

Abstract
A wealth of evidence has now demonstrated that the microenvironment in which a tumorigenic cell evolves is as critical to its evolution as the genetic mutations it accrues. However, there is still relatively little known about how signals from the microenvironment contribute to the early events in the progression to malignancy. To address this question, we used a premalignant mammary model to examine how fibroblasts, and the extracellular matrix (ECM) proteins they secrete, influence progression to malignancy. Their effect on metastatic malignant cells was also assessed for comparison. We found that carcinoma-associated fibroblasts, and the distinct aligned ECM they deposit, can cause both premalignant and malignant mammary epithelial cells to assume a mesenchymal morphology that is associated with increased dissemination and metastasis, while benign reduction mammoplasty fibroblasts favor the maintenance of an epithelial morphology and constrain early dissemination, tumor growth, and metastasis. Our results suggest that normalizing the organization of the ECM could be effective in limiting systemic dissemination and tumor growth.
Introduction
It is now very well established that all the cells and extracellular matrix (ECM) proteins within the microenvironment influence the behavior of the epithelial cells they surround and play an important role in the tumorigenic process [1]. However, there is still relatively little known about how signals from the microenvironment contribute to the early events in the progression to malignancy. In light of the importance of early intervention in successfully treating malignancies, and growing evidence that tumor cells can disseminate early [2], there is a pressing need to better understand the early events involved in the tumorigenic process so as to identify new strategies for prevention and for more effective therapeutic intervention.
Fibroblasts are among the most abundant cell type in the micro-environment of solid tumors and are responsible for the elaboration of a wide range of chemokines, growth factors, and ECM proteins found therein, all of which contribute to the evolution of the tumor [3]. The ECM is defined as a complex mixture of proteins (proteoglycans and adhesive glycoproteins such as collagens and laminins) that provides structural support for organs and tissues. In addition, the ECM plays an important role in the regulation of gene expression, growth factor activation, signal transduction, cell survival, shape, and movement [4]. Carcinoma-associated fibroblasts (CAFs) form a heterogeneous population, likely due to the diversity of sources from which they have been postulated to arise [5–7] and to the heterogeneity of the tumors in which they reside. The pivotal role of fibroblasts and their associated ECM in cancer is highlighted by the fact that an ECM signature alone can stratify primary breast tumors into different subgroups with different clinical outcomes and can also identify which patients within a “good prognosis” group (i.e., luminal) have a poorer outcome [8]. In addition, a stromal gene expression signature that includes alterations in the ECM and an “activated fibroblast” phenotype has been reported to be a more powerful predictor of prognosis than other prognostic signatures derived from whole tumor samples [9].
These data suggest that targeting non-neoplastic components within the microenvironment can lead to the development of new therapies. Hence, to gain novel insights into how we could potentially target the microenvironment therapeutically, and do so early in the development of breast cancer, we examined how fibroblasts, and the ECM proteins they secrete, influence the early events in the development of breast cancer using a series of human mammary epithelial cells (HMECs) isolated from disease-free reduction mammoplasty tissues that display premalignant properties but are not tumorigenic [10]. These include a subpopulation of HMEC in which p16INK4A expression is repressed due to promoter methylation, which we refer to as vHMEC; vHMEC expressing oncogenic Ha-rasV12, which we refer to as vHMEC-ras; and immortalized (but non-tumorigenic) vHMEC expressing oncogenic Ha-rasV12, which we refer to as vHMEC-ras0.5 [10]. We examined how fibroblasts isolated from disease-free reduction mammoplasty tissues [reduction mammoplasty fibroblasts (RMFs)] or cancer tissues (CAFs), and their ECM, influence the behavior of these premalignant cells in co-cultures in vitro as well as when co-injected in vivo. Their effect on malignant cells was also assessed for comparison.
We found that CAFs, and the distinct aligned ECM they deposit, caused both premalignant and malignant mammary epithelial cells to assume a mesenchymal morphology that was associated with increased dissemination and metastasis. In contrast, RMFs favored the maintenance of an epithelial morphology and constrained tumor growth and metastasis. Our results suggest that normalizing the organization of the ECM could be effective in limiting early dissemination and tumor growth.
Materials and Methods
Co-culture Assays
Epithelial cells were seeded at a concentration of 1 x 105 cells/well into six-well plates. Twenty-four hours later, cells were washed with serum-free growth medium and stained with the CellTracker Green 5-chloromethylfluorescein diacetate (CMFDA) probe (Life Technologies, Grand Island, NY) for 30 minutes at 37°C. The cells were then washed twice with complete growth medium and allowed to recover for 2 to 3 hours in this medium. The medium was then aspirated, and 3 x 105 fibroblasts resuspended in 3 ml of mammary epithelial cell growth medium (MEGM) + 0.5% FBS (for primary HMEC cultures) or 3 ml of Dulbecco's modified Eagle's medium + 10% FBS (for MDA-MB-231 cells) were seeded onto the CMFDA-stained epithelial cultures. Co-cultures were incubated for 3 to 4 days, cell morphology was assessed, and representative images were captured at 10x magnification with a Nikon DS-Qi1Mc monochrome camera mounted on a Nikon Eclipse TE300 microscope. The cells were then harvested and the CMFDA-positive epithelial cell population was recovered by fluorescence-activated cell sorting (FACS) for further analysis.
Matrix Overlay Assays
Fibroblasts were seeded onto collagen-coated six-well plates in complete growth medium (RPMI + 10% FBS) at 1.2 x 105 cells/well. Three to four days later, fibroblasts were removed by gently washing the cells twice with a freshly prepared solution of 20 mM NH4OH diluted in sterile water. The denuded matrix was examined microscopically to ensure complete removal of the fibroblasts, gently washed twice with sterile water, and stained with picrosirius (0.1% sirius red diluted in saturated picric acid) overnight at room temperature to better visualize the matrix. The next day, the staining solution was removed by washing three times with water, and representative images were captured at 10x magnification with a Nikon DS-Qi1Mc monochrome camera mounted on a Nikon Eclipse TE300 microscope. Replicate unstained wells were used to assess the effect of the denuded matrix on epithelial cells. For these experiments, epithelial cells were seeded on top of the denuded matrix deposited by the fibroblasts at 1 x 105 cells/well in 3 ml of complete growth medium. Two to three days later, cell morphology was assessed, and representative 10x images were captured as above.
Thy-1 and Epithelial Cell Adhesion Molecule Staining
Cells were trypsinized, washed once in phosphate-buffered saline (PBS) + 2% FBS, and incubated in MEGM + 2% FBS for 1 hour at 37°C with gentle rocking. The cells were then washed once in wash solution (PBS + 1% BSA) and incubated in 100 µl of wash solution containing allophycocyanin (APC)-conjugated anti-Thy-1 (1 µl per 1 x 106 cells; BD Pharmingen Cat. No. 559869) or fluorescein isothiocyanate-conjugated anti-epithelial cell adhesion molecule (EpCAM; 1 µl per 1 x 106 cells; Biomeda Cat. No. FM010) antibodies for 30 minutes at room temperature with shaking. Following staining, cells were washed twice with wash solution and once with PBS. Cells were then fixed in PBS + 1% paraformaldehyde and analyzed for Thy-1 or EpCAM expression with a FACSCalibur. Flow cytometry files were analyzed with the FlowJo (v8.8.02) software.
Immunoblot Analysis
Protein extracts were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, transferred onto polyvinylidene fluoride (PVDF) membranes, and probed using standard procedures. Mouse monoclonal antibodies against E-cadherin (#610181), N-cadherin (#610920), and fibronectin (Fn; #610077) were obtained from BD Biosciences (San Jose, CA). The mouse monoclonal antibody against β-actin (#5441) was obtained from Sigma-Aldrich (St Louis, MO). The rabbit polyclonal antibodies against the phosphorylated form of Smad2 (#3101), Erk1/2 (#9101), Jun (#9261), and p38 mitogen-activated protein kinase (MAPK; #9211) were obtained from Cell Signaling Technology (Danvers, MA), while those against lysyl oxidase (LOX; #NB100-2527), tenascin C (TNC; #19101) and biglycan (#54855) were from Novus (Littleton, CO), Chemicon (Billerica, MA), and Abcam (Cambridge, MA), respectively.
