergent lifespans and in one branch in all control pairs. Moreover, assuming a trivial threshold of t = 1, both 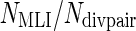 and
and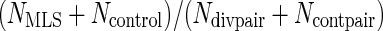 would be 1 and LSS’ would equal 0. In this study, only proteins that have a selection specificity towards MLI branches are of interest so LSS = max (0, LSS’) was used as the “longevity-specific selectivity” score. It follows that any protein with negative LSS’ has a LSS of 0, i.e., no longevity-specific selectivity. Thus, the LSS score measures the specificity of the selection in MLI branches. Although there is no minimum number of pairs for a protein to be considered, the lower the number of experimental pairs there is, the lower the potential LSS score is.
would be 1 and LSS’ would equal 0. In this study, only proteins that have a selection specificity towards MLI branches are of interest so LSS = max (0, LSS’) was used as the “longevity-specific selectivity” score. It follows that any protein with negative LSS’ has a LSS of 0, i.e., no longevity-specific selectivity. Thus, the LSS score measures the specificity of the selection in MLI branches. Although there is no minimum number of pairs for a protein to be considered, the lower the number of experimental pairs there is, the lower the potential LSS score is.
Detecting longevity-specific selectivity in functional categories
After computing the “longevity-specific selectivity” scores for all proteins, Gene Ontology (GO) annotations (Ashburner et al. 2000) were obtained in order to score each GO category by adding the LSS score of each protein within the category. To compute the significance of each GO category, the empirical distribution of the LSS scores was obtained, the proteins scores were shuffled, and the scores for the GO categories were recomputed 2,000 times. The p value for each category was computed as the number of times the simulation yielded a score for a GO category that is larger than its actual score divided by 2,000. To compare the number of significant GO categories detected in real data compared to simulated data, the proteins’ LSS scores were shuffled 2,000 times, generating 2,000 simulated datasets. Because the high score of one protein could largely influence the p value of a small GO category, only categories with at least three proteins showing specificity of selection towards MLI branches were included. After removing all GO categories with less than three proteins (out of the 15,312), the number of significant GO categories at thresholds 0.05, 0.01, 0.005, and 0.001 was computed for each simulated datasets to be averaged and compared with the number of significant GO categories from real data at the same thresholds (see Supplementary material for simulation results).
To see whether our approach was biased towards some unwanted protein properties, the distribution of protein lengths was analyzed and no correlation with score was found. Furthermore, among the top proteins in each category, none of them had an obvious splice variant that could cause a bias. Many similar phylogenetic approaches using ortholog mappings are highly sensitive to proteins with misannotated orthologs, thus an aggressive weeding strategy to remove protein sequences that are putative splice variants was used. Moreover, although phylogenetic approaches need to take into consideration the phylogenetic dependence within the species considered, our method is unaffected by this dependence as we defined disjoint longevity divergent species pairs with no common evolutionary branch. To further test our approach, it was important to verify that the selected parameters did not drastically influence the results. Using a different scoring matrix yielded similar results, and no significant differences were observed within the proteins and GO categories reported when using different thresholds for selectivity.
Results
Detecting longevity-specific selection in proteins
Under low hazard conditions, selection will favor gene changes that confer longevity. Therefore, proteins involved in the evolution of longevity are expected to be under selection in lineages where longevity evolved (Kirkwood and Austad 2000; de Magalhães and Church 2007). Moreover, we expect these proteins to be under stronger selective pressure in lineages where longevity increased (maximum lifespan increased; MLI branches) compared to lineages where longevity remained the same (maximum lifespan same; MLS branches). Proteins conferring longevity will undergo rapid evolution when longevity evolves and therefore undergo more changes in MLI branches than expected by chance. Thus, our approach aims to identify proteins under accelerated evolution in several mammalian lineages where maximum lifespan increased, in effect detecting proteins with a pattern of selectivity specific to MLI branches.
We constructed ortholog mappings of 15,350 proteins from 36 mammalian species and predicted ancestral protein sequences in accordance to the mammalian phylogenetic tree. We then computed an evolutionary pressure score for all proteins in all branches of the phylogenetic tree based on the number and type of amino acid substitutions in each branch (see “Materials and methods”). In effect, these evolutionary pressure scores measure the strength of the selective pressure on a protein in each lineage, taking into account the genome-wide average. This is similar to the method of Zhang et al. (2002) to infer selection pressure from the number of residue substitutions. We then defined nine experimental pairs of longevity divergent species plus seven control pairs of species with similar longevity (Table 1). The experimental pairs each correspond to species resulting from one MLI lineage (i.e., the longest lived lineage in the pair) and one MLS lineage stemming from a common ancestor and the control pairs to species resulting from two MLS lineages also stemming from a common ancestor.
In order to determine whether a protein in one lineage is under stronger selective pressure than in another, we compared the evolutionary pressure scores computed for the protein in both lineages using a binomial test to obtain an accelerated evolution score (see “Materials and methods”). This accelerated evolution score represents the extent of evolution acceleration of a protein in one lineage compared to another. Since both poorly and well-conserved proteins may be responsible for species divergence in aging, we defined three different accelerated evolution score thresholds to account for varying selective pressures. In other words, the different thresholds, 0.05, 0.1, and 0.2, reflect different levels of evolutionary pressure and hence molecular evolution rates on proteins. Proteins undergoing higher evolutionary pressure tend to have more species pairs satisfying the 0.05 threshold as their evolutionary pressure scores tend to fluctuate, whereas proteins that are well conserved tend to have low evolutionary pressure scores which are less likely to fluctuate between lineages.
