

Abstract
The method of window exchange umbrella sampling molecular dynamics (WEUSMD) with a pre-optimized parameter set was recently used to obtain the most probable conformations and the energetics of transmembrane (TM) helix assembly of a generic TM sequence. When applied to glycophorin A TM domain (GpA-TM) using the restraint potentials along the helix-helix distance, however, tight interfacial packing of GpA-TM resulted in insufficient conformational sampling at short helix-helix separation. This sampling issue is addressed by extending the WEUSMD into two dimensions with the restraint potentials along the helix-helix distance and crossing angle. The two-dimensional WEUSMD results demonstrate that the incomplete sampling in the one-dimensional WEUSMD arises from high barriers along the crossing angle between the GpA-TM helices. Together with the faster convergence in both the assembled conformations and the potential of mean force, the 2D-WEUSMD can be a general and efficient approach in computational studies of TM helix assembly.
Keywords: Glycophorin A, Replica Exchange Methods, First Passage Time Optimization, Potential of Mean Force
This work represents our ongoing efforts to develop general computational methods to study transmembrane (TM) helix assembly in terms of both finding most probable conformations and quantifying assembly energetics in lipid bilayer environments. Given the facts that structure determination of membrane proteins with small numbers of TM helices and their oligomers in bilayer environments remains challenging1 and the membrane proteins with a single-pass TM helix are abundant (~30% of human receptors),2 computational studies can play a crucial role in providing their TM structural models and association energetics in a given bilayer environment. However, even for the simplest TM homodimer case, there are many degrees of freedom to be explored, such as helix-helix distance (rHH) and crossing angle (Ω), tilt and rotation of each helix, and displacement of each helix along the membrane normal, assuming relatively rigid helices and no symmetry imposed.3 Such high dimensionality in TM helix assembly also makes the computational studies challenging.4
In principle, one could utilize Monte Carlo or molecular dynamics (MD) simulation methods to explore such a high dimensional conformational space, but they usually suffer from incomplete sampling (within typical simulation duration) because of deep potential wells or high barriers in the free energy landscape. These situations can be improved by employing the temperature replica exchange MD (TREXMD) methods to facilitate escapes from local energy minima or barrier crossing.5,6 However, these methods generally do not provide the potential of mean force (PMF) along certain reaction coordinate(s) of interest. The adaptive biasing force/potential methods7–9 have been known to be effective in conformational search and estimation of the PMF along chosen reaction coordinates. For example, the orthogonal space random walk (OSRW) method9 actively searches conformations along the orthogonal degrees of freedom, which has been shown to be efficient in accurate PMF calculations. Yet, since the conformational search in these approaches becomes diffusive due to the flattened effective PMF, the sampling of relevant conformations that belong to free energy minima may not be efficient and careful testing is warranted for their general applications to TM helix assembly.
To efficiently reduce (and yet to explore) the high dimensionality in TM helix assembly, we have recently demonstrated that the window exchange umbrella sampling molecular dynamics (WEUSMD),10 a variant of Hamiltonian replica exchange MD (HREXMD),11 can be a general and efficient method for TM helix assembly. Using a generic TM sequence, the WEUSMD along rHH samples other degrees of freedom much more efficiently than conventional USMD, where sampling along Ω was very limited at short rHH and strongly dependent on the initial conformations. In addition, an analytical expression for the average acceptance probability between neighboring windows was derived and combined with the first passage time optimization method12,13 to predetermine a pair of parameters, k (the window force constant) and d (the window spacing), in an optimal range,10
| (1) |
where kB is the Boltzmann constant and T is temperature. In eq 1, it is assumed that the PMF along a reaction coordinate varies much slower than the window potentials. For a given number of windows or d, eq 1 is expected to provide an optimal force constant for conventional USMD as well.