In Vivo Premalignant and Malignant Tumor Studies
For the in vivo studies with premalignant cells, the vHMEC-ras0.5 cells were engineered to express luciferase (Lux) using a lentiviral vector from System Biosciences (Mountain View, CA) encoding green fluorescent protein (GFP) and firefly Lux (pGF-CMV-EF1-Neo). In some experiments (designed to mirror in vitro co-cultures), ras0.5-GFPLux cells were injected orthotopically into the right and left No. 4 mammary fat pads of 6- to 8-week-old female severe combined immunodeficiency (SCID)-Beige mice either alone (1 x 106 cells per gland) or in combination with 3 x 106 fibroblasts. In other experiments (designed to mirror in vitro matrix overlay assays), the mammary glands of 3-week-old NOD/SCID mice were cleared of their endogenous epithelium and injected with 2.5 x 105 RMFs or CAFs at a 1:1 ratio with hTERT-immortalized RMFs, as described by Proia and Kuperwasser [11]. The fibroblasts were allowed to deposit matrix on their own in vivo for 2 to 3 weeks before the injection of 2.5 x 106 ras0.5-GFPLux cells. Cell survival was monitored biweekly by bioluminescence imaging using the Xenogen IVIS Imaging System (Caliper Life Sciences, Hopkinton, MA). Quantification of photone mission from the bioluminescent signal was performed using the acquisition and analysis software Living Image (Xenogen). By 3 to 5 weeks, the ras0.5-GFPLux cells could no longer be detected by imaging in the majority of the mice. Hence, experiments were terminated, blood was collected to quantify the number of circulating ras0.5-GFPLux cells, and mammary glands were harvested for histologic analysis and staining.
For the tumor studies, the malignant MDA-MB-231 breast cancer cells were engineered to express Lux (231-Lux) using the pLLRN retroviral Lux vector in Clontech's Pantropic Retroviral Expression System, according to the manufacturer's instructions. The 231-Lux cells were injected directly into the surgically exposed right and left No. 4 mammary fat pads of 6- to 8-week-old female SCID-Beige mice either alone (5 x 105 cells per gland) or in combination with 1.5 x 106 fibroblasts. Cell growth and survival were monitored and quantified weekly by bioluminescence imaging as described above. In addition, mammary glands were palpated weekly after cell injection for the presence of tumors. Tumor latency was recorded when palpable tumors > 15 mm3 were detected. Tumor lengths and widths were measured weekly with calipers and corresponding tumor volumes were calculated using the formula [volume = (width)2 x length/2]. Eight weeks following injection of cells, mice were anesthetized with avertin, blood was collected to measure circulating tumor cells (CTCs), and the lungs (along with other organs for comparison) were harvested and imaged ex vivo to document and quantify metastases based on the presence of luminescence in each lung. Tumors were weighed, measured with calipers to calculate final tumor volume, and cut in half. Half of each tumor was snap frozen in liquid nitrogen for subsequent analysis, while the other half of the tumor along with all the organs were fixed and processed for histologic examination and staining. All experiments involving animals were conducted in compliance with the Institutional Animal Care and Use Committee guidelines and were approved by the Institutional Animal Care and Use Committee Ethics Committee.
Quantification of CTCs
Blood was collected through cardiac puncture using a 25-G 5/8 needle attached to a 1-ml syringe precoated with acid citrate dextrose and transferred to 2.0-ml K2EDTA vacutainer tubes (BD Biosciences). Blood was stored at room temperature overnight. The next day, genomic DNA was extracted from the blood using Promega's Wizard Genomic DNA Purification Kit according to the manufacturer's instructions. The DNA was quantified and diluted to 10 ng/µl for each sample. The levels of EpCAM DNA in each sample were quantified by quantitative polymerase chain reaction (qPCR) using a custom-designed, human-specific, EpCAM primer-reporter set (Applied Biosystems, Inc, Foster City, CA). The forward and reverse primer sequences for EpCAM were 5′-GCAGGAGAATTGCCTGAATCC-3′ and 5′-GAGTGCAATGGCATCATCTCA-3′, respectively. The reporter sequence was 5′-FAM-CAACCTCCGCCTCCC-FAM-3′. Quantification of EpCAM DNA levels was used as a surrogate for the number of CTCs in the blood. This method was validated by spiking in increasing numbers of human 231-Lux cells into naïve blood samples, as shown in Figure W1. Similar results were obtained with a custom-designed firefly Lux primer-reporter set (Applied Biosystems). The forward and reverse primer sequences for Lux were 5′-GTACACGTTCGTCACATCTCATCTA-3′ and 5′-CACGATCAAAGGACTCTGGTACAAA-3′, respectively. The reporter sequence was 5′-FAM-CCTCCCGGTTTTAATG-FAM-3′.
Real-Time qPCR
Total RNA was isolated from cells and cDNA synthesized using standard methods. qPCR (Taqman) was performed on cDNA using the standard curve method with primer/probe sets (Applied Biosystems) for stromal-derived factor-1 (SDF-1; Hs00171022_m1). The expression of glucuronidase B (Integrated DNA Technologies, Inc, Coralville, IA) was used to normalize for variances in input cDNA. The forward and reverse primer sequences for glucuronidase B were 5′-CTCATTTGGAATTTTGCCGATT-3′ and 5′-CCGAGGAAGATCCCCTTTTTA-3′, respectively. The reporter sequence was 5′-FAM-TGAACAGTCACCGACGAGAGTGCTGGTA-TAM-3′. Each experiment was performed at least in triplicate. Error bars represent the standard error of the mean in a representative experiment.
Quantification of Immunohistochemical Stains
For quantification of Ki67 and cleaved caspase 3 (CC3) stains, three to four representative 20x images from each section were captured and the number of positive nuclei were quantified using the inForm Advanced Image Analysis Software (Caliper Life Sciences). Quantification of ECM alignment was carried out through automated image analysis, using a regularity index designed to capture oriented ECM fiber bundles in each sample. Differential analysis between different treatment groups was performed as described in Quantification of Regularity Index section (Figures W2–W4).
Results
Mammary Epithelial Cells Acquire a Mesenchymal Morphology When Co-cultured with CAFs but Not RMFs
To examine how signals from the microenvironment influence the behavior of epithelial cells early in breast cancer development, vHMEC, vHMEC-ras, and vHMEC-ras0.5 cells were co-cultured with RMFs or CAFs. In this assay, the epithelial cells were stained with a green tracker dye before the addition of fibroblasts so that they could be readily distinguished from the fibroblast population in the co-cultures. As shown in Figure 1A, all three epithelial cell populations maintained their epithelial morphology when co-cultured with RMFs. However, when co-cultured with CAFs, the vHMEC-ras0.5 cells assumed a mesenchymal morphology while the vHMEC and vHMEC-ras cells did not. We have previously shown that vHMEC-ras0.5 cells exhibit greater cellular plasticity and accumulate more epigenomic alterations than the vHMEC and vHMEC-ras populations [10,12], suggesting that multiple alterations may be required to render mammary epithelial cells responsive to environmental cues that promote a mesenchymal phenotype. In light of the responsiveness of vHMEC-ras0.5 cells to CAFs, we used these cells as a premalignant model for subsequent experiments in our studies.
Figure 1.
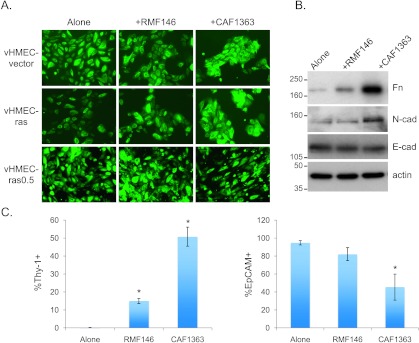
Premalignant mammary epithelial cells acquire a mesenchymal morphology when co-cultured with CAFs but not RMFs. (A) Representative 10x images of epithelial cells co-cultured with RMFs or CAFs for 3 days. (B) Immunoblot analysis of Fn, N-cadherin (N-cad), and E-cadherin (E-cad) in lysates of vHMEC-ras0.5 cells isolated by FACS after they had been cultured either alone, with RMFs, or with CAFs for 3 days. Representative immunoblots are shown. (C) Flow cytometric analysis of Thy-1 and EpCAM expression in vHMEC-ras0.5 cells cultured as above in B. Bar graphs represent the averages ± SEM of multiple experiments combined. For Thy-1: alone (n = 12), RMF146 (n = 6), CAF1363 (n = 11). For EpCAM: alone (n = 3), RMF146 (n = 3), CAF1363 (n = 2). Student's t test for Thy-1: alone versus RMF146 (P < .001), alone versus CAF1363 (P < .001); for EpCAM: alone versus RMF146 (P = .17), alone versus CAF1363 (P = .02). Statistically significant differences in expression between cells in co-culture and the cells alone are denoted with asterisk.
The transition from an epithelial to a mesenchymal morphology is often associated with up-regulation of mesenchymal markers and down-regulation of epithelial markers. Hence, we examined the expression of the mesenchymal markers Fn and N-cadherin, and the epithelial marker E-cadherin, by immunoblot analysis in vHMEC-ras0.5 cells following co-culture with the different fibroblasts. As indicated in Figure 1B, induction of the mesenchymal morphology was associated with a differential up-regulation in the expression of Fn and N-cadherin but no change in the expression of E-cadherin, suggesting that the cells were not undergoing a full epithelial-to-mesenchymal transition. To assess the induction of the mesenchymal phenotype more quantitatively, we examined the expression of two additional cell surface markers that could be readily quantified by flow cytometry, the mesenchymal marker Thy-1 and the epithelial marker EpCAM. In these experiments, we observed an increase in the percentage of vHMEC-ras0.5 cells expressing Thy-1 and a corresponding decrease in the percentage of cells expressing EpCAM following co-culture with CAFs relative to RMFs (Figure 1C).