We gave each protein three (one for each different threshold) “longevity-specific selectivity” scores (LSS) computed according to the number of experimental pairs where the protein was under stronger selective pressure in MLI branches, the number of both experimental and control pairs where the protein was under stronger selective pressure in MLS branches, and the total number of divergent lifespan species and control pairs considered (see “Materials and methods”). As such, the LSS score encapsulates how specific the selection of the protein is to lineages where maximal lifespan increased (MLI branches).
The proteins were then sorted according to their scores for each of the three levels of selective pressure (i.e., 0.05, 0.1, and 0.2). For this ranking of specificity towards MLI branches, no statistical significance test was performed as a random model for protein residue evolution at a genome-wide level, taking into consideration multiple species pairs, was not applicable. Instead, we focused on the top 153 proteins, which correspond to the top 1% of all proteins analyzed, as candidate proteins related to species difference in aging with regard to the three selectivity criteria.
Proteins with longevity-specific selectivity
After computing the LSS score for each of the 15,312 proteins in the three selectivity categories, we found that proteins generally obtained a low score, often scoring zero. When applying the relaxed selectivity threshold, only 598 proteins scored 2 or more and a mere 31 scored 4 or more; the highest score of 6.0 was attained by only one protein, FAM126B, an unstudied protein. Other high scoring proteins included COL3A1, scoring 5.29 (rank 4), TAOK3, scoring 5.0 (rank 7), and DDB1, scoring 4.83 (rank 9; see Fig. 1). Likewise, 339 proteins had a score of 2.0 or more and six with a score of 4.0 or more with CPNE5, NUP85, and RSAD2 sharing the top score of 5.0. Lastly, when applying the stringent selectivity threshold, 166 proteins had a score of 2.0 or more. The highest score was obtained by IWS1 (Protein IWS1 homolog) scoring the highest with 4.33, followed by HERC4 which shares the score of 4.0 with seven other proteins. Though some overlap between the thresholds was observed, nine proteins were ranked highly (top 20) in two categories and only one protein, FAM126B, is highly ranked in all three. The scoring is a rank-order of all proteins and thus these highly ranked proteins represent the most promising candidates for selection in long-lived species. Full results are available as Supplementary material and online (http://genomics.senescence.info/evolution/mammalian_longevity.html).
Fig. 1.

DDB1 under selection in longevity-specific lineages. Longevity drastically increased in MLI lineages (red) compared to MLS lineages (blue). Protein substitution scores are labeled next to branches when available. DDB1 has a strong signature of longevity-specific selection; its evolutionary pressure score is significantly higher in longer lived species in all experimental pairs except for Sorex araneus
As in other similar studies (de Magalhães and Church 2007; Jobson et al. 2010), proteins known to be related to aging in model systems, derived from the GenAge database, were not overrepresented in our results. Among our top hits, NUP85 is a homolog of npp-2 while ATP7A (in the top 1% hits in all threshold criteria) is a homolog of cua-1. Both npp-2 and cua-1 have been associated with longevity in Caenorhabditis elegans (Samuelson et al. 2007). POLB, involved in DNA maintenance, replication, and recombination, was among the top 1% hits at the 0.2 threshold but with a modest score of 3.3. Interestingly, three proteins among our hits were also recently reported to be under positive selection in the long-lived naked mole rat: COL3A1, PRKD2, and GSTO1 (Kim et al. 2011).
Detecting longevity-specific selection in functional categories
To identify processes and functional categories exhibiting evidence of selection specific to MLI branches, we employed GO annotations (Ashburner et al. 2000). Briefly, we gave every GO category an LSS score consisting of the sum of the LSS scores of the proteins belonging to the category. Using simulations (see “Materials and methods”), we obtained a score enrichment p value for each GO category testing against a random model. Again, for each GO category, we obtained three enrichment p values, one for each level of selective pressure.
We sorted all categories according to the geometric mean of the p values for the three thresholds of selectivity. Out of the 15,551 GO categories considered, 4,180, 4,396, and 4,443 categories had a non-zero score with respect to stringent, moderate, and relaxed selectivity pressure criteria, respectively, and 3,267 categories had non-zero scores with respect to all three criteria. We obtained around 150 GO categories with significant p value with respect to at least one selectivity criteria. This rank-order represents less than 1% of all functional groups and many of the categories are closely related to one another (see Supplementary material for full results).
By shuffling the LSS scores of the proteins, the average number of significant GO categories from a random model was compared to the actual number of significant GO categories. We found that the number of GO categories in real data was consistently higher compared to the ones of simulated data from a random model with respect to all selectivity thresholds (~20% more at 0.05 significance, ~50% at 0.01, and ~100% at 0.005 significance; see Supplementary material). Though our simulated data show significant levels of false positives, we found that, out of the top 150 GO categories with respect to each selectivity criteria, 33 categories are in the top 150 with respect to all criteria, and 76 with respect to two. Many of the top categories are related to each other without necessarily sharing proteins with high LSS score. In view of these results, we focused on categories that are found among different selectivity criteria, for which we find different related categories among our top-ranked categories and/or categories with p < 0.005.
