

Abstract
Vangl2 forms part of the planar cell polarity signalling pathway and is the gene defective in the Looptail (Lp) mouse mutant. Two previously described alleles, Lp and Lpm1Jus, segregate in a semi-dominant fashion, with heterozygotes displaying the looped-tail appearance, while homozygotes show the neural tube defect called craniorachischisis. Here, we report a novel experimentally-induced allele, Lpm2Jus, that carries a missense mutation, R259L, in Vangl2. This mutation was specific to the Lp phenotype and absent from both parental strains and 28 other inbred strains. Notably, this mutation segregates in a recessive manner with all heterozygotes appearing normal and 47% of homozygotes showing a looped-tail. Homozygous Lpm2Jus embryos showed spina bifida in 12%. Lpm2Jus genetically interacts with Lp with 77% of compound heterozygotes displaying craniorachischisis. Vangl2R259L behaved like the wild-type allele in overexpression and morpholino knockdown/rescue assays in zebrafish embryos. These data suggest that Lpm2Jus represents a new hypomorphic allele of Lp.
Keywords: Looptail, Vangl2, planar cell polarity, neural tube defects, ENU mutagenesis
INTRODUCTION
Planar cell polarity (PCP) is the process by which cells become polarized in the plane of an epithelium. This process has been well studied in the adult epithelial tissues of Drosophila where it can be observed in the distal orientation of wing hairs, the posterior orientation of the abdominal bristles and the complex organization of the ommatidia (eye units) in the adult eye. Genetic studies of a wide range of mutants affecting these highly organized structures in the fly have identified a group of so called “core” PCP genes which includes stbm/vang, frizzled (fz), dishevelled (dvl), flamingo (fmi) prickle (pk) and diego (dg) (Simons and Mlodzik, 2008; Vladar et al., 2009). In mammalian tissues, PCP is observed in the inner ear sensory organs or the fur on an animal’s back. In particular, the auditory sensory organ in the cochlea displays a precise planar organization of its sensory hair cells that is essential for its proper function. The cochlea has one row of inner hair cells and three rows of outer hair cells, which are interdigitated with non-sensory support cells. Each hair cell projects microvilli-derived stereocilia that are arranged in order of increasing height toward the edge of the apical cortex to form a “V”-shaped structure with a single primary cilium, known as the kinocilium, placed near the tallest stereocilia at the vertex of the “V”-shaped stereociliary bundle. The vertices of the hair bundles all point toward the periphery of the cochlea, thus manifesting planar polarity. This planar organization is disrupted in single and double PCP mutants (Wang et al., 2006; McNeill, 2010).
In vertebrates, PCP proteins are also involved in the morphogenetic process of convergent extension (CE) that is critical for proper gastrulation and formation of the neural tube (Wang and Nathans, 2007). During this complex process, cells elongate medio-laterally and produce oriented cytoplasmic protrusions that enable them to move directionally and to intercalate with other neighboring cells. This leads to convergence of the tissue towards the midline and its extension along the anteroposterior axis (Keller et al., 2008). Evidence for involvement of PCP signaling in CE in vertebrates has initially emerged from studies of a wide range of mutants and morpholino-oligonucleotide (MO) knockdowns of orthologs of fly PCP genes in zebrafish and frog models. Embryos with defective PCP signaling caused by gene knockdown or overexpression manifest a well described CE defect depicted by marked shortening of the anterior–posterior axis (Wallingford, 2005). In mammals, Looptail (Lp) was the first mouse mutant to implicate a role of PCP and CE in neural tube defects (Kibar et al., 2001b; Murdoch et al., 2001a). A direct study of CE in vitally labeled pre-neurulation stage Lp/Lp embryos demonstrated a defective midline extension in the axial mesoderm and neural plate of these mutant embryos (Ybot-Gonzalez et al., 2007). Mutations in other vertebrate PCP genes cause various forms of NTDs in mouse (Wang and Nathans, 2007). Rare and novel missense mutations in VANGL2 or its homologue VANGL1 are associated with NTDs in humans (Kibar et al., 2007, 2009, 2010; Lei et al., 2010).
The Lp mouse represents a well-established model for the study of neural tube defects in humans (NTDs), the most common and severe malformations of the central nervous system in humans (1-2/1000 livebirths) (Bassuk and Kibar, 2009). Lp heterozygous mice are characterized by a kinked or looped-tail appearance while homozygous embryos die in utero suffering from a severe form of NTDs called craniorachischisis where the neural tube remains open throughout the hindbrain and spinal cord region (Strong and Hollander, 1949). Lp homozygous embryos also show failure of eyelid closure, outflow tract defects in the heart, severe inner ear polarity defects, and imperforate vagina in affected females (Henderson et al. 2001; Montcouquiol et al., 2003; VandenBerg and Sassoon, 2009). Two independent alleles have been described for the Lp mice: a naturally occurring Lp and a chemically induced Lpm1Jus that both are transmitted in a semi-dominant fashion and produce identical phenotypes in heterozygotes (looped tail) and homozygous embryos (craniorachischisis) (Strong and Hollander, 1949; Kibar et al., 2001a). Positional cloning studies have identified Vangl2 as the gene mutated in Lp mice (Kibar et al., 2001b; Murdoch et al., 2001a). Vangl2 is strongly expressed in the neuroectoderm throughout neurulation and is altered in Lp where two disease-specific mutations, S464N and D255E, were identified in Lp and Lpm1Jus respectively (Kibar et al., 2001b). Vangl2 encodes a membrane protein whose predicted features include 4 transmembrane (TM) domains and a PDZ-domain binding motif at the carboxy terminus involved in protein-protein interaction (Torban et al., 2004).
Several studies were conducted to investigate the molecular mode of action of the two mutations, D255E and S464N, identified in Vangl2 in Lp mice. Since these mutations map to different parts of the protein yet they produce identical phenotypes, it was hypothesized that Vangl2 acts a rate-limiting factor in a dosage sensitive pathway (Kibar et al., 2001b). In zebrafish embryos, both Lp mutations fail to rescue the CE defect caused by a MO-knockdown of the tri gene (tri, orthologue of Vangl2) and fail to produce a CE defect when over-expressed, strongly suggesting that they present loss-of-function mutations (Reynolds et al., 2010). Both mutations affect the interaction of Vangl1/2 with the Dishevelled PCP genes, which is critical for PCP signaling (Torban et al., 2004; Suriben et al., 2009). A recent study showed that Vangl2D255E and Vangl2S464N fail to sort into COPII vesicles and are trapped in the endoplasmic reticulum (Merte et al., 2010). A parallel study showed that the targeting of the Vangl2D255E at the plasma membrane is greatly reduced where it is predominantly retained intracellularly in endoplasmic reticulum (ER) vesicles (Gravel et al., 2010). These studies highlight a critical role for D255 and S464 for normal folding and processing of Vangl proteins.
In this study, we report a new recessive missense mutation in Vangl2, R259L, that was generated in a large-scale phenotype-driven screen of mice mutagenized with N-ethyl-N-nitrosourea (ENU). Homozygous mice showed a looped tail appearance (47%), spina bifida (12%) but no craniorachischisis, and no significant PCP inner ear defects. Also, 78% of homozygous adult females exhibited imperforate vagina. All wild-type and heterozygous mice were apparently healthy. Vangl2R259L genetically interacts with Vangl2S464N, causing the severe craniorachischisis and PCP inner ear defect in compound heterozygous embryos. The Vangl2R259L variant was able to rescue the tri-MO induced CE defect and produce a severe phenotype when overexpressed. These data strongly suggest that Vangl2R259L represents a hypomorphic allele at the Lp termed Lpm2Jus, where dosage is an important modulator of the severity of the PCP and CE-related phenotypes.