We then screened RMFs isolated from 8 different individuals and CAFs isolated from 15 different individuals in co-cultures with vHMEC-ras0.5. Of the 8 RMFs examined, only one induced the mesenchymal phenotype, while of the 15 CAFs examined, 12 did. The induction of the mesenchymal phenotype did not correlate with the proliferation rate of the fibroblasts but did correlate with the induction of Thy-1 expression in the vHMEC-ras0.5 cells. As summarized in Table 1, it was induced in less than 15% of vHMEC-ras0.5 cells when the cells were co-cultured with fibroblasts that failed to induce the mesenchymal phenotype and in more than 20% of vHMEC-ras0.5 cells when they were co-cultured with fibroblasts that did. Hence, the induction of Thy-1 expression was used as a non-subjective surrogate for the induction of the mesenchymal phenotype. On the basis of this metric and an assessment of cell morphology, a subset of RMFs and CAFs with varying abilities to induce the mesenchymal phenotype was selected for further study.
Table 1.
Description of Fibroblasts Screened in Co-culture Assay.
| Cell Name | Mes | %Thy-1+ | Population Doubling Time (Days) | Age | History and Diagnosis | ER | PR | HER2 |
| RMF9 | - | 13.7 | 6.7 | 35 | ||||
| RMF13 | - | 11.9 | 8.3 | 33 | ||||
| RMF48 | - | 14.2 | 5.3 | 16 | ||||
| RMF146 | - | 13.4 ± 1.5 | 5 | 46 | ||||
| RMF146-Mes | +/- | 19.3 ± 0.9 | 46 | |||||
| RMF151 | - | 13.9 | 6.3 | 44 | ||||
| RMF156 | - | 12.4 ± 2.5 | 5.3 | 47 | ||||
| RMF163 | - | 12.4 ± 2.3 | 4.1 | 17 | ||||
| RMF166 | - | 7.8 | 7.1 | 39 | Prophylactic mastectomy | |||
| Total | 1/8 | |||||||
| CAF20 | + | 38.7 ± 9.5 | 12.5 | 53 | IDC | + | + | 1+ |
| CAF25 | + | 31.2 | 3.5 | 57 | IDC | + | + | 1+ |
| CAF26 | + | 24.4 ± 4.6 | 26 | Invasive micropapillary CA | + | + | + | |
| CAF29 | +/- | 9.1 | 49 | IDC | + | + | ||
| CAF100 | - | 9.9 | 38 | IDC | - | - | 3+ | |
| CAF126 | + | 11.1 | 67 | IDC, grade II with DCIS | + | + | - | |
| CAF149 | +/- | 52 | IDC with DCIS | - | - | - | ||
| CAF150 | - | 8.7 | 3.4 | 52 | ||||
| CAF201 | +/- | 51 | IDC with DCIS | - | - | - | ||
| CAF304 | - | 5.6 | 73 | IDC, grade III, 1.7 cm with DCIS | - | - | - | |
| CAF559 | + | 19.5 ± 1.7 | 4.4 | IDC | ||||
| CAF910 | + | 12.5 | ||||||
| CAF966 | + | 5.9 | 60 | IDC | ||||
| CAF1363 | + | 52.0 ± 4.5 | 4.4 | 63 | Mucinous CA, grade II, 3.4 cm, DCIS, grade III, 0.6 cm | + | + | 2+ |
| CAF1366 | +/- | 4.3 | 61 | IDC grade II, 1.5 cm with LVI | 3+ | 3+ | - | |
| Total | 12/15 |
ER indicates estrogen receptor; PR, progesterone receptor; LVI, lymphovascular invasion.
Description of fibroblasts screened in co-culture assays with vHMEC-ras0.5 cells detailing a qualitative assessment of whether or not they induced the mesenchymal phenotype (abbreviated as Mes), where (+) refers to induction, (+/-) refers to partial induction, and (-) refers to no induction, as well as a quantitative assessment of whether they induced the mesenchymal phenotype based on the induction of the mesenchymal marker Thy-1 indicating that the percentage of Thy-1-positive cells is less than 15% following co-culture with fibroblasts that fail to induce the mesenchymal phenotype and greater than 20% following co-culture with fibroblasts that induce the mesenchymal phenotype. Population doubling times were calculated from the growth curves of each fibroblast over several passages and indicate that induction of the mesenchymal phenotype does not correlate with proliferation rate. Where blank, values were not calculated or patient data were not available.
The ECM Deposited by CAFs Differs from that Deposited by RMFs and Contributes to the CAF-Induced Mesenchymal Phenotype Observed in Epithelial Cells
Next, we examined whether the CAF-induced mesenchymal phenotype could also be induced by CAF-conditioned media, CAF co-culture-conditioned media, or by CAFs in transwell co-cultures with vHMEC-ras0.5 cells where the two cell populations were not in direct contact with one another. Interestingly, the mesenchymal phenotype was not induced in the epithelial cells under any of those conditions, suggesting that it was not due to a factor secreted in the medium (data not shown). In light of these data, and of the fact that the epithelial cells co-cultured with CAFs appeared very elongated and regularly arranged in a parallel pattern, as though they were lining up on tracks, we examined whether the mesenchymal morphology induced in the presence of CAFs could be due to differences in the organization of the ECM deposited by the different fibroblasts. Picrosirius staining of the ECM deposited by fibroblasts revealed that, unlike the ECM deposited by the RMFs, which exhibited a random mesh-like appearance, the ECM deposited by CAFs was remarkably aligned in a parallel pattern (Figure 2A), as it has been reported to appear both in vitro [13,14] and in regions adjacent to invasive tumors in vivo [15,16]. In addition, when epithelial cells were seeded on top of the ECM deposited by CAFs (after removal of the fibroblasts), they assumed a mesenchymal morphology similar to that observed when co-cultured with CAFs (Figure 2A), and the percentage of epithelial cells expressing Thy-1 increased correspondingly (Figure 2B). These data suggest that the mesenchymal morphology assumed by epithelial cells co-cultured with CAFs is governed at least in part by the organization of the ECM that the CAFs deposit. If this is true, we reasoned that disrupting collagen deposition by co-culturing the cells in the presence of collagenase would inhibit the CAF-induced mesenchymal morphology. Indeed, both the CAF-induced mesenchymal phenotype and the CAF-induced increase in Thy-1 expression were blocked when the cells were co-cultured in the presence of collagenase. Under these conditions, the cells maintained their epithelial morphology or appeared live but rounded (Figure 2, C and D). In addition, when epithelial cells, which had acquired a mesenchymal morphology in the presence of CAFs, were isolated by FACS and replated in the absence of CAFs, they reverted back to their epithelial morphology, indicating that continued exposure to the CAFs, or the ECM they deposit, is required for maintenance of the mesenchymal phenotype (Figure W5).
Figure 2.
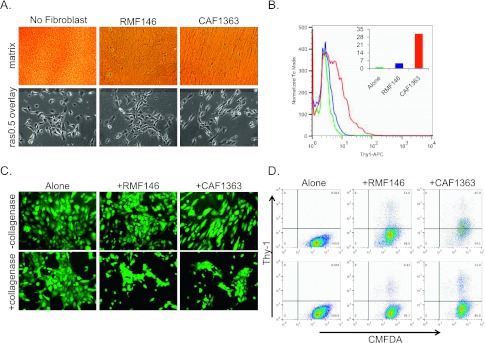
The CAF-induced mesenchymal phenotype is due to differences in the organization of the ECM deposited by CAFs. (A) Representative 10x images of ECM stained with picrosirius 4 days after seeding and removal of fibroblasts (top panels) and of vHMEC-ras0.5 cells overlaid on top of ECM (bottom panels). (B) Flow cytometric analysis of Thy-1 expression in vHMEC-ras0.5 cells overlaid on control ECM (no fibroblasts seeded) or ECM deposited by RMFs or CAFs for 2 days. Analysis of one representative experiment is shown. Inset shows percentage of Thy-1+ cells based on no antibody control for histogram shown. (C) Representative 10x images of vHMEC-ras0.5 cells co-cultured with RMFs or CAFs for 3 days in the absence or presence of collagenase. (D) Flow cytometric analysis of Thy-1 expression in the CMFDA-labeled vHMEC-ras0.5 cells of the experiment shown in C; top row, no collagenase; bottom row, + collagenase. Analysis of one representative experiment is shown.