The purpose of this work is to examine the sampling power (and the limitations and possible solutions) of WEUSMD in TM helix assembly using the TM domain of glycophorin A (GpA-TM) as a target system. GpA-TM has served as a model system for TM helix assembly4,14–18 and represents one of the tightest interfacial packing (with the well-known GxxxG motif) among known TM helix interfaces. Such tight packing makes it challenging to sample conformational space (e.g., right-handed to left-handed dimer at short rHH) and thus makes GpA-TM to be a valuable target for assessing the sampling power of WEUSMD. In addition, having a reliable sampling method is essential in quality assessment of the energy function employed in simulation; in other words, without dependable sampling, the calculated PMF is not trustworthy and it is difficult to improve the energy function. To address these issues, we performed the TREXMD as a reference for the most probable GpA-TM conformations, the one-dimensional (1D) WEUSMD along rHH of GpA-TM, and the two-dimensional (2D) WEUSMD along rHH and Ω in the IMM1 implicit membrane model19 using the pre-optimized k and d based on eq 1 and the helix restraint potentials3,20 in CHARMM21 (see Computational Methods for details).
Figure 1A shows the TREXMD results at 300 K and the 1D-WEUSMD PMFs from two initial configurations, IS1 (starting from a right-handed TM dimer, Ω < 0°) and IS2 (starting from a left-handed TM dimer, Ω > 0°). The population analysis along rHH and Ω, P(rHH,Ω), shows that its range and peak from TREXMD agree well with the global minimum of the 1D-WEUSMD PMFs (−12.53 kcal/mol) at rHH = 10.05 Å, and both the IS1 and IS2 PMFs are in excellent agreement with each other for rHH > 8 Å. The GpA-TM helix-helix interfaces in the thermally accessible regions, defined by the 2kBT region around the global PMF minimum along rHH,10 from the IS1 and IS2 1D-WEUSMDs are in good agreement with those from TREXMD (Figure S1 in SI); the RMSDs between representative structures from TREXMD and those from IS1 and IS2 1D-WEUSMDs are smaller than 1 Å. However, for rHH < 8 Å where the TREXMD-sampled population is negligible, the IS1 and IS2 PMFs diverge and the IS1 PMF has a local minimum of −11.35 kcal/mol at rHH = 6.63 Å (close to rHH = 6.59 Å of the NMR structure).22 The discrepancy between the PMFs indicates that the sampling power of 1D-WEUSMD at rHH < 8 Å is not sufficient, which is also shown in the different patterns of the average acceptance probability (Pa) between neighboring windows at rHH < 8 Å for the IS1 and IS2 1D-WEUSMDs (Figure 1B). The relatively large deviation of Pa at rHH < 8 Å from the desired value (0.3875) arises from the breakdown of the assumption in eq 1, which indicates the existence of window exchange bottlenecks around rHH = 8 Å (Figure S2). This bottleneck significantly lowers the sampling efficiency of WEUSMD, which in turn results in incomplete sampling and slower convergence of the IS1 PMF (Figure S3). The results indicate that 1D-WEUSMD with uniform window spacing is not efficient enough in conformational sampling (also in accurate PMF calculation) for tightly packed TM helix assembly, such as GpA-TM.
Figure 1.
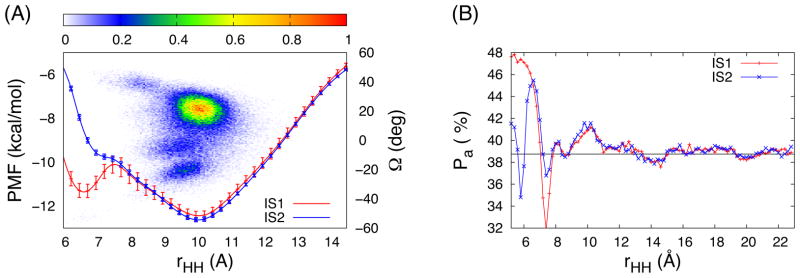
(A) The PMFs from the 1D-WEUSMDs with two initial configurations (IS1: red and IS2: blue) and the TREXMD-sampled population (density map) along rHH and Ω, P(rHH,Ω), at T = 300 K. (B) The average acceptance probability, Pa, for the IS1 and IS2 WEUSMDs with the target value of 0.3875 (black line). In (A), the PMFs at rHH = 23 Å were set to 0 (Figure S3 in SI) and the error bars were shown at every five data point for clarity.