The major functional class within the highly ranked GO terms encompassed proteins involved in muscle development along with brain development (Table 2), which showed the most significant enrichment in high LSS scores when using threshold for moderate and strong evolutionary pressures. Other GO categories related to muscle and brain development were also among our top 150 hits; besides, another GO category related to development was spermatid development which was enriched in LSS scores with respect to the three thresholds (see Supplementary material). These categories could be related to life history traits that co-evolve with longevity.
Table 2.
Top GO categories, ranked from the geometric mean of the p values at different thresholds, and their longevity-specific selection significance
| Selectivity criteria | 0.05 | 0.1 | 0.2 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| GO category | p value | # prot | Exp. | Act. | p value | # prot | Exp. | Act. | p value | # prot | Exp. | Act. |
| Postsynaptic membrane | 0.001 | 40 | 24.1 | 47.3 | 0.001 | 46 | 28.4 | 59.8 | 0.001 | 46 | 31.4 | 61.5 |
| Synapse | 0.001 | 56 | 40 | 63.4 | 0.001 | 69 | 47.1 | 83.8 | 0.001 | 64 | 52 | 84.2 |
| Muscle myosin complex | 0.001 | 5 | 1.7 | 8.3 | 0.002 | 5 | 2 | 10.3 | 0.001 | 5 | 2.2 | 10.7 |
| Sodium ion binding | 0.001 | 39 | 21.3 | 43.4 | 0.002 | 34 | 25 | 42.8 | 0.001 | 39 | 27.7 | 58.6 |
| Sodium ion transport | 0.001 | 40 | 22.7 | 43.2 | 0.004 | 36 | 26.7 | 41.5 | 0.001 | 39 | 29.5 | 55.5 |
| Endoplasmic reticulum organization | 0.001 | 3 | 1.1 | 6.9 | 0.001 | 4 | 1.3 | 7.3 | 0.011 | 3 | 1.5 | 5.8 |
| Myosin filament | 0.001 | 7 | 2.6 | 10.8 | 0.001 | 7 | 3 | 11.9 | 0.013 | 6 | 3.3 | 9.5 |
| Antiporter activity | 0.002 | 13 | 5.7 | 15.1 | 0.002 | 14 | 6.7 | 16.8 | 0.008 | 12 | 7.4 | 16.6 |
| Regulation of pH | 0.001 | 8 | 3.1 | 11.1 | 0.001 | 8 | 3.7 | 11.8 | 0.037 | 6 | 4.1 | 8.9 |
| Transmembrane transport | 0.001 | 110 | 84.6 | 118.6 | 0.006 | 124 | 99.5 | 129.0 | 0.009 | 116 | 109.9 | 141.9 |
# prot number of proteins with LSS score bigger than 0, Exp. expected sum of LSS scores, Act. actual sum of LSS scores
Furthermore, a close inspection of the highly ranked GO terms revealed several categories that may be associated with longevity evolution (Table 3). In particular, proteins involved in lipid process were among the statistically significant categories along with cholesterol catabolic process which only show significance at a high selectivity threshold. Another class of proteins comprises four functional categories involved in the proteasome–ubiquitin system which are: protein ubiquitination during ubiquitin-dependent protein catabolic process, proteasomal ubiquitin-dependent protein catabolic process, ATP-dependent peptidase activity, and lysosome organization. Interestingly, the proteins with positive LSS scores detected in these categories were non-overlapping, suggesting that these results are due to selection on these functions rather than some bias due to one or a few proteins. We also detected other GO categories with high LSS but with few highly ranked related categories. As such, they are more likely to be false positives. Both actin-binding and actin cytoskeleton proteins were ranked highly with regards to all selectivity criteria. In addition, we found that 1-phosphatidylinositol-3-kinase activity, phosphoinositide 3-kinase complex, response to food, and circadian rhythm were all highly ranked with respect to at least one selectivity criteria.
Table 3.
Selected GO categories from the top 1% of potential relevance to aging and their longevity-specific selection significance
| Selectivity criteria | 0.05 | 0.1 | 0.2 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| GO category | p value | # prot | Exp. | Act. | p value | # prot | Exp. | Act. | p value | # prot | Exp. | Act. |
| Response to food | 0.053 | 3 | 1.7 | 4.1 | 0.007 | 4 | 2.0 | 7.2 | 0.003 | 5 | 2.2 | 8.6 |
| Actin binding | 0.004 | 54 | 36.6 | 53.2 | 0.04 | 51 | 43.1 | 56.0 | 0.007 | 53 | 47.6 | 68.9 |
| Phospholipid metabolic process | 0.026 | 11 | 4.5 | 9.6 | 0.026 | 9 | 5.3 | 11.1 | 0.002 | 12 | 5.9 | 17.5 |
| Fatty acid beta-oxidation | 0.004 | 6 | 3.7 | 10.0 | 0.006 | 9 | 4.3 | 11.8 | 0.153 | 7 | 4.8 | 7.5 |
| ATP-dependent peptidase activity | 0.021 | 6 | 2.0 | 5.8 | 0.004 | 5 | 2.3 | 8.2 | 0.044 | 3 | 2.6 | 6.2 |
| 1-Phosphatidylinositol-3-kinase activity | 0.009 | 4 | 1.4 | 5.2 | 0.034 | 4 | 1.7 | 4.6 | 0.018 | 4 | 1.8 | 6.0 |
| Phosphoinositide 3-kinase complex | 0.011 | 5 | 1.7 | 5.6 | 0.016 | 5 | 2.0 | 6.0 | 0.043 | 4 | 2.2 | 6.0 |
| Peptidase activator activity | 0.067 | 2 | 0.9 | 2.4 | 0.002 | 3 | 1.0 | 5.6 | 0.068 | 3 | 1.1 | 3.2 |
| Proteasomal ubiquitin-dependent protein catabolic process | 0.185 | 6 | 2.3 | 3.7 | 0.03 | 7 | 2.7 | 6.5 | 0.004 | 5 | 3.0 | 10.4 |
| Protein ubiquitination during ubiquitin-dependent protein catabolic process | 0.039 | 3 | 2.3 | 5.4 | 0.015 | 5 | 2.7 | 7.2 | 0.042 | 4 | 3.0 | 6.9 |
| Condensed chromosome kinetochore | 0.007 | 10 | 6.2 | 14.0 | 0.012 | 8 | 7.3 | 15.0 | 0.452 | 7 | 8.1 | 8.5 |