Transforming Growth Factor-β, Erk, Jun, and Rho Signaling Are Required for the CAF-Induced Mesenchymal Phenotype
To gain insight into the mechanism by which CAFs induce the mesenchymal phenotype in epithelial cells, we examined what signaling pathways were activated in the epithelial cells after they had been cocultured with RMFs or CAFs for 3 to 4 days. For these experiments, epithelial cells were isolated from co-cultures by FACS, and the phosphorylation status of candidate signaling proteins within the epithelial cells was assessed by immunoblot analysis. Since transforming growth factor-β (TGFβ) plays a critical role in modulating cellular plasticity and in regulating ECM deposition, we examined whether the signaling pathways that have been implicated in these TGFβ-mediated functions [17–20] were differentially activated in vHMEC-ras0.5 cells following co-culture with CAFs. As shown in Figure 3A, a differential increase in the phosphorylation of Smad2, Erk1/2, and Jun was observed in vHMEC-ras0.5 cells co-cultured with CAFs relative to RMFs, while p38 phosphorylation remained unchanged. To determine whether the up-regulation in Smad, Erk1/2, and Jun signaling was required for the CAF-induced mesenchymal phenotype, vHMEC-ras0.5 cells were co-cultured with CAFs in the presence of chemical inhibitors that block each pathway. Blockade of the Smad, Erk1/2, and Jun signaling pathways, whose activity was increased by CAFs, with TGFβ receptor kinase inhibitor (TβRki), MEK inhibitor, and JNK inhibitor, respectively, inhibited the CAF-induced mesenchymal phenotype, while blockade of the p38 MAPK pathway, which was not differentially induced by CAFs, did not (Figure 3B). Inhibition of the CAF-induced mesenchymal phenotype was associated with a decrease in the expression of the mesenchymal marker Thy-1 (Figure W6). These data indicate that the up-regulation of the Smad, Erk1/2, and Jun signaling pathways in the vHMEC-ras0.5 cells exposed to CAFs is necessary for the acquisition of the mesenchymal phenotype.
Figure 3.

The CAF-induced mesenchymal phenotype is dependent on the activation of the Smad, Erk, Jun, and Rho signaling pathways. (A) Representative immunoblots of signaling proteins expressed in vHMEC-ras0.5 cells after they were cultured either alone (lane 1) or with RMFs (lane 2) or CAFs (lane 3) for 3 days. (B) Representative 10x images of vHMEC-ras0.5 cells co-cultured with CAFs in the absence or presence of different pathway kinase inhibitors, as indicated: TβRKi (LY364947, 10 µM), MEK inhibitor (U0126, 10 µM), JNK inhibitor II (10 µM), p38MAPKi (SB203580, 10 µM), PI3K inhibitor (LY294002, 10 µM), mTORi (rapamycin, 10 nM), and ROCK inhibitor (Y27632, 40 µM).
Since Rho-associated kinase (ROCK) signaling plays a fundamental role in regulating cell morphology, adhesion, and motility [21,22], we also investigated whether blockade of ROCK signaling could inhibit the CAF-induced mesenchymal phenotype. Indeed, it did (consistent with its important role in cytoskeletal reorganization), while blockade of the phosphatidylinositol 3-kinase (PI3K) pathway with either LY294002 (PI3K inhibitor) or rapamycin mammalian target of rapamycin inhibitor (mTORi) did not (Figure 3B). The fact that not all of the signaling pathways examined affected the acquisition of the mesenchymal morphology points to the contribution of specific signaling pathways.
CAFs Promote the Dissemination of Premalignant Cells In Vivo
The acquisition of a mesenchymal phenotype is often associated with increased motility. In addition, studies have shown that tumor cells can move on collagen fibers and use these fibers as tramlines to rapidly move through the stroma away from the tumor mass [23]. Hence, we reasoned that if the mesenchymal phenotype we were observing in vitro was biologically relevant in vivo, the aligned ECM deposited by CAFs might promote the dissemination of vHMECras0.5 cells in vivo. To test this hypothesis, vHMEC-ras0.5 cells were genetically engineered to express GFP-Lux (ras0.5-GFPLux) to allow their direct visualization by bioluminescence imaging in vivo. The cells were injected either alone or in combination with RMFs or CAFs into the number four mammary glands of immunocompromised mice. Three weeks following injection, the ECM content in their mammary glands was assessed by picrosirius staining to determine whether the aligned ECM deposited by CAFs in vitro could also be observed in vivo. In addition, blood was collected and the number of circulating ras0.5-GFPLux cells was quantified by qPCR by measuring the levels of human EpCAM in genomic DNA extracted from the blood.
As shown in Figures 4A and W7, the ras0.5-GFPLux cells injected into the mammary glands could be detected both by bioluminescence imaging and histologically (Figure 4A, first and second rows). The histologic detection of these human cells in the mouse mammary gland was confirmed using a human-specific lamin A/C stain (Figure 4A, third row). Assessment of the ECM by picrosirius staining revealed that the ECM deposited in the presence of CAFs was not only more abundant but also more aligned than that observed in the presence of RMFs or when the cells were injected alone [Figures 4, A (fourth row) and B, and W7]. These data suggest that the aligned ECM deposited by the CAFs in vitro can also be detected in vivo and could thus potentially be used as tracks to disseminate. Indeed, co-injection with CAFs caused a 15% increase in the number of circulating ras0.5-GFPLux cells, while co-injection with RMFs caused a 38% decrease in the number of circulating ras0.5-GFPLux cells relative to the cells injected alone (Figure 4C). An even greater differential increase in dissemination in the presence of CAFs relative to RMFs (2.4-fold) was observed when fibroblasts were injected into the cleared mammary fat pads of mice before injection of the ras0.5-GFPLux cells (to mimic our in vitro matrix overlay experiments; Figure 4D).
Figure 4.

CAFs deposit aligned matrix and promote the dissemination of premalignant cells in vivo. (A) Bioluminescence imaging, hematoxylin and eosin (H&E), lamin A/C, and picrosirius staining of mammary glands harvested 3 weeks following orthotopic injection of 1 x 106 ras0.5-GFPLux alone or in combination with 3 x 106 fibroblasts. All images were captured at 10x (higher 40x magnification images are available in Figure W7). The location of ras0.5-GFPLux cells is indicated by arrows in the H&E and outlined with a white dashed line in the picrosirius image. (B) Bar graphs represent the averages ± SEM of mammary ECM abundance and alignment obtained through automated image analysis (alone, n = 18; RMF163, n = 14; CAF559, n = 18). Student's t test comparing ECM abundance: alone versus RMF163 (P = .4), alone versus CAF559 (P < .001), RMF163 versus CAF559 (P = .03); comparing ECM alignment: alone versus RMF163 (P = .4), alone versus CAF559 (P < .001), RMF163 versus CAF559 (P < .001). (C and D) qPCR analysis of EpCAM levels in genomic DNA extracted from blood collected 3 weeks following: (C) injection of 1 x 106 ras0.5-GFPLux cells alone (n = 4), or in combination with 3 x 106 RMF163 (n = 4), or3 x 106 CAF559 (n = 8). Student's t test comparing qPCR replicates from alone versus RMF163 (P = .04), alone versus CAF559 (P = .4), RMF163 versus CAF559 (P = .003); and (D) injection of 2.5 x 106 ras0.5-GFPLux cells into RMF163 (n = 4) or CAF559 (n = 4) pre-injected fat pads. Student's t test comparing qPCR replicate values of nanogram EpCAM per milliliter of blood collected from mice in the RMF163 group versus CAF559 group (P = .03). Statistically significant differences between groups are denoted with asterisk.
To determine whether a CAF's ability to promote dissemination correlated with its ability to induce the aligned mesenchymal phenotype, we examined whether a CAF (CAF304) that failed to induce the mesenchymal phenotype in co-cultures with ras0.5-GFPLux cells could enhance dissemination. For these experiments, ras0.5-GFPLux cells were injected orthotopically either alone or in combination with CAF304, and blood was collected 2 and 5 weeks following injection of the cells. No difference in the number of circulating ras0.5-GFPLux cells was detected between the two groups at either time point (Figure W8), suggesting that dissemination correlates with the induction of the aligned mesenchymal phenotype and that the behavior of CAFs in vitro is biologically relevant in vivo.
RMFs Suppress, while CAFs Promote, the Tumorigenic and Metastatic Potential of Malignant Cells In Vivo
Having observed that RMFs could suppress, and CAFs could promote, the dissemination of premalignant cells in vivo, we examined whether they could similarly alter the tumorigenic and metastatic potential of malignant cells in vivo. To address this question, we chose to use the metastatic MDA-MB-231 breast cancer cells as a malignant model. Although these cells lack E-cadherin expression and are already somewhat mesenchymal, they became elongated and aligned in a parallel pattern as though they were lining up on tracks when they were co-cultured with CAFs (just like the vHMEC-ras0.5 cells did; see Figure W9). Hence, we reasoned that if the MDA-MB-231 cells could use the aligned ECM deposited by CAFs as tramlines to disseminate, their metastatic potential could be enhanced. To test this, we engineered the MDA-MB-231 cells to express Lux (231-Lux) so that their behavior could be monitored by live bioluminescence imaging. The 231-Lux cells were then injected orthotopically either alone or in combination with RMFs isolated from four different individuals or CAFs isolated from three different individuals. All but 2 of the 131 mice injected developed tumors and did so with a latency of ∼3 to 4 weeks (Table 2). Hence, the fibroblasts did not have much of an effect on tumor incidence or latency. However, all four RMFs significantly inhibited primary tumor growth (Figure 5A) resulting in an approximate 30% to 50% decrease in final tumor volume and weight (Figure 5B). In contrast, each CAF had a different effect on primary tumor growth (Figure 5A). Specifically, CAF1366 had little or no effect on final tumor volume, yet it caused a small (15%) but statistically significant (P = .02) increase in final tumor weight(Figure 5B). CAF559 caused an ∼30% increase, while CAF1363 caused an ∼30% decrease, in final tumor volume and weight (Figure 5B).