A multi-dimensional WEUSMD is a natural extension to overcome the sampling issue in the framework of the REX method, where the window potentials along the additional degrees of freedom (DOF) allow better sampling around the hidden barriers because the window exchange along the multiple DOF can bypass such barriers. As long as the exchange between windows is allowed only along the same reaction coordinate, eq 1 can be readily applied to the multidimensional WEUSMD for the choice of a parameter set. In this study, we consider Ω as an additional DOF because the sampling along other DOF can be either restrained by the TM helix size and sequence and the membrane thickness or can be much easier compared to that along Ω in WEUSMD. For example, the sampling along tilt angles of TM helices was more or less the same for all the windows in 1D-WEUSMDs, and the sampling along rotation angles of TM helices can be much easier even at short rHH compared to that along Ω.
Figure 2A shows the 2D-PMF,
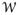 (rHH,Ω), as a function of rHH and Ω, calculated by WHAM23 from the 2D-WEUSMD. The first two lowest PMF minima are consistent with P(rHH,Ω) from TREXMD (Figure 1A) and the third one represents the local minimum in the IS1 1D-PMF. In addition, high barriers exist along Ω at short rHH, separating two possible pathways found by the modified string method24: L1 for the right-handed configuration leading to the IS1 case and L2 for the left-handed configuration leading to the IS2 case. This is also reflected in 2D-Pa shown in Figure 2B, which indicates that the replica exchange along Ω at short rHH is still not optimal due to the bottlenecks around Ω = −20° and Ω = 20°. However, in 2D-Pa, there exists a channel with relatively even Pa values close to the desired one (0.3875) (the area inside the black dashed circle in Figure 2B) that connects the IS1 local minimum to the other region, which acts as a route to bypass the barriers along Ω. Therefore, the sampling in 2D-WEUSMD is more efficient than that in 1D-WEUSMD, and the PMF converges faster (Figure S4C) and becomes accurate compared to the 1D-WEUSMDs. As shown below, this 2D-PMF can also be used to better understand the results from TREXMD and 1D-WEUSMDs.
(rHH,Ω), as a function of rHH and Ω, calculated by WHAM23 from the 2D-WEUSMD. The first two lowest PMF minima are consistent with P(rHH,Ω) from TREXMD (Figure 1A) and the third one represents the local minimum in the IS1 1D-PMF. In addition, high barriers exist along Ω at short rHH, separating two possible pathways found by the modified string method24: L1 for the right-handed configuration leading to the IS1 case and L2 for the left-handed configuration leading to the IS2 case. This is also reflected in 2D-Pa shown in Figure 2B, which indicates that the replica exchange along Ω at short rHH is still not optimal due to the bottlenecks around Ω = −20° and Ω = 20°. However, in 2D-Pa, there exists a channel with relatively even Pa values close to the desired one (0.3875) (the area inside the black dashed circle in Figure 2B) that connects the IS1 local minimum to the other region, which acts as a route to bypass the barriers along Ω. Therefore, the sampling in 2D-WEUSMD is more efficient than that in 1D-WEUSMD, and the PMF converges faster (Figure S4C) and becomes accurate compared to the 1D-WEUSMDs. As shown below, this 2D-PMF can also be used to better understand the results from TREXMD and 1D-WEUSMDs.
Figure 2.
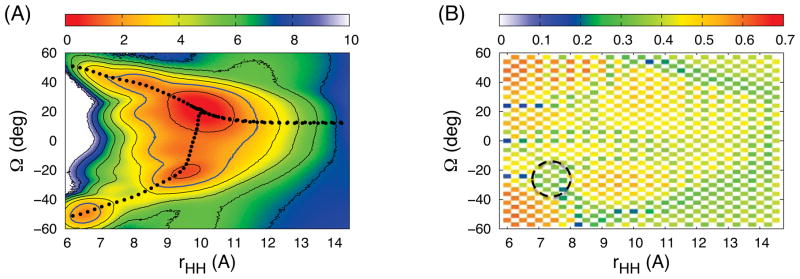
The results from 2D-WEUSMD simulations: (A) The 2D-PMF and two probable pathways (black circles) for GpA-TM assembly in the IMM1 implicit membrane model and (B) Pa along rHH and Ω. In (B), the area inside the black dashed circle is a channel that allows the exchanges between windows in the IS1 local minimum and in the other region. The contour lines for the 2D-PMF are shown at every 1.2 kcal/mol and those at 2.4 kcal/mol (thicker blue lines) represent the boundary of two probable basins for sampling in TREXMD.