| Circadian rhythm | 0.042 | 8 | 5.4 | 9.8 | 0.01 | 10 | 6.3 | 13.6 | 0.096 | 9 | 7.0 | 11.2 |
| Lipid biosynthetic process | 0.107 | 5 | 4.0 | 6.7 | 0.016 | 7 | 4.7 | 10.9 | 0.036 | 6 | 5.2 | 10.5 |
Full results are available in the Supplementary material and online (http://genomics.senescence.info/evolution/mammalian_longevity.html)
# prot number of proteins with LSS score bigger than 0, Exp. expected sum of LSS scores, Act. actual sum of LSS scores
Discussion
Proteins related to longevity in model organisms appear to be well conserved and might even tend to be better conserved than expected by chance, suggesting that the genetic mechanisms for longevity regulation within species are not the same that determine species differences in longevity (de Magalhães and Church 2007). Therefore, our method is conceptually different than previous comparative genomic methods employed to study the evolution of longevity which primarily focused on known aging-related genes and pathways (de Magalhães and Church 2007; Jobson et al. 2010). Instead, our genome-wide analysis was unbiased, and we wanted to detect selection in proteins with different evolutionary rates but having specificity towards phylogenetic branches where maximal longevity significantly increased. Our LSS score thus measures both the number of MLI branches under selective pressure and its specificity. Although long-lived species may have different substitution rates due to population effects or generation times, we normalize the protein substitution scores by the genome-wide average for each branch and hence control for the phylogenetic distance. Therefore, our method detects outliers with substitution scores in MLI branches much higher than average, and such proteins can thus be seen as candidates for playing a role in longevity evolution.
Because some proteins tend to be under higher selective pressures, and thus evolve faster, than others, we employed three different thresholds (stringent, moderate, and relaxed). Employing these different thresholds allowed us to detect proteins with longevity-associated signatures that evolved at different paces. Based on the LSS scores of proteins belonging to GO categories, we also obtained LSS scores for processes and functions. Interestingly, the shuffling of real data shows that we obtained more functional categories with high LSS scores than expected by chance alone, which as far as we know is the first evidence that natural selection acts on species when longevity evolves.
There are different reasons why some proteins may undergo accelerated evolution in MLI lineages. Our assumption is that an increased number of substitutions in MLI branches indicates that a given protein is under selection for longevity. One caveat, however, is that changes in mutation rates, relaxed purifying selection, and positive selection could lead to accelerate evolution of proteins (Zhang et al. 2002). Although we cannot exclude these alternative explanations, given the patterns detected in our work when compared to a random model and the functions detected from the GO analyses, we think our results are better explained by genetic changes driven by selection. One related issue, however, is whether selection is acting on longevity or on another correlated life history trait. In our analysis, we found many proteins involved in development and growth resulting in functional categories such as muscle development, postsynaptic density, and spermatid development. These results, among others (see Supplementary material for full results), could be interpreted as proteins that were selected for phenotypes that correlate longevity, such as body size or brain size (de Magalhães et al. 2007; Austad 2009; Ricklefs 2010). Therefore, some of our results may be due to selection on other life history traits apart from longevity. For example, one of our top proteins was IWS1 (rank 1; stringent) which, as far as we know, has not been studied in mammals. A recent study in Arabidopsis, however, shows that IWS1 is involved in plant steroid hormone and a loss of function mutations in AtIWS1 leads to overall dwarfism (Li et al. 2010) and thus may have been detected in our work due to a putative role in the evolution of body size. Nonetheless, as detailed below, the protective nature of some of the categories identified leads us to hypothesize that some of these selection patterns are due to selection for longevity.
Candidate proteins related to longevity evolution
Though it is doubtful that changes in the same protein are responsible for the evolution of longevity in all mammalian lineages, proteins with accelerated evolution in lineages where longevity increased are candidates for being involved in species divergence in aging. The rank-order of the LSS scores provides us with the proteins having the most number of pairs where selective pressure occurred in MLI branches while having a small number of pairs where selective pressure occurred in lineages where longevity stayed the same. Thus, highly ranked proteins represent candidate targets of selection due to the evolution of longevity.