Table 2.
Summary of the Effect of Fibroblasts on Primary Tumor Formation.
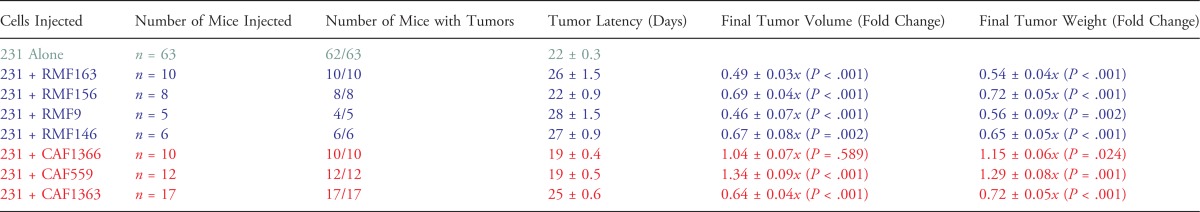 |
231-Lux cells were injected orthotopically either alone (5 x 105) or in combination with 1.5 x 106 fibroblasts, as indicated, and tumors were harvested 8 weeks following injection. P values correspond to Student's t test comparing the fold change in final tumor volume or weight x SEM in the presence of each fibroblast relative to the 231-Lux cells alone.
Figure 5.
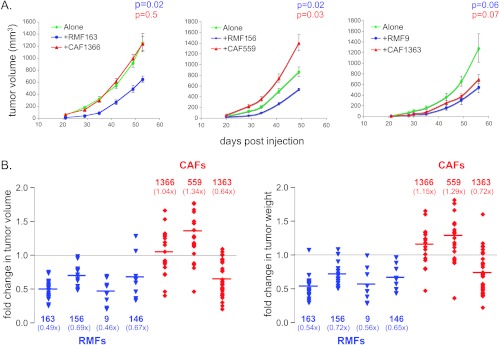
RMFs and CAFs differentially modulate primary tumor growth. (A) Tumor growth curves of 231-Lux cells alone (green) or in combination with RMFs (blue) or CAFs (red). Representative experiments performed with RMFs and CAFs isolated from three different individuals each are shown. P values correspond to Student's t test comparing growth curves in the presence of each fibroblast relative to 231-Lux cells alone. Experiments with each RMF and CAF were performed at least twice. (B) Graph of fold change in final tumor volume and weight in the presence of RMFs isolated from four different individuals (in blue from left to right: 163, 156, 9, 146) or CAFs isolated from three different individuals (in red from left to right: 1366, 559, 1363) relative to the 231-Lux cells alone for all experiments combined. The number of mice in each group and the P values comparing the effects of each RMF and CAF on tumor volume and weight relative to the 231-Lux cells alone are shown in Table 2.
In a previous study where CAFs were injected in combination with the MCF7-ras breast cancer cell line, all CAFs examined stimulated tumor growth and did so through their ability to secrete SDF-1 [24]. Hence, we examined the expression of SDF-1 in our fibroblasts and found that it was expressed at low levels in all the fibroblasts except in CAF559, which was the only CAF that stimulated the growth of 231-Lux cells in our experiments (Figure W10). Hence, our results are in agreement with previous reports and, in addition, highlight the fact that there is considerable heterogeneity between CAFs, consistent with the intratumoral and intertumoral heterogeneity observed in human breast cancer [25].
Because 231-Lux cells metastasize predominantly to the lung, to assess the impact of the fibroblasts on metastatic potential, lungs were harvested 8 weeks following injection of the tumor cells and imaged ex vivo to accurately quantify total metastatic burden by bioluminescence. In addition, surface lung metastases were counted using a dissection scope (Figure W11). Although the number of mice that developed lung metastases was similar between groups, the number of metastases per lung differed. As shown in Figure 6A, and summarized in Table 3, two of the four RMFs (RMF163 and RMF9) significantly decreased the number of surface metastases per lung, while all three CAFs increased the number of surface metastases per lung. Quantification of lung luminescence indicated that all CAFs caused a four-fold to seven-fold increase in total pulmonary metastatic burden relative to tumor cells injected alone (Figure 6B and Table 3). Remarkably, even CAF1363, which caused a decrease in final tumor weight and volume, increased both the number of surface lung metastases (2.3-fold, P = .022) and total pulmonary metastatic burden (6.9-fold, P = .015).
Figure 6.

RMFs suppress, while CAFs enhance, the metastatic potential of malignant cells in vivo. Graphs of fold change in (A) the number of metastatic foci per lung or (B) lung luminescence as a measure of total pulmonary metastatic burden in the presence of RMFs (in blue from left to right: 163, 156, 9, 146) or CAFs (in red from left to right: 1366, 559, 1363) relative to the 231-Lux cells alone for all experiments combined. The number of mice in each group and the P values comparing the effects of each RMF and CAF on metastatic burden relative to the 231-Lux cells alone are shown in Table 3.
Table 3.
Summary of the Effect of Fibroblasts on Lung Metastasis.
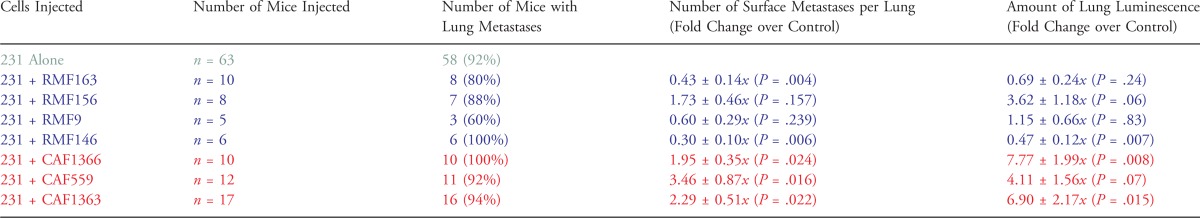 |
231-Lux cells were injected orthotopically either alone (5 x 105) or in combination with 1.5 x 106 fibroblasts, as indicated, and lungs were harvested for analysis 8 weeks following injection. P values correspond to the results of Student's t test comparing the fold change in the number of metastatic foci per lung ± SEM in the presence of each fibroblast relative to the 231-Lux cells alone as well as the fold change in lung luminescence ± SEM as a measure of total pulmonary metastatic burden in the presence of each fibroblast relative to the 231-Lux cells alone.
In light of these data, we wondered whether the primary tumors that grew in the presence of CAF1363 might be smaller not because CAF1363 inhibited their proliferation or survival, but rather because it actively promoted their dissemination into the circulation. To address this question, we 1) assessed the proliferative status of the primary tumors by Ki67 staining and apoptotic cell death by CC3 staining, 2) examined whether the aligned ECM deposited by CAFs in vitro could also be detected in vivo, and 3) collected blood 8 weeks following injection of 231-Lux cells with or without CAF1363 to quantify the number of CTCs. A group of mice injected with RMF146 was used for comparison in these analyses. Quantification of Ki67 and CC3 staining indicated that CAF1363 did not inhibit the proliferation of tumors nor did it promote apoptosis (Figure 7, A and B). In contrast, RMF146 promoted apoptosis but had no effect on proliferation. Picrosirius staining of the tumors indicated that aligned ECM could, in fact, be detected in tumors that arose in the presence of CAF1363 and differed from the sparser more randomly organized ECM present in tumors that arose when the cells were injected alone or in the presence of RMF146 (Figure 7C), suggesting that the tumor cells could potentially use the aligned ECM to migrate away from the tumors and into the circulation. Indeed, CAF1363 caused a 2.1-fold increase in the number of CTCs relative to the cells injected alone, while RMF146 caused a decrease in the number of CTCs, but this decrease was not statistically significant (Figure 7D). Collectively, these data suggest that the tumors that grew in the presence of CAF1363 may be smaller due to increased dissemination facilitated by the aligned ECM present in these tumors.
Figure 7.