In TREXMD, the sampling around the IS1 1D-PMF local minimum was negligible (Figure 1A), which can be explained quantitatively by the quotient between the weights for two basins B1 and B2 in the 2D-PMF. These basins are defined as the areas enclosed by contour lines at
(rHH,Ω) = 2.4 kcal/mol (~4kBT) in Figure 2A, so that B1 represents the majority of the sampled region from TREXMD (Figures 1A and S4F) and B2 represents the region around the IS1 1D-PMF local minimum. The quotient Q between the weights of both basins is given by
| (2) |
The calculated value of Q = 0.0128 (1.3%) is consistent with the negligible sampling in B2 during TREXMD.
Based on the 2D-PMF (Figure 2A), one can describe what would happen in the course of the IS1 and IS2 1D-WEUSMDs. For windows at rHH > 9.5 Å, the GpA-TM conformation could be in either left-handed or right-handed dimer due to negligible barriers along Ω (Figure S4D). For those at rHH < 9 Å, the pathways for the GpA-TM assembly would be separated into L1 and L2 because the barriers along Ω becomes higher with decreasing rHH (Figure 3A). Consequently, depending on the initial configurations in 1D-WEUSMD, the GpA-TM conformations would be trapped in either L1 or L2 without exchanges along Ω, resulting in incomplete sampling (along Ω) at rHH < 8 Å (Figures S3C–D). Specifically, sampling of right-handed GpA-TM conformations at rHH < 8 Å would be suppressed due to the preference for the left-handed conformations at rHH = 7.5–8.5 Å (Figure 3A). In other words, at rHH < 8 Å, it would be difficult to have the right-handed conformations originated from the windows at rHH > 8.5 Å, which is the case for the IS2 WEUSMD. For the IS1 WEUSMD, despite the preference for left-handed conformations at rHH < 8 Å, sampling of such conformations is very limited due to the high energy barriers along Ω (Figure 3A), resulting in conformational separation (right-handed at rHH < 8 Å and left-handed at rHH > 8 Å). Thus, the inconsistent sampling between the IS1 and IS2 1D-WEUSMDs can be attributed to the exchange bottlenecks (Figure S2) due to the high barriers along a hidden variable (Ω in this case).
Figure 3.
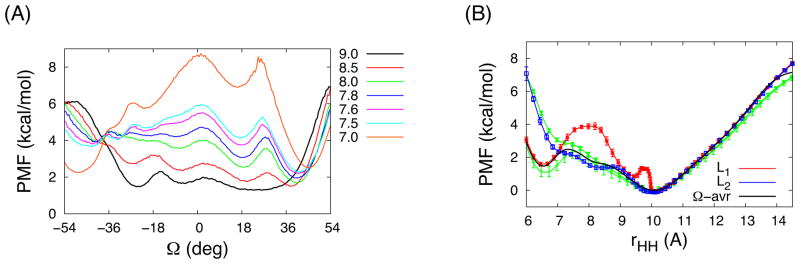
(A) The PMFs along Ω at several rHH (shown in legends) and (B) the PMFs as a function of rHH along the two probable pathways L1 and L2 in Figure 2A and the Ω-averaged 2D-PMF (black) obtained by eq 3. The error bars in the L1 and L2 PMFs were calculated from the average pathway ensembles obtained from the modified string method (Figure S5). In (B), the green lines are the IS1 and IS2 PMFs, whose minima are set to 0.
One can also interpret the IS1 and IS2 1D-PMFs in terms of the 2D-PMF. For rHH < 7 Å, the samplings from the IS1 and IS2 1D-WEUSMDs were localized in the vicinity of L1 and L2, respectively. Thus, the L1 and L2 PMFs are in a good agreement with the IS1 and IS2 PMFs (Figure 3B). For rHH > 7 Å, since both 1D-WEUSMDs were able to sample both right-handed and left-handed conformations of GpA-TM separately, as shown in Figure 3B, we can reconstruct the 1D-PMF by the Ω-averaged 2D-PMF as a function of rHH,
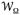 (rHH),
(rHH),
| (3) |
The fact that the 2D-PMF can explain all the results from TREXMD and 1D-WEUSMDs indicates that the sampling in the 2D-WEUSMD covers both the IS1 and IS2 1D-WEUSMDs. Therefore, the 2D-WEUSMD along rHH and Ω is sufficient even for tightly packed TM helix assembly, such as GpA-TM.