RESULTS
Molecular studies of Lpm2Jus
The open reading frame and the exon-intron junctions of Vangl2 were sequenced in Lpm2Jus mouse with a looped tail phenotype and a single homozygous mutation, c.776 G>T, which changes arginine 259 to leucine (R259L), was identified. This mutation was absent from both parental strains and 28 other inbred strains, confirming its specificity to the Lp phenotype (and not a rare variant). The variant R259L resides in the predicted cytoplasmic domain of Vangl2 (Fig. 1A), just three amino acids away from the Vangl2D255E mutation previously identified in Lpm1Jus. R259 is invariant in all known orthologs except in zebrafish Vangl1 where it is replaced by histidine. Substitution of histidine preserves the basic nature of arginine, a physical property that is completely lost by the non conservative R259L substitution (Fig. 1B).
Figure 1.
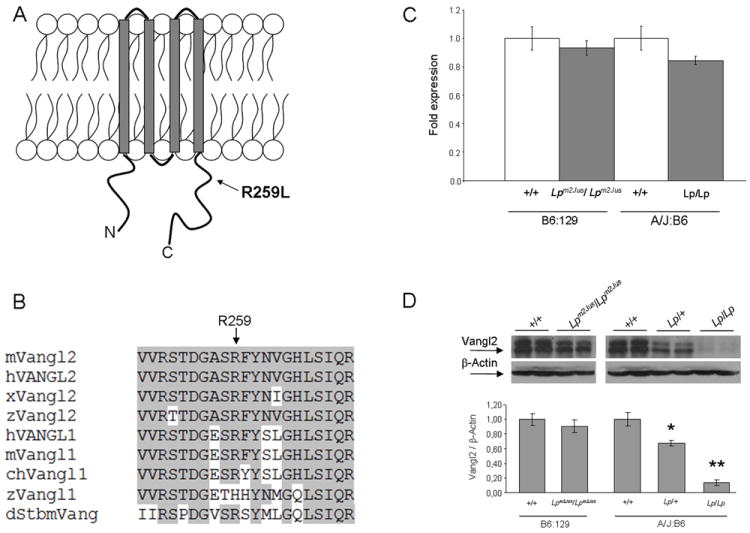
Molecular characterization of the Lpm2Jus mutant. Panel A. Diagram of the Vangl2 protein structure with the approximate position of the R259L mutation identified in Lpm2Jus indicated by an arrow. Panel B. Partial alignment of mouse Vangl2 with 8 other Vangl/Stbm sequences. Residues conserved between Vangl2 and other family members are highlighted. The R259L variant affects a highly conserved amino acid residue (indicated by an arrow). Accession numbers: mouse Vangl2 (mVangl2), NP_277044; human VANGL2 (hVANGL2), NP_065068; frog Vangl2 (xVangl2), AAK70879; zebrafishVangl2 (zVangl2), NP_705960; human VANGL1 (hVANGL1), NP_620409; mouse Vangl1 (mVangl1), NP_808213; chicken Vangl1 (chVangl1), XP_001232441; zebrafishVangl1 (zVangl1), AAQ84560; and Drosophila Stbm (dStbm), NP_477177. Panel C. Real- time PCR showing the relative levels of Vangl2 mRNA compared with Hprt transcripts in brain tissues of E16.5 wild-type and mutant embryos. Panel D. Western analysis of Vangl2 protein levels in brain tissues of E16.5 wild-type and mutant embryos. β-actin was used as an internal control. Top, western blots of two representative embryos per group are shown. Bottom, a histogram showing the relative levels of Vangl2 protein compared with β-actin. A significant difference was detected between wild-type and Lp/+ (*P<0.001) and between wild-type and Lp/Lp embryos (**P<0.0001).
To determine whether R259L has any pathogenic effect on Vangl2 gene expression, real-time RT-PCR was conducted to determine Vangl2 mRNA levels in brain tissues of wild-type and homozygous mutant E16.5 embryos. The levels of Vangl2 mRNA in mutant embryos were not significantly different from wild-type levels (Fig. 1C). To investigate whether the missense mutation R259L causes a decrease in Vangl2 protein levels, we performed western blot analysis on brain tissues from wild-type and homozygous mutant Lpm2Jus E16.5 embryos, who are on a mixed B6 and 129 background. As reference and additional controls, we included wild-type, heterozygous Lp/+ and homozygous Lp/Lp mice, which are on a mixed A/J and B6 background. In brain, Lpm2Jus/ Lpm2Jus had 90% of Vangl2 protein as compared to their wild-type littermates (t-test, P=0.06) (Fig. 1D). However, protein levels were reduced by 32% in Lp/+ heterozygotes (t-test, P<0.001) and almost completely eliminated in Lp/Lp homozygous embryos (t-test, P<0.0001), as compared to their wild-type littermates (Fig.1D). A very faint band could be detected in Lp/Lp homozygous brain tissues upon longer exposure (data not shown).
We next examined Vangl2 protein expression pattern in the neural tube of E9.5 wild type and Lpm2Jus/ Lpm2Jus embryos. Vangl2 was detected around the membrane of neuroepithelial cells, and no clear difference in expression pattern or levels was observed in the mutant embryos (Fig. 2).
Figure 2.
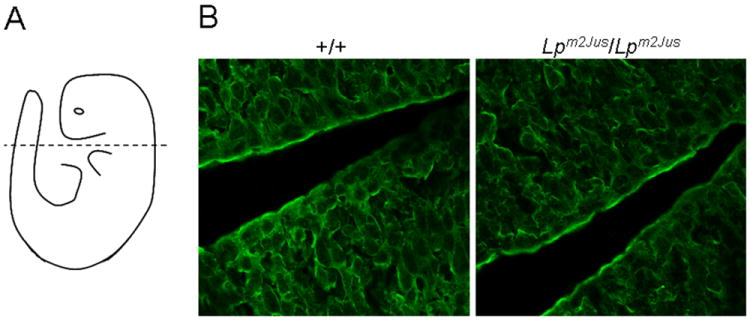
Localization of the Vangl2 protein in neural tube of wild-type and Lpm2Jus/Lpm2Jus embryos. (A) A diagrammatic representation of a mouse embryo indicating the level and plane of section. (B) Vangl2 protein is present and membrane-associated in the neural tube of wild-type (+/+) and of homozygous (Lpm2Jus/Lpm2Jus) mouse embryos. 630x magnification.
Genotype-phenotype studies of the Lpm2Jus allele
Lpm2Jus N1F1 mutants were archived as frozen sperm and were consequently recovered at The Jackson Laboratory (http://jaxmice.jax.org/services/) by using the Assisted In Vitro Fertilization (AIVF) method. A total of 13 female and 15 male mice were generated following this procedure; all were heterozygotes for the R259L mutation and all had a normal appearance and breeding performance. These founder mice were used for expansion and maintenance of the Lpm2Jus mutant line. In addition to these 28 founder mice, a total of 213 mice were generated over 4 generations and were examined for the presence of NTDs (Table 1). Crosses between parents that were heterozygotes at the mutant locus (Lpm2Jus/+) generated a total of 164 litters including 36 wild-type, 85 heterozygotes and 43 homozygotes at the R259L mutation. The distribution of genotypes in these litters did not significantly differ from the expected Hardy-Weinberg ratio of 1:2:1 (χ2 test, P=0.66). Matings between heterozygote and homozygote parents at the mutant locus generated 49 animals including 28 heterozygote and 21 homozygote mice. This genotype distribution was also not significantly different from the expected Hardy-Weinberg ratio of 1:1 (χ2 test, P=0.39), suggesting that homozygosity for Vangl2R259L has no effect on viability or embryonic development. The Lpm2Jus R259L mutation showed a recessive mode of inheritance where all wild-type and heterozygote mice were phenotypically normal and 47% (30/64) of homozygous adult mice had a looped or kinky tail appearance (Table 1). These results suggest incomplete penetrance of the R259L mutation on the mixed C57BL/6J; 129S6/SvEvTac genetic background.
Table 1.
Genotype-phenotype studies of the Lpm2Jus looptail mutant
| R259L genotype at the Vangl2 locus | Phenotype
|
||||
|---|---|---|---|---|---|
| Looped or kinky tail | Spina bifida | Imperforate vagina | Male infertility | ||
| Adult (n=241) | 36 +/+, 141 +/- (99 ♀, 78 ♂) | 0 | NA | 0 | 0b |
| 64 -/- (27 ♀, 37 ♂) | 30 (47%) | NA | 21 (78%) | 12 (32%) | |
| E16.5-E18.5 (n=156) | 37 +/+, 69 +/- | 0 | 0 | NA | NA |
| 50 -/- | 16 (32%) a | 6 (12%) | NA | NA | |
This frequency could be underestimated due to the difficulty in visual examination of the looped or kinky tail in embryos.