Candidate proteins from the high stringency threshold include HERC4, a probable E3 ubiquitin–protein ligase by sequence similarity (Wu et al. 2006), and NUP85, a component of the nuclear pore complex thought to play a role in phosphatidyl-inositol-3-kinase-dependent pathways (Terashima et al. 2005). CAPNS1, scoring 4.0 (rank 7; moderate), belongs to a well-conserved family of calcium-dependent, cysteine proteases whose link to cellular senescence and DNA damage response has been studied (Demarchi and Schneider 2007). COL3A1 (rank 4; relaxed) is a collagen-type protein whose expression is downregulated with age across tissues (de Magalhães et al. 2009b) and has also been reported to be under positive selection in the long-lived naked mole rat (Kim et al. 2011), making it a top candidate for selection in long-lived lineages. Also, TAOK3 (rank 7; relaxed) is a serine/threonine-protein kinase whose overexpression may activate ERK1/ERK2 and JNK/SAPK (Zhang et al. 2000). Furthermore, TAOK3 is thought to be phosphorylated upon DNA damage possibly by ATM or ATR (Matsuoka et al. 2007). And finally, the damage-specific DNA binding protein DDB1 (rank 11; relaxed) is a well-studied protein and is a subunit of the DDB1-CUL4-X (DCX) box which can form many different complexes that are involved in different DNA damage response pathways.
Other examples of high scoring proteins comprise PIK3C2A, scoring 3.0 (rank 26; stringent) and SMC1A, scoring 2.77 (rank 74; stringent). PIK3C2A is a protein belonging to the PI3/PI4-kinase family and is believed to play a role in the EGF signaling pathway (Arcaro et al. 2000). Moreover, Didichenko et al. (2003) showed that H. sapiens PIK3C2A is phosphorylated upon exposure of cells to UV irradiation and is also a target of stress-induced phosphorylation during the G2/M transition of cell cycle by the JNK/SAPK pathway. These authors further found that the phosphorylation seems to lead to the proteasome-dependent degradation of PIK3C2A (Didichenko et al. 2003). SMC1A is a protein involved in chromosome cohesion during cell division as well as in DNA repair. More precisely, SMC1A is related to the cohesion between sister chromatids during DNA replication and, at least in yeast, the cohesin complex also has functions in DNA repair and is essential for efficient double-strand break repair in mitotic cells (Sjögren and Nasmyth 2001).
Candidate pathways related to longevity evolution: lipid metabolism, DNA repair, and the proteasome–ubiquitin system
It would be surprising if the evolution of longevity in all mammalian lineages could be explained by adaptive changes to the same few proteins. A more intuitive explanation of the evolution of longevity is due to selection of proteins in common pathways and biological processes, and thus, our analysis of GO categories provides novel clues about the processes involved in species differences in aging. Interestingly, we found functional categories showing specificity of selection in MLI branches which have been previously associated to aging such as actin cytoskeleton (Gourlay and Ayscough 2005), 1-phosphatidylinositol-3-kinase activity, phosphoinositide 3-kinase complex, response to food, and circadian rhythm (Wyse et al. 2010). Moreover, many of our highly ranked functional categories may have been selected for due to their contribution to the evolution of longevity.
Our finding of phospholipid metabolic process proteins corroborates previous results suggesting a role of lipid metabolism in species differences in longevity (Hulbert 2008; Jobson et al. 2010). It has been reported that membrane fatty acid composition is correlated with the maximal lifespans of mammals through the reduction of oxidative damage caused by products of lipooxidation (Hulbert 2008). Proteins belonging to the lipid biosynthetic process were also identified in our analysis which have previously been linked to the peroxidative damage via increased saturation and to the control of mitochondrial ROS production by reducing membrane potential and increasing the efficiency of ETC uncoupling (Kua 2006; Jobson et al. 2010). Moreover, cholesterol catabolic process-related proteins were also identified, and these findings fit studies of the protein APOE well, even though APOE did not show longevity-specific selectivity in our study. The identification of cholesterol catabolic process in our analysis and the evolutionary pressure detected on proteins involved in lipid metabolic process lead us to believe that lipid metabolism and cholesterol catabolism may have been important in the evolution of mammalian longevity.
Many high ranked proteins at different levels of selection are involved in cellular responses to damage. With respect to the stringent selectivity criteria, PIK3C2A (rank 26) and SMC1A (rank 74) are two proteins thought to be sensitive to external stress or DNA damage. In the moderate selectivity criteria category, CAPNS1 (rank 7) has been connected to DNA damage response, and in the relaxed selectivity criteria category, we have TOAK3 (rank 7) and DDB1 (rank 9; Fig. 1) which have been shown to respond to DNA damage. DDB1 in particular is involved in many distinct DNA response and DNA repair pathways. For example, DDB1 binds to SKP2 and plays a role in the ubiquitination of CDKN1B, a cyclin-dependent kinase inhibitor (Nishitani et al. 2006) and may recruit nucleotide excision repair proteins in order to repair DNA damage (Li et al. 2006). Deficiency in DDB1 is associated with xeroderma pigmentosum (Kapetanaki et al. 2006). Mutational inactivation of DDB1 is also associated with Cockayne syndrome (Groisman et al. 2003) which is characterized by premature and accelerated aging in addition to neurodegeneration (Weidenheim et al. 2009). In this context, it is interesting to note that differences in expression in DDB2 between rodents and primates have been reported to play a role in protection against DNA damage and carcinogenesis (Alekseev et al. 2005). Although DNA repair was not among our top GO categories, we speculate that the evolutionary pressure on a few specific proteins involved in DNA repair or DNA damage response could be an optimization leading to a better regulation of damage, cell cycle, and genome stability and hence to a longer lifespan, in line with results suggesting higher DNA repair in longer-lived mammals (Hart and Setlow 1974; Freitas and de Magalhães 2011).