In summary, we applied WEUSMD for the GpA-TM assembly in the IMM1 implicit membrane model to examine its sampling power, which is crucial for accurate PMF calculations in a given energy function. The 1D-WEUSMD along rHH suffered from incomplete sampling at short rHH due to the high barriers along Ω. In order to improve the sampling power, we extended WESUMD into two dimensions along rHH and Ω. The 2D-WEUSMD substantially improved the conformational sampling, which addressed all the issues caused by the inconsistency between the TREXMD and 1D-WEUSMD results: the lack of sampling in the B2 in TREXMD and the inconsistent sampling and PMFs in the IS1 and IS2 1D-WEUSMDs. This result indicates that the sampling power of 2D-WEUSMD is sufficient even for GpA-TM (i.e., tightly packed TM helix assembly) and 2D-WEUSMD can be an effective and general tool for modeling TM helix assembly. Another implication of the 2D-WEUSMD result is that the quality of the IMM1 implicit membrane model is not so bad in that the free energy difference between the global minimum and the B2 local minimum is only about 1.60 kcal/mol. Therefore, the current sampling technique and trajectories will be useful to improve the quality of any type of implicit membrane models in combination of the PMF reweighting scheme.25 In addition, for practical applications of WEUSMD in explicit membranes, it would be most effective to combine 2D-WEUSEMD (at rHH < 9) and 1D-WEUSEMD (at rHH > 9 or 10 Å).
In the framework of the first passage time optimization, the WEUSMD performance is ideal when Pa along the window space is uniform with Pa = 0.3875.10,12,13 However, as reflected in the deviation of Pa from the desired value, the performance was not optimal, suggesting that further optimization could be possible. The optimal performance can be achieved either by flattening the effective PMF along the reaction coordinate or by adjusting the window force constant and spacing to control Pa systematically. The former optimization can be accomplished by the combination of adaptive biasing force (or potential) methods7,8 and WEUSMD. The latter one can be realized within the framework of WEUSMD by using a parameter set obtained from the first passage time optimization with the generalized Pa for different restraint force constants for different window pairs. Such optimizations are important for further developments of WEUSMD.
COMPUTATIONAL METHODS
The GpA-TM is defined as 72EITLI IFGVM AGVIG TILLI SYGIR96, in which we have chosen Cα atoms of residues 76–92 to define the helix principal axis for rHH and Ω.3,12 For the NMR structure (PDB:1AFO),22 the calculated rHH and Ω are 6.59 Å and −39.2°, respectively.26 To get the reference data, a 200-ns TREXMD simulation was performed with 16 replicas in a temperature range of 300–550 K starting from the configuration whose rHH = 20 Å and Ω = −39.2°. For the WEUSMD simulations, the initial configurations from the NMR structure were generated by translating each helix along rHH and then by rotating the helices along the helix-helix distance vector for a desired Ω.
For 1D-WEUSMD, a total of 90 windows were generated in a rHH range of 5.2–23 Å with ΔrHH = 0.2 Å and the restraint force constant of 22 kcal/(mol·Å2) determined by eq 1 was used. To examine the WEUSMD sampling power, two sets of initial configurations were used: one with right-handed helix-dimer interfaces (IS1, Ω = −39.2°) and the other with left-handed interfaces (IS2, Ω = 39.2°). For the 2D-WEUSMD simulations, a total of 360 (18×20) windows were generated in a region of (rHH,Ω) bounded by [6 Å, 14.5 Å] × [−57°, 57°] with ΔrHH = 0.5 Å and ΔΩ = 6°. The restraint force constants determined from eq 1 were 4 kcal/(mol·Å2) and 82 kcal/(mol·rad2), respectively. We used the NMR structure as the initial configuration for the windows up to the second nearest neighbor of the window whose target rHH = 6.5 Å and Ω = −39°.