Absence of male infertility is confirmed only in 4 +/+ and 29 +/- males included in the breeding scheme.
In addition, 78% (21/27) of homozygous adult females showed imperforate vagina and 32% (12/37) of homozygote adult males appeared to be infertile (Table 1). All infertile Lpm2Jus male were able to set copulatory plugs. A whole body necropsy analysis showed no histological differences between Lpm2Jus homozygous infertile mice (n=2) and their wild-type littermates (n=2) (data not shown). Sperm assessment revealed no evidence for defective spermatogenesis and spermiogenesis since adequate and even superior sperm quality was observed in of Lpm2Jus homozygous infertile mice as compared to wild-type littermates (data not shown).
The two previously described mutations in Vangl2, D225E and S464N, cause severe embryonic lethal craniorachischisis and failure of eyelid closure in homozygous embryos (Kibar et al., 2001a,b). Consequently, we examined Lpm2Jus/ Lpm2Jus embryos for these two phenotypes. A total of 156 E12.5-E18.5 embryos were generated which included 37 wild-type, 69 heterozygous and 50 homozygous embryos for the R259L mutation. Interestingly, 12% (6/50) of the R259L homozygous embryos showed spina bifida characterized by an open neural tube in the lumbosacral region of the spinal cord (Table 1, Fig.3A). Histological sections across spinal regions of affected embryos revealed widely spaced neural folds characteristic of spina bifida (data not shown). In PCP mouse mutants, including Lp, a defective CE leads to a broad embryonic midline thus preventing the onset of neurulation through wide spacing of the neural folds (Ybot-Gonzalez et al., 2007). None of R259L homozygous embryos showed the more severe craniorachischisis NTD. In addition, all homozygous Lpm2Jus/ Lpm2Jus embryos had normal eyelid closure (Fig. 3A). All wild-type and heterozygous embryos were apparently normal (Table 1).
Figure 3.
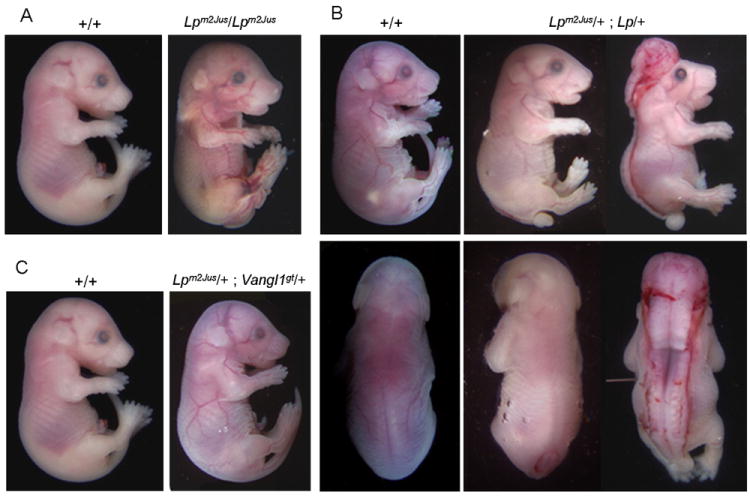
Morphological appearance of Lpm2Jus homozygotes (panel A), Lpm2Jus/Lp (panel B) double heterozygotes and Lpm2Jus/Vangl1gt (panel C) at E18.5.
Complementation studies with the Lp and Vangl1gt alleles
Phenotype complementation studies were conducted between Lpm2Jus and the previously described Lp Jackson allele caused by the Vangl2 mutation S464N. Heterozygous Lp/+ mice were crossed to Lpm2Jus/+ mice and embryos were recovered at various stages and examined macroscopically for the presence of NTDs and eyelid closure defects. A total of 13 compound heterozygous Lpm2Jus/+; Lp/+ embryos were generated. Of these, 10 embryos exhibited the full-blown phenotype of craniorachischisis seen in Lp/Lp embryos and 2 embryos showed spina bifida, resulting in a penetrance of 92% (12/13) of the NTD phenotype in these compound heterozygotes for the S464N and R259L mutations at the Vangl2 locus (Fig. 3B). Eyelid closure phenotype was examined in E18.5 embryos. Both compound heterozygotes with spina bifida had normal eye lid closure, while 60% (3/5) of compound heterozygotes with craniorachischisis exhibited unfused eyelids. These results demonstrate that Lpm2Jus genetically interacts with the original Lp allele and contributes to the development of the severe NTD. These complementation studies together with the disease-specific nature of R259L strongly suggest that Lpm2Jus is a new allele at the Lp locus.
Phenotype complementation studies were also conducted between Lpm2Jus and the Vangl1gt gene trap allele. Vangl1gt heterozygotes and Vangl1gt/gt homozygotes were viable and fertile, as previously described. Lpm2Jus/ Lpm2Jus mice were mated to Vangl1gt/gt homozygotes and a total of 23 double heterozygous embryos were generated and examined macroscopically for the presence of NTDs and eyelid closure defects. All embryos were apparently normal with no looped tail or NTD or eyelid closure defect detected (Fig. 3C).
Analysis of the stereociliary bundle orientation in Lpm2Jus
We next analyzed the effect of the R259L mutation on PCP regulation in vivo in the inner ear in Lpm2Jus mutants and Lpm2Jus/+; Lp/+ compound heterozygotes. We examined cochleae isolated from wild-type, Lpm2Jus /+, Lpm2Jus/ Lpm2Jus and Lpm2Jus/+; Lp/+ E18.5 embryos. Upon gross examination, the cochlear size of all mutant animals did not differ from those of the wild-type (data not shown). Examination of the inner (IHCs) and outer hair cells (OHCs) at the apical regions of the organ of Corti revealed no statistically significant difference between wild-type and each of the Lpm2Jus/+ (data not shown) and Lpm2Jus/ Lpm2Jus cochlea (Fig. 4). In Lpm2Jus+/-; Lp+/-compound heterozygotes, bundle orientation in OHC2 and OHC3 at the apical regions of the organ of Corti was severely affected (P=0.005) as compared to wild-type controls (Fig.4), confirming genetic interaction between Lp and Lpm2Jus.
Figure 4.
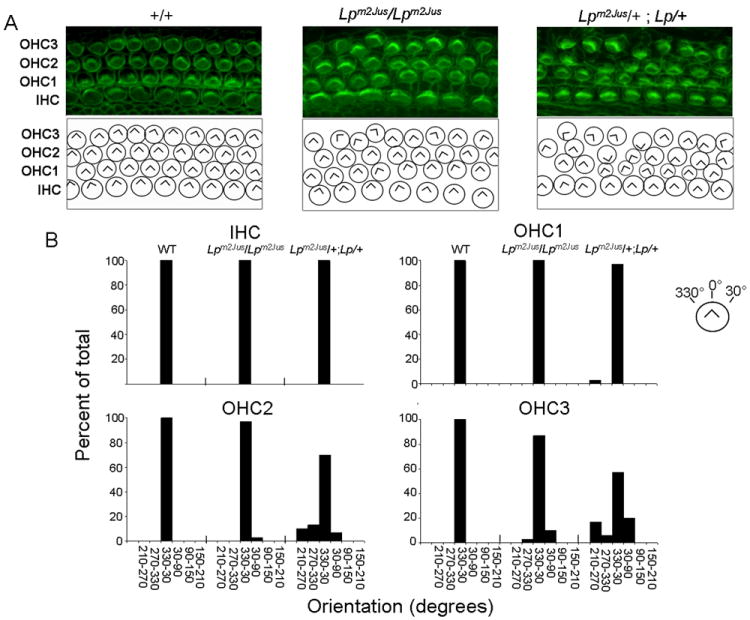
Comparison of hair bundle orientation defects at the apical region of the organ of Corti in WT, Lpm2Jus/ Lpm2Jus and Lpm2Jus/+; Lp/+ at E18.5. Panel A, Top, Stereocilia labeled with phalloidin (green). Bottom, Diagrams showing the scoring of hair bundle orientation for the images above. IHC, Inner hair cells; OHC1, Inner row of outer hair cells; OHC2, central row of outer hair cells; OHC3, outer row of outer hair cells. The genotype is indicated above each column of panels. Panel B. Quantification of the IHC, OHC1, OHC2, and OHC3 bundle orientations for each of the three genotypes indicated above based on phalloidin staining. The convention for angular measurements is shown in the top right corner of the panel. Statistical analysis was done by the Kolmogorov - Smirnov test (P=0.005 for OHC2 and OHC3 of the Lpm2Jus/+; Lp/+ as compared to the wild-type).