CAFs deposit aligned matrix and promote dissemination of malignant cells in vivo. Representative 20x images of tumors stained with (A) Ki67, (B) CC3, or (C) picrosirius, grown in the absence (alone) or presence of RMF146 or CAF1363, as indicated. Staining was quantified using three to four randomly chosen fields from each tumor section. Alone (n = 34, 15, 10), RMF146 (n = 12, 8, 8), and CAF1363 (n = 22, 12, 7) for stains shown in A, B, and C, respectively. Quantification of stains is shown in bar graphs as average ± SEM for each group. P values correspond to Student's t test comparing scores of tumors grown in the presence or absence of fibroblasts. For Ki67: alone versus RMF146 (P = .11), alone versus CAF1363 (P = .72); for CC3: alone versus RMF146 (P <.001), alone versus CAF1363 (P = .8); for ECM abundance: alone versus RMF146 (P = .09), alone versus CAF1363 (P < .001); for ECM alignment: alone versus RMF146 (P = .04), alone versus CAF1363 (P < .001). (D) qPCR analysis of EpCAM levels in genomic DNA extracted from blood collected 8 weeks following injection of 5 x 105 231-Lux cells alone (n = 9) or in combination with 1.5 x 106 RMF146 (n = 4), or 1.5 x 106 CAF1363 (n = 7). Student's t test comparing qPCR replicates from alone versus RMF146 (P = .15), alone versus CAF1363 (P = .04), and RMF146 versus CAF1363 (P = .01). (E) Representative immunoblots of Fn, TNC, biglycan, and LOX expression in MDA-MB-231 cells (231), RMFs (163, 156, 009, 146, Mes), and CAFs (1366, 559, 1363). “Mes” denotes an RMF that induced the aligned mesenchymal phenotype. Statistically significant differences between endpoints measured in tumors grown alone versus with fibroblasts are denoted with asterisk.
Consistent with these latter observations, immunoblot analysis of ECM assembly proteins expressed in RMFs and CAFs revealed that, relative to all the fibroblasts examined, CAF1363 secreted the highest levels of 1) Fn, 2) biglycan, a matrix assembly protein whose targeted disruption results in abnormal collagen fibril morphology [26], and 3) LOX, an enzyme that plays a critical role in the cross-linking of ECM proteins, the formation of invadopodia, migration, and metastasis [27] (Figure 7E). Hence, given their role in ECM assembly and metastasis, the high levels of biglycan and LOX expression in CAF1363 could account for the increased dissemination and abundance of aligned ECM present in the tumors that arose in the presence of CAF1363.
In addition to being most highly expressed in CAF1363, Fn, LOX, as well as the matrix protein, TNC, were also more highly expressed in the fibroblasts that induced the aligned mesenchymal phenotype relative to those that did not. This was true not only for the CAFs that induced the aligned mesenchymal phenotype but also for the one RMF (referred to as RMF-Mes) that induced the mesenchymal phenotype (Table 1 and Figures 7E and 8A).
Figure 8.
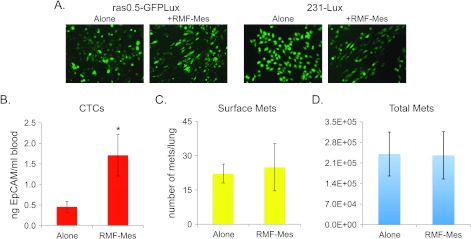
Induction of the aligned mesenchymal phenotype by an RMF enhances dissemination but is not sufficient to promote metastasis. (A) Representative 10x images of ras0.5-GFPLux and 231-Lux cells cultured in the absence (alone) or presence of RMF-Mes (+RMF-Mes) for 3 days. (B) qPCR analysis of EpCAM levels in genomic DNA extracted from blood shown as average ± SEM. (C) Graph depicting the average number of surface metastases per lung ± SEM. (D) Graph depicting the quantification of lung luminescence ± SEM as a measure of total metastatic burden. All measurements shown in B to D were taken 8 weeks following injection of 5 x 105 231-Lux cells alone (n =17) or in combination with 1.5 x 106 RMF-Mes (n = 12). Student's t test comparing results obtained in the presence of RMF-Mes relative to the 231-Lux cells alone (CTCs, P = .007; surface metastases, P = .8; total metastatic burden, P = .9). Statistically significant difference is denoted with asterisk.
An RMF that Induces an Aligned Mesenchymal Phenotype Can Enhance Dissemination but Not Metastasis
Having observed that CAFs that can induce an aligned mesenchymal phenotype in vitro could promote the dissemination and metastasis of tumor cells in vivo, we then examined whether an RMF that induced an aligned mesenchymal phenotype in vitro (Figure 8A) could also promote dissemination and metastasis. To address this question, 231-Lux cells were injected orthotopically either alone or in combination with RMF-Mes. Eight weeks following injection, blood was collected for the quantification of CTCs, and lungs were harvested for the quantification of surface metastases and lung bioluminescence. In these experiments, RMF-Mes enhanced the dissemination of 231-Lux cells as evidenced by the detection of a 3.8-fold higher number of CTCs in the blood of mice injected with RMF-Mes relative to those injected with 231-Lux cells alone (Figure 8B). However, the presence of RMF-Mes did not increase the number of surface metastases per lung or total pulmonary metastatic burden (Figure 8, C and D). These data indicate that the induction of an aligned mesenchymal phenotype alone is sufficient to promote early dissemination but not sufficient to enhance the formation of overt metastases.
Discussion
In this report, we have shown that both premalignant (vHMEC-ras0.5) and malignant (MDA-MB-231) mammary epithelial cells can assume an aligned mesenchymal morphology when co-cultured with CAFs but not RMFs. This phenotype differs from the classic epithelial-to-mesenchymal transition that is accompanied by a down-regulation of E-cadherin. Instead, this mesenchymal phenotype appears to be governed at least in part by the organization of the ECM deposited by CAFs in that it was not induced by CAF-conditioned media or CAFs in transwell co-cultures with the epithelial cells where the two cell populations were not in contact with one another, but it was induced when the epithelial cells were overlaid on top of ECM deposited by CAFs, and it was blocked in co-cultures when deposition of ECM was blocked. Thus, while it is well known that some CAF-induced phenotypes can be transmitted by secreted factors [24], this particular CAF-induced phenotype requires contact with deposited ECM proteins.
Importantly, our data indicate that the phenotype induced by CAFs in vitro is biologically relevant in vivo in that the aligned ECM deposited by CAFs in vitro could be detected in the mammary glands of mice following co-injection with premalignant cells as well as in tumors following co-injection with malignant cells. This ECM phenotype is also clinically relevant in that studies have shown that the radial alignment of collagen fibers relative to tumors facilitates local invasion [15], and correlates with poor disease-free survival, suggesting that quantifying collagen alignment could be a viable, novel strategy for the prediction of human breast cancer survival [16]. Our results reinforce this notion and, in addition, suggest that the presence of aligned collagen could also predict dissemination in the earliest stages of breast cancer development.
There is increasing evidence that in some cancers, tumor cells start spreading long before the primary tumor is detected and removed [2,28]. Our data suggest that alterations in the composition and organization of the ECM surrounding premalignant lesions could account for this. Alterations in the ECM component of the stroma that resemble those observed in malignant lesions are often observed in the context of wound healing, radiation response, pregnancy-associated involution, and high mammographic density [29–33]. These alterations manifest even in the absence of malignancy and may thus provide a favorable environment for early dissemination of premalignant cells. This might explain why certain cancers that are driven by alterations in ECM, such as postpartum breast cancers, have such poor prognosis [32] and why metastatic dissemination can occur in women with small tumors.
Our data suggest that alterations in stromal ECM that result in the induction of an aligned mesenchymal phenotype are both necessary and sufficient to enhance dissemination because a CAF that failed to induce the phenotype did not enhance dissemination (Figure W8) and an RMF that did induce the mesenchymal phenotype did (Figure 8). However, unlike the induction of the aligned mesenchymal phenotype by a CAF, which enhanced both dissemination and metastasis, the aligned mesenchymal phenotype induced by an RMF enhanced dissemination but not metastasis. These data indicate that reorganization of the ECM plays a key role in the early stages of the metastatic cascade but is not sufficient to bestow on tumor cells a fully enhanced metastatic potential and suggest that CAFs induce additional alterations in tumor cells that endow them with the ability to metastasize more efficiently. Notably, the alterations that drive metastatic dissemination are uncoupled from those that drive primary tumor growth as all three CAFs co-injected with tumor cells increased pulmonary metastatic burden (Figure 6 and Table 3) whether they stimulated, inhibited, or failed to affect primary tumor growth (Figure 5 and Table 2). These data highlight the heterogeneity of signals emanating from the tumor microenvironment.