For computational efficiency, the IMM1 implicit membrane model19 was used with the hydrophobic thickness of 23 Å to mimic a dimyristoylphosphatidylcholine (DMPC) lipid bilayer. For each system, 200-ns and 100-ns Langevin dynamics simulations were performed for 1D- and 2D-WEUSMD, respectively. All the simulations were performed using CHARMM21 with the default IMM1 options and the window/replica exchange was controlled by the MMTSB toolset.27 With the SHAKE algorithm a time step of 2 fs was used for all the simulation. The analysis was performed for the last 170-ns (TREXMD), 120-ns (1D-WEUSMD), and 60-ns (2D-WEUSMD) trajectories, respectively.
Supplementary Material
Acknowledgments
The authors are grateful to Huan Rui for help with the modified string method. This work was supported by NIH R01-GM092950 and TeraGrid/XSEDE resources (TG-MCB070009).
ABBREVIATIONS
- TM
transmembrane helix
- GpA-TM
transmembrane domain of glycophorin A
- MD
molecular dynamics
- REX
replica exchange
- TREXMD
temperature REXMD
- HREXMD
Hamiltonian REXMD
- USMD
umbrella sampling MD
- WEUSMD
window exchange USMD
- PMF
potential of mean force
Footnotes
The authors declare no competing financial interest.
Supporting Information. S1: Sampling of the TM-TM interfaces near the global PMF minimum in 1D-WEUSMD. S2: Exchange bottlenecks in 1D-WEUSMD. S3: Additional results from 2D-WEUSMD. S4: Two most probable pathways along rHH and Ω. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Bill RM, Henderson PJ, Iwata S, Kunji ER, Michel H, Neutze R, Newstead S, Poolman B, Tate CG, Vogel H. Overcoming barriers to membrane protein structure determination. Nat Biotechnol. 2011;29:335–340. doi: 10.1038/nbt.1833. [DOI] [PubMed] [Google Scholar]
- 2.Almen MS, Nordstrom KJ, Fredriksson R, Schioth HB. Mapping the human membrane proteome: a majority of the human membrane proteins can be classified according to function and evolutionary origin. BMC Biol. 2009;7:50. doi: 10.1186/1741-7007-7-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Im W, Lee J, Kim T, Rui H. Novel free energy calculations to explore mechanisms and energetics of membrane protein structure and function. J Comput Chem. 2009;30:1622–1633. doi: 10.1002/jcc.21320. [DOI] [PubMed] [Google Scholar]
- 4.Dell’Orco D, De Benedetti PG, Fanelli F. In silico screening of mutational effects on transmembrane helix dimerization: Insights from rigid-body docking and molecular dynamics simulations. J Phys Chem B. 2007;111:9114–9124. doi: 10.1021/jp071383r. [DOI] [PubMed] [Google Scholar]
- 5.Swendsen RH, Wang JS. Replica Monte Carlo simulation of spin glasses. Phys Rev Lett. 1986;57:2607–2609. doi: 10.1103/PhysRevLett.57.2607. [DOI] [PubMed] [Google Scholar]
- 6.Sugita Y, Okamoto Y. Replica-exchange molecular dynamics method for protein folding. Chem Phys Lett. 1999;314:141–151. [Google Scholar]
- 7.Laio A, Gervasio FL. Metadynamics: a method to simulate rare events and reconstruct the free energy in biophysics, chemistry and material science. Rep Prog Phys. 2008;71:126601. [Google Scholar]
- 8.Darve E, Rodriguez-Gomez D, Pohorille A. Adaptive biasing force method for scalar and vector free energy calculations. J Chem Phys. 2008;128:144120. doi: 10.1063/1.2829861. [DOI] [PubMed] [Google Scholar]
- 9.Zheng LQ, Chen MG, Yang W. Random walk in orthogonal space to achieve efficient free-energy simulation of complex systems. Proc Natl Acad Sci USA. 2008;105:20227–20232. doi: 10.1073/pnas.0810631106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Park S, Kim T, Im W. Transmembrane helix assembly by window exchange umbrella sampling. Phys Rev Lett. 2012;108:108102. doi: 10.1103/PhysRevLett.108.108102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sugita Y, Kitao A, Okamoto Y. Multidimensional replica-exchange method for free-energy calculations. J Chem Phys. 2000;113:6042–6051. [Google Scholar]