Examination of the inner ear of Lpm2Jus/+; Vangl1gt/+ double heterozygotes revealed no difference from the wild-type (data not shown), further demonstrating that both alleles fail to interact in these mutants.
Functional studies of VANGL2R259L in the zebrafish model
We next validated the potential pathogenic effect of the R259L mutation on protein function in vivo by investigating its effect on convergent extension (CE) in zebrafish. Overexpression of tri impairs CE in zebrafish embryos and causes a reduction in axial length (Jessen et al., 2002; Park and Moon, 2002). We have shown previously that both Lp mutations D255E and S464N failed to induce a CE defect, as compared to wild-type, providing strong evidence that they act as loss-of-function alleles (Reynolds et al., 2010). We carried out an overexpression assay with wild-type VANGL2, VANGL2R259L, and as additional references, VANGL2D255E and VANGL2S464N. When wild-type VANGL2 was overexpressed, a severe reduction in axial length was observed as expected (P<0.001) (Fig. 5). Surprisingly, VANGL2R259L behaved as the wild-type VANGL2 and induced a severe CE defect as well (P<0.001) (Fig. 5A,B). Injection of wild-type Vangl2 or VANGL2R259L at lower doses of 50 and 100 pg also caused a similar severe CE defect (P<0.01 and P<0.001 respectively) (Supplementary Fig.1). As expected, VANGL2D255E and VANGL2S464N failed to induce a CE defect and were statistically significant from VANGL2 alone (P<0.001) (Fig. 5). Injection of 200 pg wild-type VANGL2 alone produced a range of phenotypes that were clustered into three groups based on the phenotype severity (Fig. 5C). We examined the distributions of these three clusters among all 5 groups described above. The distribution of the clusters obtained with wild-type VANGL2 or VANGL2R259L was significantly different from uninjected wild-type fish (P<0.05) (Fig.5C). On the other hand, the distribution of the 3 clusters obtained from overexpression of either VANGL2D255E or VANGL2S464N was similar to uninjected fish and was significantly different than that obtained with wild-type VANGL2 (P<0.05).
Figure 5.
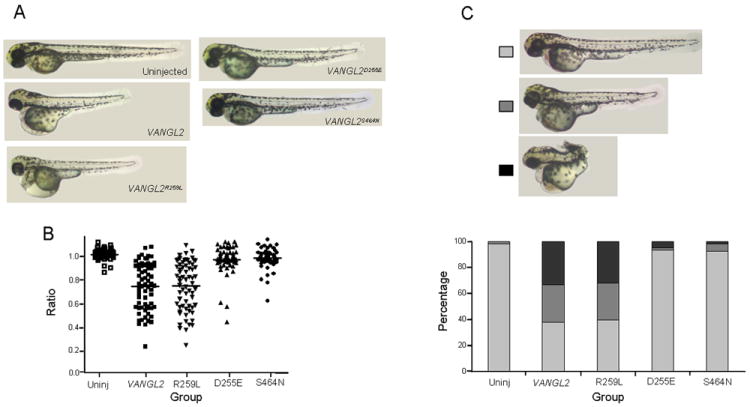
Effect of overexpression of VANGL2R259L variant on convergent extension in zebrafish embryos. Panel A, lateral views of 2 days post fertilization zebrafish including uninjected wild-type and fish injected with 200 pg of either VANGL2 or VANGL2R259L or VANGL2D255E or VANGL2S464N. Panel B, statistical analyses of the body length measurements of all 5 groups indicated above. The Kruskal-Wallis test was used to compare differences between experimental groups (P<0.001 for VANGL2 and VANGL2R259L as compared to the uninjected fish and P<0.001 for VANGL2D255E or VANGL2S464N as compared to VANGL2). Panel C, Top, lateral views of 2 days post fertilization zebrafish from the 3 clusters identified based on phenotype severity. Bottom, the distribution of the 3 clusters in each of the 5 experimental groups (χ2 test, P<0.05 for VANGL2 and VANGL2R259L as compared to the uninjected fish and P<0.05 for VANGL2D255E or VANGL2S464N as compared to VANGL2).
To substantiate the data obtained with the overexpression of VANGL2R259L, a tri-MO knockdown/rescue assay was conducted (Fig. 6). When embryos were injected with tri-MO and raised until 2 days post fertilization, a severe (43%) decrease in length of the anterior-posterior axis in tri-MO fish was observed (P<0.001) (Fig. 6), consistent with previous findings (Jessen et al., 2002; Park and Moon, 2002). When co-injecting embryos with tri-MO and human VANGL2 RNA, the average body length of tri-MO/VANGL2 fish was significantly increased (P<0.05) (Fig. 6). Co-injection of tri-MO with VANGL2R259L also was also able to rescue the CE defect suggesting that this variant is still functional (P<0.01) (Fig.6).
Figure 6.
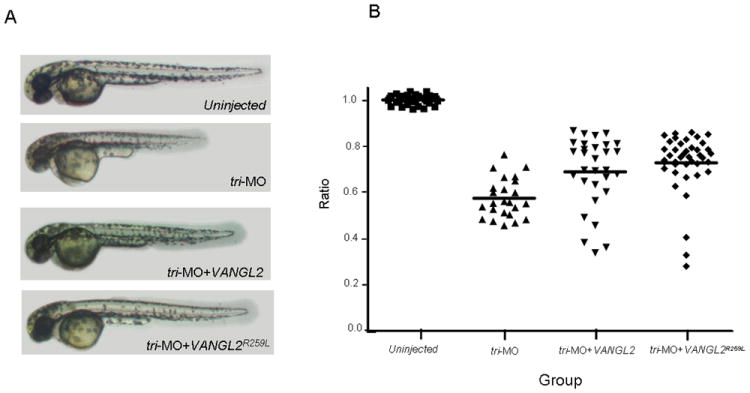
Ability of VANGL2R259L variant to rescue the convergent extension defect in zebrafish embryos treated with tri-MO. Panel A, lateral views of 2 days post fertilization zebrafish including uninjected wild-type and fish injected with either tri-MO alone, or tri-MO+VANGL2 or tri-MO+VANGL2R259L. Panel B, statistical analyses of the body length measurements of all 4 groups indicated above. The Kruskal-Wallis test was used to compare differences between experimental groups (P<0.001 for tri-MO vs uninjected; P<0.05 and P<0.01 for VANGL2 and VANGL2R259L as compared to the tri-MO alone respectively).
DISCUSSION
In this study, we identified and characterized a novel ENU-induced allele (Lpm2Jus) at the Vangl2 locus caused by the R259L missense mutation. This mutant represents a hypomorphic allele with a recessive mode of inheritance, as compared to the two other previously described Lp alleles (Lp and Lpm1Jus) that segregated in a semi-dominant fashion (Kibar et al., 2001a,b). Vangl2R259L caused a milder NTD defect in Lpm2Jus mutants manifested by a looped or kinky tail in 47% of adult homozygotes and spina bifida in 12% of homozygous embryos as compared to the two previously described Vangl2 mutations, Vangl2S464N in Lp and Vangl2D255E in Lpm1Jus. In fact, two copies of the Vangl2R259L mutation were needed to exert this phenotype that is usually caused by one copy of either of the Vangl2S464N or Vangl2D255E mutation. No craniorachischisis or eyelid closure defect was detected in Vangl2R259L homozygotes. Examination of the inner ear also revealed no PCP defects in homozygous Vangl2R259L embryos, similar to Lp heterozygous mice. This mild NTD phenotype and the absence of a PCP phenotype could be explained by a milder functional defect of the Vangl2R259L where the dosage is less limiting than that of either the two other Vangl2 mutations and/or that there may be different genetic modifiers in the different backgrounds of the 3 mutants.