Importantly, the composition and organization of the ECM cannot only promote the tumorigenic and metastatic potential of malignant cells, but it can also constrain it, as RMFs isolated from four different disease-free individuals inhibited primary tumor growth (Figure 5 and Table 2) and one of these also inhibited metastatic spread to the lung (Figure 6 and Table 3). In addition, RMFs also inhibited the dissemination of premalignant cells (Figure 4). These data imply that maintaining a normal ECM environment can be therapeutically beneficial. Along these lines, others have shown that normalizing a matrix-rich desmoplastic stroma can enhance the delivery and efficacy of chemotherapy [34,35]. In one study, this was achieved with the antihypertensive drug losartan, which depletes ECM in part by suppressing active TGFβ levels [35,36]. Consistent with this, we observed that blocking TGFβ signaling as well as other signaling pathways through which TGFβ induces a mesenchymal phenotype and ECM remodeling prevented the CAF-induced mesenchymal phenotype in co-culture assays. TGFβ's role in promoting metastasis is well established; however, targeting TGFβ in the clinic has been a challenge due to the complex nature of TGFβ's tumor suppressive and tumor promoting actions. Losartan may represent a safe alternative to indirectly inhibit some of the ECM-related pro-metastatic effects of TGFβ. In fact, losartan has been used clinically to abrogate excess TGFβ in patients with Marfan's syndrome, a genetic disorder caused by mutations in the ECM protein fibrillin-1, which binds to the large latent TGFβ complex [37,38]. Antihypertensive therapeutics such as losartan could potentially also be used prophylactically, as there is evidence that women on anti-hypertensive therapy have a lower risk of developing breast cancer [39].
In conclusion, our data underscore the potential of the ECM as a prognostic and therapeutic target. Targeting ECM proteins, such as biglycan and LOX, that are involved in the organization and cross-linking of collagen may prove effective in limiting dissemination, an event that may occur much earlier than previously appreciated. Hence, the possibility that cells may have disseminated long before the detection of the primary tumor must be considered when designing experiments and testing new therapeutics. To make personalized cancer medicine a success, companion diagnostics aimed at identifying the right patient populations for the right therapeutics should also include markers to assess early systemic cancer spread.
Supplementary Materials
Quantification of Regularity Index
This section summarizes the computational protocol for capturing intrinsic geometric patterns in collagen formation for treatment groups. Toward this goal, we derived two indices corresponding to the amount of collagen and its regularity in each sample because both of these indices can be associated to invasive properties. The main challenge is quantifying the regularity index, because 1) there could be multiple collagen bundles in a sample, 2) collagen bundles are often perceptual and lack continuity as a result of either sample preparation or microscopy, and 3) each bundle may have a different density and/or strength. Therefore, we have designed a protocol with filters that 1) fill perceptual gaps, 2) compute regularity locally, through steerable filters, and then aggregate the local filter responses over the entire image, and 3) are rotationally invariant so that the fiber bundles in orthogonal directions remain additive. The details of these computational steps can be found in our earlier publications and those of others. The protocol is a cascade of operators, shown in Figure W2, and consists of:
Converting the original image from RGB space to grayscale space. This step simplifies 1) the collagen content into one dimension and 2) the application of steerable filters for local feature extraction.
Detecting collagen signals through Otsu thresholding [1]. This step differentiates the signal (foreground) from the background, and it enables the computation of percentage of collagen content per image and per treatment group.
Applying perceptual grouping and regularization through tangential voting [2], where voting is only applied to the thresholded foreground pixels. As a result, only the foreground pixels can cast their information (e.g., orientation) into a local neighborhood. The net result is a more regularized structure, where small gaps are filled.
- Calculating the regularity index for each foreground pixel from the responses from the steerable filter bank [2], where the filters are oriented (e.g., having an ellipsoidal shape) as first or second derivatives of Gaussians. The regularity index, RI for a pixel, p, is defined as follows:
where std{…} corresponds to the standard deviation of the local responses; MSFRNθi(p) is the mean steerable filter response along orientation θi, for pixel p, within a local neighborhood of N; and k is the total number of orientations forthe steerablefilterbank. The rationale for this design index is that random (irregular) structures do not have preferred orientation, which leads to relatively invariant filter responses among different orientations; thus, the regularity index is low. This concept is demonstrated in Figure W3.(1) - Calculating the regularity composition index per image, where regular and irregular structures are differentiated through a simple thresholding policy. The threshold, T, is determined empirically by learning from the spatial distribution of the fiber bundles. In addition, the regularity composition index is simply the percentage of regular signals in an image, e.g.,
(2)
Having validated our approach on synthetic images, we then apply our approach on the real data. The parameter settings were kept constant for all experimental factors.
Local neighborhood: N = 300 by 300 pixels;
Number of orientations: k = 16;
Scale for steerable filter: σ = 6.0;
Threshold for regularity: T = 600.
Several examples are shown in Figure W4.
Acknowledgments
We thank members of the Tlsty laboratory, particularly, Philippe Gascard, for thoughtful discussions and assistance with animal studies as well as Alfred Au (University of California, San Francisco), Stefanie Jeffrey (Stanford), and the staff of Great Lakes Pathology (Milwaukee, WI) for their assistance with tumor procurement. We also thank Somdutta Roy, Mahvash Sigaroudinia, and Tara Rambaldo for assistance with cell sorting, Jianxin Zhao for performing the lamin stain, and the University of California, San Francisco and Gladstone Institute Immunohistochemistry Core Facilities for performing all other immunohistochemical stains.
Abbreviations
- ECM
extracellular matrix
- CAFs
carcinoma-associated fibroblasts
- RMFs
reduction mammoplasty fibroblasts
- HMECs
human mammary epithelial cells
- vHMEC
variant HMEC with repressed p16INK4A
- DCIS
ductal carcinoma in situ
- IDC
invasive ductal carcinoma
- FACS
fluorescence-activated cell sorting
- GFP
green fluorescent protein
- Lux
luciferase
- CTCs
circulating tumor cells
- EpCAM
epithelial cell adhesion molecule
- TGFβ
transforming growth factor-β
- TβRKi
TGFβ receptor kinase inhibitor
- ROCK
Rho-associated kinase
- SDF-1
stromal-derived factor-1
- CC3
cleaved caspase 3
- Fn
fibronectin
- LOX
lysyl oxidase
- TNC
tenascin C
Footnotes
This work was supported by the Susan G. Komen Foundation, postdoctoral award PDF124906 to N.D., National Institutes of Health/National Cancer Institute PO1 CA107584 to T.D.T. and B.P. (under LBNL contract DE-AC02-05CH11231), NIH/NCI R01 CA097214 to T.D.T., and NIH/NCI U54 CA143803 to T.D.T. and B.P. (University of California, Riverside).
This article refers to supplementary materials, which are designated by Figures W1 to W11 and are available online at www.neoplasia.com.
References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Husemann Y, Geigl JB, Schubert F, Musiani P, Meyer M, Burghart E, Forni G, Eils R, Fehm T, Riethmuller G, et al. Systemic spread is an early step in breast cancer. Cancer Cell. 2008;13:58–68. doi: 10.1016/j.ccr.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 3.Allen M, Louise Jones J. Jekyll and Hyde: the role of the micro-environment on the progression of cancer. J Pathol. 2011;223:162–176. doi: 10.1002/path.2803. [DOI] [PubMed] [Google Scholar]
- 4.Hynes RO. The extracellular matrix: not just pretty fibrils. Science. 2009;326:1216–1219. doi: 10.1126/science.1176009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Haviv I, Polyak K, Qiu W, Hu M, Campbell I. Origin of carcinoma associated fibroblasts. Cell Cycle. 2009;8:589–595. doi: 10.4161/cc.8.4.7669. [DOI] [PubMed] [Google Scholar]
- 6.Zeisberg EM, Potenta S, Xie L, Zeisberg M, Kalluri R. Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res. 2007;67:10123–10128. doi: 10.1158/0008-5472.CAN-07-3127. [DOI] [PubMed] [Google Scholar]