- 12.Nadler W, Hansmann UH. Generalized ensemble and tempering simulations: a unified view. Phys Rev E. 2007;75:026109. doi: 10.1103/PhysRevE.75.026109. [DOI] [PubMed] [Google Scholar]
- 13.Lingenheil M, Denschlag R, Mathias G, Tavan P. Efficiency of exchange schemes in replica exchange. Chem Phys Lett. 2009;478:80–84. [Google Scholar]
- 14.Henin J, Pohorille A, Chipot C. Insights into the recognition and association of transmembrane alpha-helices. The free energy of alpha-helix dimerization in glycophorin A. J Am Chem Soc. 2005;127:8478–8484. doi: 10.1021/ja050581y. [DOI] [PubMed] [Google Scholar]
- 15.Zhang JM, Lazaridis T. Calculating the free energy of association of transmembrane helices. Biophys J. 2006;91:1710–1723. doi: 10.1529/biophysj.106.081224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Psachoulia E, Marshall DP, Sansom MS. Molecular dynamics simulations of the dimerization of transmembrane alpha-helices. Acc Chem Res. 2010;43:388–396. doi: 10.1021/ar900211k. [DOI] [PubMed] [Google Scholar]
- 17.Polyansky AA, Volynsky PE, Efremov RG. Structural, dynamic, and functional aspects of helix association in membranes: a computational view. Adv Protein Chem Struct Biol. 2011;83:129–161. doi: 10.1016/B978-0-12-381262-9.00004-5. [DOI] [PubMed] [Google Scholar]
- 18.Hong H, Bowie JU. Dramatic destabilization of transmembrane helix interactions by features of natural membrane environments. J Am Chem Soc. 2011;133:11389–11398. doi: 10.1021/ja204524c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lazaridis T. Effective energy function for proteins in lipid membranes. Proteins: Struct, Funct Genet. 2003;52:176–192. doi: 10.1002/prot.10410. [DOI] [PubMed] [Google Scholar]
- 20.Lee J, Im W. Implementation and application of helix-helix distance and crossing angle restraint potentials. J Comput Chem. 2007;28:669–680. doi: 10.1002/jcc.20614. [DOI] [PubMed] [Google Scholar]
- 21.Brooks BR, Brooks CL, 3rd, Mackerell AD, Jr, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, Caflisch A, Caves L, Cui Q, Dinner AR, Feig M, Fischer S, Gao J, Hodoscek M, Im W, Kuczera K, Lazaridis T, Ma J, Ovchinnikov V, Paci E, Pastor RW, Post CB, Pu JZ, Schaefer M, Tidor B, Venable RM, Woodcock HL, Wu X, Yang W, York DM, Karplus M. CHARMM: the biomolecular simulation program. J Comput Chem. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.MacKenzie KR, Prestegard JH, Engelman DM. A transmembrane helix dimer: Structure and implications. Science. 1997;276:131–133. doi: 10.1126/science.276.5309.131. [DOI] [PubMed] [Google Scholar]
- 23.Kumar S, Bouzida D, Swendsen RH, Kollman PA, Rosenberg JM. The Weighted Histogram Analysis Method for Free-Energy Calculations on Biomolecules .1. The Method. J Comput Chem. 1992;13:1011–1021. [Google Scholar]
- 24.Jo S, Rui H, Lim JB, Klauda JB, Im W. Cholesterol flip-flop: insights from free energy simulation studies. J Phys Chem B. 2010;114:13342–13348. doi: 10.1021/jp108166k. [DOI] [PubMed] [Google Scholar]
- 25.Konig G, Boresch S. Non-Boltzmann Sampling and Bennett’s Acceptance Ratio Method: How to Profit from Bending the Rules. J Comput Chem. 2011;32:1082–1090. doi: 10.1002/jcc.21687. [DOI] [PubMed] [Google Scholar]
- 26.Chothia C, Levitt M, Richardson D. Helix to helix packing in proteins. J Mol Biol. 1981;145:215–250. doi: 10.1016/0022-2836(81)90341-7. [DOI] [PubMed] [Google Scholar]
- 27.Feig M, Karanicolas J, Brooks CL., 3rd MMTSB Tool Set: enhanced sampling multiscale modeling methods for applications in structural biology. J Mol Graphics Modell. 2004;22:377–395. doi: 10.1016/j.jmgm.2003.12.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.