The phenotype detected in the mutant argues against a dominant negative mode of action for the R259L mutation since heterozygotes do not have an NTD phenotype. Our zebrafish validation data favor the hypothesis of a weak hypomorphic allele at Lpm2Jus where Vangl2R259L behaves like the wild-type in overexpression (even at low doses) and tri-MO knockdown/ rescue assays. Hence the zebrafish assays may not be sensitive to slight losses of protein function, and/or the mutation affects a functional aspect of the protein which cannot be revealed in the zebrafish test. However, this does not exclude the presence of modifiers that are exaggerating this mild functional defect. In fact, a study by Wang et al. (2006) showed that the NTD defect and PCP defect in the cochlea can be modified by genetic background variation. They generated Dvl1−/−; Dvl2−/− double knockouts to show that although both defects were nearly 100% penetrant in an inbred 129 SvEv background, Dvl1−/−; Dvl2−/− mutants showed incompletely penetrant neural tube closure defects in a mixed 129 SvEv/C57B6 background. They hypothesized that multiple dominant modifiers from the C57B6 background act together to rescue the NTD phenotype in their Dvl1−/−; Dvl2−/− mutants. While Lpm2Jus is on a mixed 129:B6 background, Lp and Lpm2Jus are on a mixed A/J:B6 and C3H:101 backgrounds respectively. Careful genotyping of Lp and Lpm2Jus to determine the exact contribution of B6, followed by serial backcrosses to put each of the 2 mutations, Vangl2S464N and Vangl2R259L, on a pure B6 background, are needed to investigate the presence of genetic modifiers of the NTD and PCP phenotypes in B6.
In addition to the looped/kinky tail phenotype, 78% of adult Lpm2Jus/Lpm2Jus females exhibited imperforate vagina, a phenotype previously reported for the Lp allele in female heterozygotes but at a much lower frequency (30-50%) (Strong and Hollander, 1949; Vandenberg and Sassoon, 2009). A recent study demonstrated an important role for Vangl2 and non-canonical PCP signaling in establishment and maintenance cell polarity through development of the female reproductive tract (FRT) (Vandenberg and Sassoon, 2009). This remarkable increase in penetrance of the imperforate vagina in this specific Lpm2Jus genetic background as compared to the Lp allele suggests that distinct and tissue-specific molecular mechanisms might act in parallel or interact with PCP signaling to regulate FRT development. This mutant represents an important tool to dissect such mechanisms.
Around one third of Lpm2Jus/ Lpm2Jus males were infertile. These mice had normal mating abilities assessed by the presence of copulation plugs and did not display any abnormal reproductive tract histology or defective spermiogenesis or spermatogenesis. A reduced fertility was reported in Fz3+/-; Fz6-/- male mice; however, this was due to a marked reduction in number of sperm cells in the vas deferens suggesting that these genes may play a redundant role in sperm development and/or endocrine processes that regulate male fertility (Wang et al., 2006). In the same study, a reduced fertility in Lp/+ males was reported with no detailed reproductive analyses or hypothesized causes. The reduced fertility in Lpm2Jus/ Lpm2Jus males remains unexplained and requires in depth structural and molecular studies of the reproductive system.
Compound Lpm2Jus: Lp heterozygous embryos develop craniorachischisis and show quantitatively significant PCP inner ear defect, demonstrating that both alleles genetically interact. Interestingly, a small percentage of Lpm2Jus homozygotes (12%) and Lpm2Jus: Lp compound heterozygotes (15%) showed spina bifida. A variable phenotypic behavior for Vangl2 was previously reported when interacting with other PCP mutants. For example, while Vangl2: Scribble1 and Vangl2:Dvl3 double heterozygotes develop craniorachischisis (Murdoch et al., 2001b; Etheridge et al; 2008), Vangl2:PTK7 double heterozygotes develop spina bifida and Vangl2: Cordon bleu double heterozygotes develop exencephaly (Carroll et al., 2003; Lu et al., 2004). In this study, we show a variable phenotype caused by distinct mutations in the same Vangl2 gene but in different genetic backgrounds. This variability is most likely caused by modifiers affecting the site of neural tube closure defect.
On the other hand, Lpm2Jus: Vangl1gt double heterozygotes showed no CE or PCP defect in any of the organs studies including neural tube, eyelid and inner ear. Vangl1gt genetically interacts with Vangl2S464N, with double heterozygotes developing craniorachischisis and inner ear defects (Torban et al., 2008). These data further support the importance of dosage in the Vangl signaling pathway as a modifier of the severity of the resulting phenotype. It was hypothesized that loss of 50% of Vangl2 puts the pathway in a “sensitized” mode eliciting a phenotype from partial loss of Vangl1 (Torban et al., 2008). However in the case of the mild Lpm2Jus allele, we hypothesize that the loss of Vangl2 activity in these double heterozygotes is not at 50% and the minimum threshold level of combined Vangl activity required for neural tube closure is still achieved. Additional experiments are needed to test this hypothesis and to assess genetic interaction between Lpm2Jus and Vangl1gt when two copies of each allele are mutated for example in Lpm2Jus/ Lpm2Jus; Vangl1gt/+ or Lpm2Jus/+; Vangl1gt/ Vangl1gt mutants.
The variability in NTD phenotype incidence and severity encountered in the Vangl mouse mutants reflect to a certain extent a similar situation in human NTDs. Rare and novel missense mutations in VANGL2 or VANGL1 were associated with various forms of NTDs in humans, and were shown to affect the protein function in vivo, strongly implicating a defective PCP signaling in the pathogenesis of these malformations. When analyzed within the family, the same mutation was detected in one unaffected parent or in other moderately affected family members. It was hypothesized that mutations in these genes are of low penetrance that must interact with other undetermined genetic or environmental factors or modifiers to modulate the phenotype (Kibar et al. 2007; 2009; 2010).
We showed that the mutation R259L has no effect on gene expression and only a slight non significant effect on protein levels in brains of E16.5 homozygous embryos. We also showed that Vangl2R259L is still present at the plasma membrane of the neuroepithelium of Lpm2Jus/Lpm2Jus embryos although we cannot exclude the possibility of slight alterations that were not detected in our assay. A parallel study recently published showed that the S464N and R259L variants fail to reach the plasma membrane in polarized MDCK kidney cells, and like D255E, the mutant variants are retained intracellularly in the ER, and rapidly degraded by the proteasome (Iliescu et al., 2010). In agreement with previous findings on the effect of the S464N mutation onVangl2 protein levels and membrane targeting (Torban et al., 2004; Montcouquiol et al., 2006; Devenport and Fuchs, 2008), our western blot analysis supports a similar pathogenic effect for the S464N mutation where the protein levels are reduced by one third in the brain of Vangl2S464N (Lp) heterozygotes and almost completely eliminated in homozygotes. However, we did not detect any similar effect of the R259L mutation on Vangl2 protein levels by western analysis of embryonic brain tissues or on localization to plasma membrane by immunostaining of neuroepithelial cells of mutant embryos. Our results are compatible with the mild phenotype detected in the mouse Lpm2Jus mutant and the lack of a pathogenic effect of Vangl2R259L on CE in zebrafish embryos.
The dramatic effects of the Vangl2R259L variant detected in the polarized MDCK kidney cells as compared to their milder effects in western and immunofluorescent studies of affected tissues could be due to the lack of in vivo compensatory mechanisms in cell models or to the fact that stable expression of the protein over-saturates the system with high levels of the protein that do not reflect its true physiological level. These findings stress the importance of using complementary approaches to dissect the molecular mechanisms of pathogenically- hypothesized gene variants.