We found that proteins involved in the proteasome–ubiquitin system have high LSS scores. More precisely, we found four highly ranked GO categories related to the proteasome–ubiquitin system which are protein ubiquitination during ubiquitin-dependent protein catabolic process, proteasomal ubiquitin-dependent protein catabolic process, ATP-dependent peptidase activity, and lysosome organization. Interestingly, these GO categories do not share a single protein that contributed to their score which provides strong evidence that the system was the target of evolutionary pressure in lineages where longevity increased. The proteasome has been extensively linked to aging. Interestingly, Pérez et al. found that, compared to mice, naked mole rats show resistance to protein unfolding and attenuated accumulation of ubiquitinated proteins and a sustained proteasomal function during aging (Pérez et al. 2009), suggesting that these mechanistic differences may contribute to species divergence in aging and that the maintenance of protein stability is of great importance to successful aging. Proteasome activity was also shown to participate in DNA repair at different levels (Brégégère et al. 2006). In sum, the ubiquitination process and the proteasome complex are active components of cellular response to stress and damage, and their evolutionary selection might have contributed to a lifespan increase in mammals.
Overall, our analysis identified several proteins involved in damage response and repair pathways that are highly specific in their accelerated evolution to lineages where longevity evolved. Taken together, these results suggest that our approach was able to detect proteins that are connected to species differences in aging. Moreover, these findings support the view that the evolution of longevity requires optimization of repair pathways, one of the tenants of the evolutionary theory of aging (Kirkwood and Austad 2000).
Concluding remarks
In this work, we present a novel approach to study the evolution of lifespans in mammalian species by looking for selection specificity in lineages where longevity is thought to have considerably increased. We identify several candidate proteins, including COL3A1, DDB1, and CAPNS1, that are candidates for further studies. Among the significant genes and categories, some may be related to selection on life history traits that co-evolve with longevity while others may be related to the evolution of longevity, including proteins involved in DNA damage response and repair. Better protein degradation and turnover mechanisms via the optimization of proteins involved in the proteasome–ubiquitin system might also have contributed to the evolution of longevity. To our knowledge, the identification of these categories is the first evidence of selection associated with the evolution of longevity detected on a whole-genome level. As more genomes are sequenced, analyses employing this and similar approaches will become more powerful and our work is only one more step into unraveling the adaptive genetic changes involved in the evolution of long lifespans.
Electronic supplementary material
(XLS 220 kb)
(XLS 178 kb)
(DOC 73 kb)
Acknowledgments
YL was supported by a Postgraduate Scholarship from the Natural Sciences and Engineering Research Council of Canada. JPM thanks the BBSRC (BB/G024774/1 & BB/H008497/1), the Ellison Medical Foundation, and a Marie Curie International Reintegration Grant within EC-FP7 for supporting work in his lab.
References
- Aledo JC, Li Y, de Magalhães JP, Ruíz-Camacho M, Pérez-Claros JA. Mitochondrially encoded methionine is inversely related to longevity in mammals. Aging Cell. 2011;10:198–207. doi: 10.1111/j.1474-9726.2010.00657.x. [DOI] [PubMed] [Google Scholar]
- Alekseev S, Kool H, Rebel H, Fousteri M, Moser J, Backendorf C, de Gruijl FR, Vrieling H, Mullenders LH. Enhanced DDB2 expression protects mice from carcinogenic effects of chronic UV-B irradiation. Cancer Res. 2005;65:10298–10306. doi: 10.1158/0008-5472.CAN-05-2295. [DOI] [PubMed] [Google Scholar]
- Arcaro A, Zvelebil MJ, Wallasch C, Ullrich A, Waterfield MD, Domin J. Class II phosphoinositide 3-kinases are downstream targets of activated polypeptide growth factor receptors. Mol Cel Bio. 2000;20:3817–3830. doi: 10.1128/MCB.20.11.3817-3830.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austad SN. Diverse aging rates in metazoans: targets for functional genomics. Mech Ageing Dev. 2005;126:43–49. doi: 10.1016/j.mad.2004.09.022. [DOI] [PubMed] [Google Scholar]
- Austad SN. Comparative biology of aging. J Gerontol A Biol Sci Med Sci. 2009;64:199–201. doi: 10.1093/gerona/gln060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brégégère F, Milner Y, Friguet B. The ubiquitin–proteasome system at the crossroads of stress-response and ageing pathways: a handle for skin care? Ageing Res Rev. 2006;5:60–90. doi: 10.1016/j.arr.2005.09.002. [DOI] [PubMed] [Google Scholar]