- 7.Mishra PJ, Humeniuk R, Medina DJ, Alexe G, Mesirov JP, Ganesan S, Glod JW, Banerjee D. Carcinoma-associated fibroblast-like differentiation of human mesenchymal stem cells. Cancer Res. 2008;68:4331–4339. doi: 10.1158/0008-5472.CAN-08-0943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bergamaschi A, Tagliabue E, Sorlie T, Naume B, Triulzi T, Orlandi R, Russnes HG, Nesland JM, Tammi R, Auvinen P, et al. Extracellular matrix signature identifies breast cancer subgroups with different clinical outcome. J Pathol. 2008;214:357–367. doi: 10.1002/path.2278. [DOI] [PubMed] [Google Scholar]
- 9.Finak G, Bertos N, Pepin F, Sadekova S, Souleimanova M, Zhao H, Chen H, Omeroglu G, Meterissian S, Omeroglu A, et al. Stromal gene expression predicts clinical outcome in breast cancer. Nat Med. 2008;14:518–527. doi: 10.1038/nm1764. [DOI] [PubMed] [Google Scholar]
- 10.Dumont N, Crawford YG, Sigaroudinia M, Nagrani SS, Wilson MB, Buehring GC, Turashvili G, Aparicio S, Gauthier ML, Fordyce CA, et al. Human mammary cancer progression model recapitulates methylation events associated with breast premalignancy. Breast Cancer Res. 2009;11:R87. doi: 10.1186/bcr2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Proia DA, Kuperwasser C. Reconstruction of human mammary tissues in a mouse model. Nat Protoc. 2006;1:206–214. doi: 10.1038/nprot.2006.31. [DOI] [PubMed] [Google Scholar]
- 12.Dumont N, Wilson MB, Crawford YG, Reynolds PA, Sigaroudinia M, Tlsty TD. Sustained induction of epithelial to mesenchymal transition activates DNA methylation of genes silenced in basal-like breast cancers. Proc Natl Acad Sci USA. 2008;105:14867–14872. doi: 10.1073/pnas.0807146105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Amatangelo MD, Bassi DE, Klein-Szanto AJ, Cukierman E. Stromaderived three-dimensional matrices are necessary and sufficient to promote desmoplastic differentiation of normal fibroblasts. Am J Pathol. 2005;167:475–488. doi: 10.1016/S0002-9440(10)62991-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Damianova R, Stefanova N, Cukierman E, Momchilova A, Pankov R. Three-dimensional matrix induces sustained activation of ERK1/2 via Src/Ras/Raf signaling pathway. Cell Biol Int. 2008;32:229–234. doi: 10.1016/j.cellbi.2007.08.029. [DOI] [PubMed] [Google Scholar]
- 15.Provenzano PP, Eliceiri KW, Campbell JM, Inman DR, White JG, Keely PJ. Collagen reorganization at the tumor-stromal interface facilitates local invasion. BMC Med. 2006;4:38. doi: 10.1186/1741-7015-4-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Conklin MW, Eickhoff JC, Riching KM, Pehlke CA, Eliceiri KW, Provenzano PP, Friedl A, Keely PJ. Aligned collagen is a prognostic signature for survival in human breast carcinoma. Am J Pathol. 2011;178:1221–1232. doi: 10.1016/j.ajpath.2010.11.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Davies M, Robinson M, Smith E, Huntley S, Prime S, Paterson I. Induction of an epithelial to mesenchymal transition in human immortal and malignant keratinocytes by TGF-β1 involves MAPK, Smad and AP-1 signalling pathways. J Cell Biochem. 2005;95:918–931. doi: 10.1002/jcb.20458. [DOI] [PubMed] [Google Scholar]
- 18.Alcorn JF, Guala AS, van der Velden J, McElhinney B, Irvin CG, Davis RJ, Janssen-Heininger YM. Jun N-terminal kinase 1 regulates epithelial-to-mesenchymal transition induced by TGF-β1. J Cell Sci. 2008;121:1036–1045. doi: 10.1242/jcs.019455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hocevar BA, Brown TL, Howe PH. TGF-β induces fibronectin synthesis through a c-Jun N-terminal kinase-dependent, Smad4-independent pathway. EMBO J. 1999;18:1345–1356. doi: 10.1093/emboj/18.5.1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rodriguez-Barbero A, Obreo J, Yuste L, Montero JC, Rodriguez-Pena A, Pandiella A, Bernabeu C, Lopez-Novoa JM. Transforming growth factor-β1 induces collagen synthesis and accumulation via p38 mitogen-activated protein kinase (MAPK) pathway in cultured L6E9 myoblasts. FEBS Lett. 2002;513:282–288. doi: 10.1016/s0014-5793(02)02337-2. [DOI] [PubMed] [Google Scholar]
- 21.Lane J, Martin TA, Watkins G, Mansel RE, Jiang WG. The expression and prognostic value of ROCK I and ROCK II and their role in human breast cancer. Int J Oncol. 2008;33:585–593. [PubMed] [Google Scholar]
- 22.Liu S, Goldstein RH, Scepansky EM, Rosenblatt M. Inhibition of rho-associated kinase signaling prevents breast cancer metastasis to human bone. Cancer Res. 2009;69:8742–8751. doi: 10.1158/0008-5472.CAN-09-1541. [DOI] [PubMed] [Google Scholar]
- 23.Wang W, Wyckoff JB, Frohlich VC, Oleynikov Y, Huttelmaier S, Zavadil J, Cermak L, Bottinger EP, Singer RH, White JG, et al. Single cell behavior in metastatic primary mammary tumors correlated with gene expression patterns revealed by molecular profiling. Cancer Res. 2002;62:6278–6288. [PubMed] [Google Scholar]
- 24.Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, Carey VJ, Richardson AL, Weinberg RA. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 2005;121:335–348. doi: 10.1016/j.cell.2005.02.034. [DOI] [PubMed] [Google Scholar]
- 25.Berman HK, Gauthier ML, Tlsty TD. Premalignant breast neoplasia: a paradigm of interlesional and intralesional molecular heterogeneity and its biological and clinical ramifications. Cancer Prev Res (Phila) 2010;3:579–587. doi: 10.1158/1940-6207.CAPR-10-0073. [DOI] [PubMed] [Google Scholar]
- 26.Ameye L, Aria D, Jepsen K, Oldberg A, Xu T, Young MF. Abnormal collagen fibrils in tendons of biglycan/fibromodulin-deficient mice lead to gait impairment, ectopic ossification, and osteoarthritis. FASEB J. 2002;16:673–680. doi: 10.1096/fj.01-0848com. [DOI] [PubMed] [Google Scholar]
- 27.Xiao Q, Ge G. Lysyl oxidase, extracellular matrix remodeling and cancer metastasis. Cancer Microenviron. 2012;5:261–273. doi: 10.1007/s12307-012-0105-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pantel K, Alix-Panabieres C, Riethdorf S. Cancer micrometastases. Nat Rev Clin Oncol. 2009;6:339–351. doi: 10.1038/nrclinonc.2009.44. [DOI] [PubMed] [Google Scholar]
- 29.Tlsty TD, Coussens LM. Tumor stroma and regulation of cancer development. Annu Rev Pathol. 2006;1:119–150. doi: 10.1146/annurev.pathol.1.110304.100224. [DOI] [PubMed] [Google Scholar]
- 30.Schedin P, O'Brien J, Rudolph M, Stein T, Borges V. Microenvironment of the involuting mammary gland mediates mammary cancer progression. J Mammary Gland Biol Neoplasia. 2007;12:71–82. doi: 10.1007/s10911-007-9039-3. [DOI] [PubMed] [Google Scholar]
- 31.Guo YP, Martin LJ, Hanna W, Banerjee D, Miller N, Fishell E, Khokha R, Boyd NF. Growth factors and stromal matrix proteins associated with mammographic densities. Cancer Epidemiol Biomarkers Prev. 2001;10:243–248. [PubMed] [Google Scholar]
- 32.Lyons TR, O'Brien J, Borges VF, Conklin MW, Keely PJ, Eliceiri KW, Marusyk A, Tan AC, Schedin P. Postpartum mammary gland involution drives progression of ductal carcinoma in situ through collagen and COX-2. Nat Med. 2011;17:1109–1115. doi: 10.1038/nm.2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.DeFilippis RA, Chang H, Dumont N, Rabban JT, Chen YY, Fontenay GV, Berman HK, Gauthier ML, Zhao J, Hu D, et al. CD36 repression activates a multicellular stromal program shared by high mammographic density and tumor tissues. Cancer Discov. 2012;2:826–839. doi: 10.1158/2159-8290.CD-12-0107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, Madhu B, Goldgraben MA, Caldwell ME, Allard D, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324:1457–1461. doi: 10.1126/science.1171362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Diop-Frimpong B, Chauhan VP, Krane S, Boucher Y, Jain RK. Losartan inhibits collagen I synthesis and improves the distribution and efficacy of nanotherapeutics in tumors. Proc Natl Acad Sci USA. 2011;108:2909–2914. doi: 10.1073/pnas.1018892108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Arnold SA, Rivera LB, Carbon JG, Toombs JE, Chang CL, Bradshaw AD, Brekken RA. Losartan slows pancreatic tumor progression and extends survival of SPARC-null mice by abrogating aberrant TGFβ activation. PLoS One. 2012;7:e31384. doi: 10.1371/journal.pone.0031384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brooke BS, Habashi JP, Judge DP, Patel N, Loeys B, Dietz HC., III Angiotensin II blockade and aortic-root dilation in Marfan's syndrome. N Engl J Med. 2008;358:2787–2795. doi: 10.1056/NEJMoa0706585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kalluri R, Han Y. Targeting TGF-β and the extracellular matrix in Marfan's syndrome. Dev Cell. 2008;15:1–2. doi: 10.1016/j.devcel.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 39.Lever AF, Hole DJ, Gillis CR, McCallum IR, McInnes GT, MacKinnon PL, Meredith PA, Murray LS, Reid JL, Robertson JW. Do inhibitors of angiotensin-I-converting enzyme protect against risk of cancer? Lancet. 1998;352:179–184. doi: 10.1016/S0140-6736(98)03228-0. [DOI] [PubMed] [Google Scholar]
References
- 1.Otsu N. A threshold selection method from gray-level histograms. IEEE Trans Syst Man Cyber. 1979;9(1):62–66. [Google Scholar]
- 2.Han J, Chang H, Andarawewa K, Yaswen P, Barcellos-Hoff MH, Parvin B. Multidimensional profiling of cell surface proteins and nuclear markers. IEEE/ACM Trans Comput Biol Bioinform. 2010;7(1):80–90. doi: 10.1109/TCBB.2008.134. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.