ENU-induced mutagenesis represents a powerful tool for the study of gene function and generation of human disease models (Clark et al., 2004). The allelic series of mutations in the Vangl2 gene represent an extremely valuable genetic resource for understanding this gene function in PCP signaling and for identification of genetic modifiers of the PCP and CE phenotypes in the developing neural tube, inner ear, eyelid and female reproductive tract.
MATERIALS AND METHODS
Maintenance and phenotyping of Lpm2Jus mice
The Lpm2Jus (MGI:3038827) mouse mutant with a looptail phenotype was generated and identified as part of the chromosome 4 balancer mutagenesis screens at the Mouse Mutagenesis and Phenotyping Center for Developmental Defects in Texas Medical Center in Houston (http://www.mouse-genome.bcm.tmc.edu). The design of the chromosome 4 balancer chromosomes and the mutagenesis screens have been described elsewhere in details (Hentges et al., 2006). Briefly, mice homozygous for the chromosome 4 balancer chromosome (on a C57BL/6BrdTyr-/- B6-albino genetic background), which could be visually recognized by their dark brown coat because of the presence of tyrosinase and K14-agouti transgenes at the inversion breakpoints were crossed to ENU-injected C57BL/6BrdTyr-/- (albino) mice. G1 pups that were heterozygous for a potential mutation and the balancer (light brown coat) were backcrossed to balancer homozygotes (dark brown coat). G2 mice that were heterozygous for the inversion (light brown coat) were intercrossed and the offspring (G3) were analyzed. Lpm2Jus was identified as a recessive mouse mutant with a looped tail phenotype in the G3 cross that failed to segregate to Chromosome 4. Lpm2Jus homozygous G3 mice were outcrossed to 129S6/SvEvTac and intercrossed to regenerate the mutant phenotype at N1F1. Lpm2Jus N1F1 mutants were archived as frozen sperm and were consequently recovered at The Jackson Laboratory (http://jaxmice.jax.org/services/) by using the Assisted In Vitro Fertilization (AIVF) method with C57BL/6J oocytes. The Lpm2Jus mutant stock was generated and maintained by backcross and brother-sister matings. Mice were examined macroscopically for the presence of a severely “kinked” or “looped” tail. Embryos were recovered at various stages and examined for the presence of NTDs.
Two Lp/+ males on a mixed A/J:B6 background and four Vangl1gt/ Vangl1gt mice (2 males, 2 females) on a mixed 129:B6 background were obtained from Philippe Gros (McGill University, Montreal, Quebec). Both Lp and Vangl1gt colonies were maintained by crosses to C57BL/6J mice.
DNA extraction and Genotyping
Genomic DNA was extracted from frozen liver or brain tissue using phenol-chloroform standard protocol, and from mouse tail or yolk sac using the DNeasy Blood & Tissue Kit (QIAGEN). Genotypes were confirmed for all mice included in the study. For Lp and Lpm2Jus mice, genotyping was done by PCR amplification on genomic DNA for mVangl2-x3 (171 bp) and mVangl2-x7 (360bp) with these primers: mVangl2-x3F (5′-CACTATCTGGCCGTAGTTCT-3′), mVangl2-x3R (5′-CATCTGGGTCTCATCTTTGTC-3’), mVangl2-x7F (5’-GCTGTGTCACAGAGGATGAAG-3′), mVangl2-x7R (5′-ACATCCCTCCTCCGCGGCT-3′). PCR amplification was done in a total volume of 30μL containing 25-50ng genomic DNA, 80μM dNTPs, 0.25μM for each primer, 1x PCR buffer minus Mg, 1.5mM MgCl2 and 1U Taq DNA polymerase (Invitrogen). PCR conditions consisted of 5 min at 94°C followed by 35 cycles with each cycle consisting of 30s at 94°C, 30s at (59°C for mVangl2-x3, 58°C for mVangl2-x7), 30s at 68°C, followed by one cycle of 5 min at 68°C. The PCR products were visualized on 1.2% agarose gel. Direct dye terminator sequencing of PCR products was done using Applied Biosystem’s 3730xl DNA Analyzer technology. The R259L mutation was also detected by AciI digestion on mVangl2-exon3 PCR product and visualized on 2% agarose gel.
For Vangl1gt mice, genotyping was done by PCR amplification on genomic DNA with 3 primers: mVangl1-exon3F (5′- AGAACAAGAGAAAGACACAAATCAC -3′), mVangl1-Intron3-R (5′- CACCAGATTTCCATGCTTGCCA -3′), GT1-R (5′-CATACTTTCGGTTCCTCTTCC CATG-3′). PCR conditions consisted of 5 min at 94°C followed by 35 cycles with each cycle consisting of 30s at 94°C, 30s at 60°C, 30s at 68°C, followed by one cycle of 5 min at 68°C. The PCR products were run on 1.2% agarose gel. Homozygous embryos showed one band at 410bp, wild-type showed one band at 744bp and heterozygous showed both bands.
Real-time PCR for mRNA quantification
Total RNA was extracted from E16.5 mouse brain with TRIzol Reagent protocol (Invitrogen). Reverse transcription were done with 1μg of total mouse brain RNA, 200 units of SuperScript II reverse transcriptase (Invitrogen), 1 mM dNTPs (Invitrogen), 300ng random primer (Invitrogen), 500ng Olido(dT)12-18 primer and 40 units RNase Out (Invitrogen). Vangl2 transcript levels were determined by real time PCR using the Roche Light Cycler and Taqman gene expression (ABI). We used probes specifically complementing mouse cDNA sequence of Vangl2 (Assay ID: Mm00473768_m1; Applied BioSystems). All qPCR reactions were done in triplicate for each sample with Taqman Fast qPCR Master Mix (Applied Biosystems). Vangl2 levels were determined in fold changes, as compared with Hprt levels using the Roche Light Cycler software.
Western Blot analysis
E16.5 mouse brain was isolated from Lpm2Jus/ Lpm2Jus embryos (n=7) and their wild-type littermates (n=7) and from Lp/+ (n=7) and Lp/Lp (n=6) and their wild-type littermates (n=7). Brain tissue was washed in PBS and crushed in liquid nitrogen. Protein extractions were done using RIPA buffer technique. Briefly, cold RIPA buffer (50mM Tris-HCl pH 7.4, 1% triton x-100, 0.25% Na-deochycholate, 150 mM NaCl, 1mM EDTA, 1mM phenylmethanesulfonylfluoride (PMSF), 1μg/mL of aprotinin, leupeptin and pepstatin) was added to the crushed brain and incubated at -80°C for 10 minutes. A 15 min centrifugation at 12 000 RPM was done and the surnageant, containing the protein, was recuperated. A total of 35μg of total protein was boiled in loading buffer, separated by SDS-PAGE and transferred on PVDF membrane. Membrane was blocked in 5% milk/TBS/ 0.1% tween 20 and incubated overnight at 4°C with primary antibody. Washes in TBS/0.1% tween 20 were done followed by one hour incubation at room temperature with appropriated secondary HRP-conjugate antibody (Abcam). After washes in TBP/0.1% tween 20, the membrane was incubated with SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific) and exposed on film. Western blot analysis was performed with rabbit polyclonal Vangl2 (provided by PG; 1:200) (Torban et al., 2004) and mouse monoclonal β actin (Novus Biologicals; 1:10 000) antibodies. For the Vangl2 antibody, a segment corresponding to amino acids 12-64 at the N terminus of the Vangl2 protein was used to construct a GST fusion immunogen to produce anti-Vangl2 rabbit polyclonal antisera. This antibody was demonstrated to be isoform-specific where it does not cross-react with the Vangl1 protein (Torban et al., 2004). Quantification of the protein levels was done using the program ImageJ. The Student’s t-test was used to compare protein levels between wild-type and mutant embryos.