- Cutler RG. Evolution of human longevity: a critical overview. Mech Ageing Dev. 1979;9:337–354. doi: 10.1016/0047-6374(79)90110-6. [DOI] [PubMed] [Google Scholar]
- de Magalhães JP, Church GM. Analyses of human–chimpanzee orthologous gene pairs to explore evolutionary hypotheses of aging. Mech Ageing Dev. 2007;128:355–364. doi: 10.1016/j.mad.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Magalhães JP, Costa J. A database of vertebrate longevity records and their relation to other life-history traits. J Evol Biol. 2009;22:1770–1774. doi: 10.1111/j.1420-9101.2009.01783.x. [DOI] [PubMed] [Google Scholar]
- de Magalhães JP, Toussaint O. The evolution of mammalian aging. Exp Gerontol. 2002;37:769–775. doi: 10.1016/S0531-5565(02)00008-6. [DOI] [PubMed] [Google Scholar]
- de Magalhães JP, Costa J, Church GM. An analysis of the relationship between metabolism, developmental schedules and longevity using phylogenetic independent contrasts. J Gerontol A Biol Sci Med Sci. 2007;62:149–160. doi: 10.1093/gerona/62.2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Magalhães JP, Budovsky A, Lehmann G, Costa J, Li Y, Fraifeld V, Chuch GM. The Human Ageing Genomic Resources: online databases and tools for biogerontologists. Aging Cell. 2009;8:65–72. doi: 10.1111/j.1474-9726.2008.00442.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Magalhães JP, Curado J, Chuch GM. Meta-analysis of age-related gene expression profiles identifies common signatures of aging. Bioinformatics. 2009;25:875–881. doi: 10.1093/bioinformatics/btp073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demarchi F, Schneider C. The calpain system as a modulator of stress/damage response. Cell Cycle. 2007;6:136–138. doi: 10.4161/cc.6.2.3759. [DOI] [PubMed] [Google Scholar]
- Didichenko SA, Fragoso CM, Thelen M. Mitotic and stress-induced phosphorylation of HsPI3K-C2alpha targets the protein for degradation. J Biol Chem. 2003;278:26055–26064. doi: 10.1074/jbc.M301657200. [DOI] [PubMed] [Google Scholar]
- Edwards RJ, Shields DC (2004) GASP: Gapped Ancestral Sequence Prediction for proteins. BMC Bioinformatics 5:123 [DOI] [PMC free article] [PubMed]
- Finch CE. Longevity, senescence, and the genome. Chicago: University of Chicago Press; 1990. [Google Scholar]
- Finch CE, Stanford CB. Meat-adaptive genes and the evolution of slower aging in humans. Q Rev Biol. 2004;79:3–50. doi: 10.1086/381662. [DOI] [PubMed] [Google Scholar]
- Freitas AA, de Magalhães JP. A review and appraisal of the DNA damage theory of ageing. Mutat Res. 2011;728:12–22. doi: 10.1016/j.mrrev.2011.05.001. [DOI] [PubMed] [Google Scholar]
- Gourlay CW, Ayscough KR. The actin cytoskeleton: a key regulator of apoptosis and ageing? Nat Rev Mol Cell Biol. 2005;6:583–589. doi: 10.1038/nrm1682. [DOI] [PubMed] [Google Scholar]
- Grantham R. Amino acid difference formula to help explain protein evolution. Science. 1974;185:862–864. doi: 10.1126/science.185.4154.862. [DOI] [PubMed] [Google Scholar]
- Groisman R, Polanowska J, Kuraoka I, Sawada J, Saijo M, Drapkin R, Kisselev AF, Tanaka K, Nakatani Y. The ubiquitin ligase activity in the DDB2 and CSA complexes is differentially regulated by the COP9 signalosome in response to DNA damage. Cell. 2003;113:357–367. doi: 10.1016/S0092-8674(03)00316-7. [DOI] [PubMed] [Google Scholar]
- Harper JM, Salmon AB, Leiser SF, Galecki AT, Miller RA. Skin-derived fibroblasts from long-lived species are resistant to some, but not all, lethal stresses and to the mitochondrial inhibitor rotenone. Aging Cell. 2007;6:1–13. doi: 10.1111/j.1474-9726.2006.00255.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart RW, Setlow RB. Correlation between deoxyribonucleic acid excision-repair and life-span in a number of mammalian species. Proc Natl Acad Sci USA. 1974;71:2169–2173. doi: 10.1073/pnas.71.6.2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulbert AJ. Explaining longevity of different animals: is membrane fatty acid composition the missing link? Age (Dordr) 2008;30:89–97. doi: 10.1007/s11357-008-9055-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jobson RW, Nabholz B, Galtier N. An evolutionary genome scan for longevity-related natural selection in mammals. Mol Biol Evol. 2010;27:840–847. doi: 10.1093/molbev/msp293. [DOI] [PubMed] [Google Scholar]
- Kapetanaki MG, Guerrero-Santoro J, Bisi DC, Hsieh CL, Rapic-Otrin V, Levine AS. The DDB1-CUL4ADDB2 ubiquitin ligase is deficient in xeroderma pigmentosum group E and targets histone H2A at UV-damaged DNA sites. Proc Natl Acad Sci USA. 2006;103:2588–2593. doi: 10.1073/pnas.0511160103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EB, Fang X, Fushan AA, Huang Z, Lobanov AV, Han L, Marino SM, Sun X, Turanov AA, Yang P, Yim SH, Zhao X, Kasaikina MV, Stoletzki N, Peng C, Polak P, Xiong Z, Kiezun A, Zhu Y, Chen Y, Kryukov GV, Zhang Q, Peshkin L, Yang L, Bronson RT, Buffenstein R, Wang B, Han C, Li Q, Chen L, Zhao W, Sunyaev SR, Park TJ, Zhang G, Wang J, Gladyshev VN. Genome sequencing reveals insights into physiology and longevity of the naked mole rat. Nature. 2011;479:223–227. doi: 10.1038/nature10533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkwood TB, Austad SN. Why do we age? Nature. 2000;408:233–238. doi: 10.1038/35041682. [DOI] [PubMed] [Google Scholar]