Immunofluorescence of the neural tube
E9.5 wild-type and Lpm2Jus/ Lpm2Jus embryos (n=3 for each genotype) were isolated and fixed in 4% paraformaldehyde (PFA) at 4°C during overnight, followed by 3 washes in phosphate-buffered saline (PBS) at room temperature (RT). Overnight incubation in 30% sucrose/PBS at 4°C was done, followed by 2 hours incubation at room temperature in OCT : 30% sucrose/PBS (1:1) with gentle rocking. Tissues were then embedded in OCT and conserved at -80°C. The tissues, cut into 14μm cryosections, were fixed in 4% PFA, boiled for 6 min in 10mM citric buffer, pH 6.0 and blocked in a solution containing 10% normal inactivated goat serum/0.4% Triton in PBS for 1 hour at RT. The tissues were incubated with goat polyclonal Vangl2 (N-13) antibody (Sigma; 1:50) for 1 hour, followed by an incubation with an anti-goat alexa fluor 488 (Molecular Probes; 1:250). Sections were mounted with Prolong Gold. For negative control, PBS was used instead of the primary antibody.
Cochlear immunohistochemistry and analysis of stereociliary bundle orientation
Cochlea from E18.5 embryos were isolated under microscope and flat mounted, then fixed in 4% paraformaldehyde for 1 hour at room temperature, and washed 3X in PBS. The cochlea were then microdissected in PBS containing 0.1% triton x-100, and incubated in PBS with 5% goat serum and 0.1% triton x-100 (blocking solution) for 1 hour. This step was followed by overnight incubation with the antibody Alexa Fluor 488-conjugated phalloidin (1:40) at 4°C.Tissues were then washed 3 times in PBS with 0.1% triton x-100 at room temperature and mounted with Prolong Gold. At least 30 cells for each row at the apical region of the organ of Corti were used for quantification per sample. To determine stereociliary bundle orientation, a line was drawn through the middle of the “V”-shaped stereocilia (bisecting line). The angle formed between this line and the line parallel to the mediolateral axis was used for quantifications. In wild-type animals, this angle is close to 0 °C. Stereocilia with a deviation from normal larger than 30°C were considered to be misoriented. For statistical analysis, the Kolmogorov - Smirnov test was used to compare the distributions of the various subgroups of hair cells.
Zebrafish experiments
The open reading frame (ORF) of VANGL2 (Accesssion nb: NM_020335) was amplified from total human RNA by RT-PCR and the resulting fragment was cloned into pCS2+ using ClaI and XhoI sites built in the PCR primers. The R259L variant introduced in the human VANGL2 ORF cloned into pCS2+. The mutagenic primer used was: 5′-GACGGCGCCAGCCTCTTCTACAACGTTG-3′. All constructs were verified by sequencing. Sense capped RNA was synthesized using the mMESSAGE mMACHINE transcription kit (Ambion) as described by the manufacturer.
Longfin zebrafish were raised from a colony maintained according to established procedures in compliance with guidelines set out by the Canadian Council for Animal Care. Injections were performed in oocytes at the one to four cell stage. The vital dye fast-green was incorporated to the injection vehicle to monitor injection quality (0.1%; Sigma). For the overexpression assay, 50 or 100 or 200pg of human VANGL2 RNA was used. Body length was assessed at 2 days post fertilization. To knockdown translation of the zebrafish tri/VANGL2, ~7 ng of the previously described tri-MO antisense morpholino oligonucleotide was injected. To rescue the phenotype, 50 pg of human VANGL2 RNA was co-injected with the tri-MO.
For statistical analyses, since the measurements of zebrafish body length were not normally distributed, the Kruskal-Wallis test was used to compare differences between experimental groups, followed by Dunn’s comparison as a post-hoc test (GraphPad Prism). We considered groups statistically significantly different from the control group if the P value was less than 0.05. Cluster analysis was done using the SSPS statistics software. A Chi-square test was used to analyze the difference in distribution of the clusters among the experimental groups.
Supplementary Material
Effect of overexpression of wild-type VANGL2 and VANGL2R259L variant on convergent extension in zebrafish embryos at 50 pg and 100 pg doses. The Kruskal-Wallis test was used to compare differences between experimental groups (P<0.01 for 50 pg VANGL2 vs. uninjected; P<0.001 for each of the groups: 50 pg VANGL2R259L vs. uninjected; 100pg VANGL2 vs. uninjected; and 100 pg VANGL2R259L vs. uninjected).
Acknowledgments
We would like to thank Farida Kibar for her help in statistical analyses, Stephanie Lachance and Cynthia Horth for their technical assistance and Edna Brustein for the technical support on zebrafish experiments.
Grant Sponsor: the Canadian Institutes for Health Research; Grants number: 86520 (Z.K.) and MT-13425 (P.G.). Grant Sponsor: the Natural Science and Engineering Research Council, Genome Quebec and Genome Canada (P.D.). Grant Sponsor: the National Institutes of Health; Grants number: U01 HD39372 and R01 CA115503 (M.J.).
References
- Bassuk AG, Kibar A. Genetic basis of neural tube defects. Semin Pediatr Neurol. 2009;16:101–110. doi: 10.1016/j.spen.2009.06.001. [DOI] [PubMed] [Google Scholar]
- Carroll EA, Gerrelli D, Gasca S, Berg E, Beier DR, Copp AJ, Klingensmith J. Cordon-bleu is a conserved gene involved in neural tube formation. Dev Biol. 2003;262:16–31. doi: 10.1016/s0012-1606(03)00323-3. [DOI] [PubMed] [Google Scholar]
- Devenport D, Fuchs E. Planar polarization in embryonic epidermis orchestrates global asymmetric morphogenesis of hair follicles. Nat Cell Biol. 2008;10:1257–1268. doi: 10.1038/ncb1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etheridge SL, Ray S, Li S, Hamblet NS, Lijam N, Tsang M, Greer J, Kardos N, Wang J, Sussman DJ, Chen P, Wynshaw-Boris A. Murine dishevelled 3 functions in redundant pathways with dishevelled 1 and 2 in normal cardiac outflow tract, cochlea, and neural tube development. PLoS Genet. 2008;4:e1000259. doi: 10.1371/journal.pgen.1000259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AT, Goldowitz D, Takahashi JS, Vitaterna MH, Siepka SM, Peters LL, Frankel WN, Carlson GA, Rossant J, Nadeau JH, Justice MJ. Implementing large-scale ENU mutagenesis screens in North America. Genetica. 2004;122:51–64. doi: 10.1007/s10709-004-1436-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gravel M, Iliescu A, Horth C, Apuzzo S, Gros P. Molecular and cellular mechanisms underlying neural tube defects in the loop-tail mutant mouse. Biochemistry. 2010;49:3445–3455. doi: 10.1021/bi902180m. [DOI] [PubMed] [Google Scholar]
- Henderson DJ, Conway SJ, Greene ND, Gerrelli D, Murdoch JN, Anderson RH, Copp AJ. Cardiovascular defects associated with abnormalities in midline development in the Loop-tail mouse mutant. Circ Res. 2001;89:6–12. doi: 10.1161/hh1301.092497. [DOI] [PubMed] [Google Scholar]
- Hentges KE, Nakamura H, Furuta Y, Yu Y, Thompson DM, O’Brien W, Bradley A, Justice MJ. Novel lethal mouse mutants produced in balancer chromosome screens. Gene Expr Patterns. 2006;6:653–665. doi: 10.1016/j.modgep.2005.11.015. [DOI] [PubMed] [Google Scholar]
- Iliescu A, Gravel M, Horth C, Kibar Z, Gros P. Loss of membrane targeting of vangl proteins causes neural tube defects. Biochemistry. 2010 doi: 10.1021/bi101286d. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Jessen JR, Topczewski J, Bingham S, Sepich DS, Marlow F, Chandrasekhar A, Solnica-Krezel L. Zebrafish trilobite identifies new roles for Strabismus in gastrulation and neuronal movements. Nat Cell Biol. 2002;4:610–615. doi: 10.1038/ncb828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller R, Shook D, Skoglund P. The forces that shape embryos: physical aspects of convergent extension by cell intercalation. Phys Biol. 2008;5:015007. doi: 10.1088/1478-3975/5/1/015007. [DOI] [PubMed] [Google Scholar]
- Kibar Z, Underhill DA, Canonne-Hergaux F, Gauthier S, Justice MJ, Gros P. Identification of a new chemically induced allele (Lpm1Jus) at the loop-tail locus: morphology, histology, and genetic mapping. Genomics. 2001a;72:331–337. doi: 10.1006/geno.2000.6493. [DOI] [PubMed] [Google Scholar]
- Kibar Z, Vogan KJ, Groulx N, Justice MJ, Underhill DA, Gros P. Ltap, a mammalian homolog of Drosophila Strabismus/Van Gogh, is altered in the mouse neural tube mutant Loop-tail. Nat Genet. 2001b;28:251–255. doi: 10.1038/90081. [DOI] [PubMed] [Google Scholar]
- Kibar Z, Torban E, McDearmid JR, Reynolds A, Berghout J, Mathieu M, Kirillova I, De Marco P, Merello E, Hayes JM, Wallingford JB, Drapeau P, Capra V, Gros P. Mutations in VANGL1 are associated with neural tube defects in humans. New Engl J Med. 2007;35:1432–1437. doi: 10.1056/NEJMoa060651. [DOI] [PubMed] [Google Scholar]
- Kibar Z, Bosoi CM, Kooistra M, Salem S, Finnell RH, De Marco P, Merello E, Bassuk AG, Capra V, Gros P. Novel mutations in VANGL1 in neural tube defects. Hum Mutat. 2009;30:E706–715. doi: 10.1002/humu.21026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kibar Z, Salem S, Bosoi CM, Pauwels E, De Marco P, Merello E, Bassuk AG, Capra V, Gros P. Contribution of VANGL2 mutations to isolated neural tube defects. Clin Genet. 2010 doi: 10.1111/j.1399-0004.2010.01515.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei YP, Zhang T, Li H, Wu BL, Jin L, Wang HY. VANGL2 mutations in human cranial neural-tube defects. N Engl J Med. 2010;62:2232–2235. doi: 10.1056/NEJMc0910820. [DOI] [PubMed] [Google Scholar]
- Lu X, Borchers AG, Jolicoeur C, Rayburn H, Baker JC, Tessier-Lavigne M. PTK7/CCK-4 is a novel regulator of planar cell polarity in vertebrates. Nature. 2004;430:93–98. doi: 10.1038/nature02677. [DOI] [PubMed] [Google Scholar]
- McNeill H. Planar cell polarity: keeping hairs straight is not so simple. Cold Spring Harb Perspect Biol. 2010;2:a003376. doi: 10.1101/cshperspect.a003376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merte J, Jensen D, Wright K, Sarsfield S, Wang Y, Schekman R, Ginty DD. Sec24b selectively sorts Vangl2 to regulate planar cell polarity during neural tube closure. Nat Cell Biol. 2010;12:41–46. doi: 10.1038/ncb2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montcouquiol M, Rachel RA, Lanford PJ, Copeland NG, Jenkins NA, Kelley MW. Identification of Vangl2 and Scrb1 as planar polarity genes in mammals. Nature. 2003;423:173–177. doi: 10.1038/nature01618. [DOI] [PubMed] [Google Scholar]
- Montcouquiol M, Sans N, Huss D, Kach J, Dickman JD, Forge A, Rachel RA, Copeland NG, Jenkins NA, Bogani D, Murdoch J, Warchol ME, Wenthold RJ, Kelley MW. Asymmetric localization of Vangl2 and Fz3 indicate novel mechanisms for planar cell polarity in mammals. J Neurosci. 2006;26:5265–5275. doi: 10.1523/JNEUROSCI.4680-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murdoch JN, Doudney K, Paternotte C, Copp AJ, Stanier P. Severe neural tube defects in the loop-tail mouse result from mutation of Lpp1, a novel gene involved in floor plate specification. Hum Mol Genet. 2001a;10:2593–2601. doi: 10.1093/hmg/10.22.2593. [DOI] [PubMed] [Google Scholar]
- Murdoch JN, Rachel RA, Shah S, Beermann F, Stanier P, Mason CA, Copp AJ. Circletail, a new mouse mutant with severe neural tube defects: chromosomal localization and interaction with the loop-tail mutation. Genomics. 2001b;78:55–63. doi: 10.1006/geno.2001.6638. [DOI] [PubMed] [Google Scholar]
- Park M, Moon RT. The planar cell-polarity gene Stbm regulates cell behavior and cell fate in vertebrate embryos. Nat Cell Biol. 2002;4:20–25. doi: 10.1038/ncb716. [DOI] [PubMed] [Google Scholar]
- Reynolds A, McDearmid JR, Lachance S, De Marco P, Merello E, Capra V, Gros P, Drapeau P, Kibar Z. VANGL1 rare variants associated with neural tube defects affect convergent extension in zebrafish. Mech Dev. 2010;127:385–392. doi: 10.1016/j.mod.2009.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons M, Mlodzik M. Planar cell polarity signaling: from fly development to human disease. Annu Rev Genet. 2008;42:517–540. doi: 10.1146/annurev.genet.42.110807.091432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strong LC, Hollander WF. Hereditary Loop-tail in the house mouse. J Hered. 1949;40:329–334. [Google Scholar]
- Suriben R, Kivimäe S, Fisher DA, Moon RT, Cheyette BN. Posterior malformations in Dact1 mutant mice arise through misregulated Vangl2 at the primitive streak. Nat Genet. 2009;41:977–985. doi: 10.1038/ng.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torban E, Wang H-J, Groulx N, Gros P. Independent mutations in mouse Vangl2 that cause neural tube defects in looptail mice impair interaction with members of the Dishevelled family. J Biol Chem. 2004;279:52703–52713. doi: 10.1074/jbc.M408675200. [DOI] [PubMed] [Google Scholar]
- Torban E, Patenaude AM, Leclerc S, Rakowiecki S, Gauthier S, Andelfinger G, Epstein DJ, Gros P. Genetic interaction between members of the Vangl family causes neural tube defects in mice. Proc Natl Acad Sci U S A. 2008;105:3449–3454. doi: 10.1073/pnas.0712126105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenberg AL, Sassoon DA. Non-canonical Wnt signaling regulates cell polarity in female reproductive tract development via van gogh-like 2. Development. 2009;136:1559–1570. doi: 10.1242/dev.034066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vladar EK, Antic D, Axelrod JD. Planar Cell Polarity Signaling: The Developing Cell’s Compass. Cold Spring Harb Perspect Biol. 2009;1:a002964. doi: 10.1101/cshperspect.a002964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallingford JB. Neural tube closure and neural tube defects: studies in animal models reveal known knowns and known unknowns. Am J Med Genet C Semin Med Genet. 2005;135:59–68. doi: 10.1002/ajmg.c.30054. [DOI] [PubMed] [Google Scholar]
- Wang J, Hamblet NS, Mark S, Dickinson ME, Brinkman BC, Segil N, Fraser SE, Chen P, Wallingford JB, Wynshaw-Boris A. Dishevelled genes mediate a conserved mammalian PCP pathway to regulate convergent extension during neurulation. Development. 2006;133:1767–1778. doi: 10.1242/dev.02347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Guo N, Nathans J. The role of Frizzled3 and Frizzled6 in neural tube closure and in the planar polarity of inner-ear sensory hair cells. J Neurosci. 2006;26:2147–2156. doi: 10.1523/JNEUROSCI.4698-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Nathans J. Tissue/planar cell polarity in vertebrates: new insights and new questions. Development. 2007;134:647–658. doi: 10.1242/dev.02772. [DOI] [PubMed] [Google Scholar]
- Ybot-Gonzalez P, Savery D, Gerrelli D, Signore M, Mitchell CE, Faux CH, Greene ND, Copp AJ. Convergent extension, planar-cell-polarity signalling and initiation of mouse neural tube closure. Development. 2007;134:789–799. doi: 10.1242/dev.000380. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Effect of overexpression of wild-type VANGL2 and VANGL2R259L variant on convergent extension in zebrafish embryos at 50 pg and 100 pg doses. The Kruskal-Wallis test was used to compare differences between experimental groups (P<0.01 for 50 pg VANGL2 vs. uninjected; P<0.001 for each of the groups: 50 pg VANGL2R259L vs. uninjected; 100pg VANGL2 vs. uninjected; and 100 pg VANGL2R259L vs. uninjected).