- Kua C-H. Uncoupling the relationship between fatty acids and longevity. IUBMB life. 2006;58:153–155. doi: 10.1080/15216540600644812. [DOI] [PubMed] [Google Scholar]
- Li J, Wang Q-E, Zhu Q, El-Mahdy MA, Wani G, Praetorius-Ibba M, Wani AA. DNA damage binding protein component DDB1 participates in nucleotide excision repair through DDB2 DNA-binding and cullin 4A ubiquitin ligase activity. Cancer Res. 2006;66:8590–8597. doi: 10.1158/0008-5472.CAN-06-1115. [DOI] [PubMed] [Google Scholar]
- Li L, Ye H, Guo H, Yin Y. Arabidopsis IWS1 interacts with transcription factor BES1 and is involved in plant steroid hormone brassinosteroid regulated gene expression. Proc Natl Acad Sci USA. 2010;107:3918–3923. doi: 10.1073/pnas.0909198107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini N, Lerenthal Y, Shiloh Y, Gygi SP, Elledge SJ. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316:1160–1166. doi: 10.1126/science.1140321. [DOI] [PubMed] [Google Scholar]
- Miller RA (2001) A position paper on longevity genes. Sci Aging Knowledge Environ 2001:vp6 [DOI] [PubMed]
- Nishitani H, Sugimoto N, Roukos V, Nakanishi Y, Saijo M, Obuse C, Tsurimoto T, Nakayama KI, Nakayama K, Fujita M, Lygerou Z, Nishimoto T. Two E3 ubiquitin ligases, SCF-Skp2 and DDB1-Cul4 target human Cdt1 for proteolysis. EMBO J. 2006;25:1126–1136. doi: 10.1038/sj.emboj.7601002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez VI, Buffenstein R, Masamsetti V, Leonard S, Salmon AB, Mele J, Andziak B, Yang T, Edrey Y, Friguet B, Ward W, Richardson A, Chaudhuri A. Protein stability and resistance to oxidative stress are determinants of longevity in the longest-living rodent the naked mole-rat. Proc Natl Acad Sci USA. 2009;106:3059–3064. doi: 10.1073/pnas.0809620106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricklefs RE. Life-history connections to rates of aging in terrestrial vertebrates. Proc Natl Acad Sci USA. 2010;107:10314–10319. doi: 10.1073/pnas.1005862107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmon AB, Leonard S, Masamsetti V, Pierce A, Podlutsky AJ, Podlutskaya N, Richardson A, Austad SN, Chaudhuri AR. The long lifespan of two bat species is correlated with resistance to protein oxidation and enhanced protein homeostasis. FASEB J. 2009;23:2317–2326. doi: 10.1096/fj.08-122523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuelson AV, Carr CE, Ruvkun G. Gene activities that mediate increased life span of C. elegans insulin-like signaling mutants. Genes Dev. 2007;21:2976–2994. doi: 10.1101/gad.1588907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjögren C, Nasmyth K. Sister chromatid cohesion is required for postreplicative double-strand break repair in Saccharomyces cerevisiae. Curr Biol. 2001;11:991–995. doi: 10.1016/S0960-9822(01)00271-8. [DOI] [PubMed] [Google Scholar]
- Terashima Y, Onai N, Murai M, Enomoto M, Poonpiriya V, Hamada T, Motomura K, Suwa M, Ezaki T, Haga T, Kanegasaki S, Matsushima K. Pivotal function for cytoplasmic protein FROUNT in CCR2-mediated monocyte chemotaxis. Nat Immunol. 2005;6:827–835. doi: 10.1038/ni1222. [DOI] [PubMed] [Google Scholar]
- Weidenheim KM, Dickson DW, Rapin I. Neuropathology of Cockayne syndrome: evidence for impaired development, premature aging and neurodegeneration. Mech Ageing Dev. 2009;130:619–636. doi: 10.1016/j.mad.2009.07.006. [DOI] [PubMed] [Google Scholar]
- Wu CH, Apweiler R, Bairoch A, Natale DA, Barker WC, Boeckmann B, Ferro S, Gasteiger E, Huang H, Lopez R, Magrane M, Martin MJ, Mazumder R, O’Donovan C, Redaschi N, Suzek B. The Universal Protein Resource (UniProt): an expanding universe of protein information. Nucleic Acids Res. 2006;34:D187–D191. doi: 10.1093/nar/gkj161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyse CA, Coogan AN, Selman C, Hazlerigg DG, Speakman JR. Association between mammalian lifespan and circadian free-running period: the circadian resonance hypothesis revisited. Biol Lett. 2010;6:696–698. doi: 10.1098/rsbl.2010.0152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Chen T, Wan T, He L, Li N, Yuan Z, Cao X. Cloning of DPK a novel dendritic cell-derived protein kinase activating the ERK1/ERK2 and JNK/SAPK pathways. Biochem Biophys Res Commun. 2000;274:872–879. doi: 10.1006/bbrc.2000.3244. [DOI] [PubMed] [Google Scholar]
- Zhang J, Webb DM, Podlaha O. Accelerated protein evolution and origins of human-specific features: Foxp2 as an example. Genetics. 2002;162:1825–1835. doi: 10.1093/genetics/162.4.1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(XLS 220 kb)
(XLS 178 kb)
(DOC 73 kb)