

Abstract
Aim
We sought to define the mechanism of total body irradiation (TBI)-induced seizures in NOS1−/− mice and amelioration by intra-esophageal manganese superoxide dismutase-plasmid liposomes (MnSOD-PL).
Materials and Methods
We evaluated the role of vagus nerve pathways in irradiation-induced seizures using biochemical, physiologic, and histopathologic techniques.
Results
Heterozygous NOS1+/− mice demonstrated radioresistance similar to wild-type C57BL/6NHsd mice (p=0.9269). Irradiation-induced lipid peroxidation in fetal brain cultures from NOS1−/− or wild-type mice was reduced by MnSOD-PL. Right-sided vagotomy did not alter the TBI radiation response of wild-type or reverse the radiosensitivity of NOS1−/− mice. Excised esophagus from irradiated NOS1−/− mice demonstrated an increased histopathologic inflammatory response compared to C57BL/6NHsd mice.
Conclusion
NOS1−/− mice represent a model system for dissecting the developmental abnormalities leading to esophageal-mediated TBI-induced seizures.
Keywords: NOS1−/− mice, vagus nerve, irradiation-induced seizures
Nitric oxide synthase-1 homozygous recombinant negative (NOS1−/−) mice demonstrate multiple congenital abnormalities associated with developmental deficiency of NOS1 neurons (1), including abnormalities in the cerebellum, adrenal medulla, and a pattern of esophageal dilation similar to pediatric pyloric stenosis (1). In addition, NOS1−/− mice demonstrate behavioral abnormalities and propensity for seizures (1). We recently reported that NOS1−/− mice demonstrate upper body or total body irradiation-induced rapid death, not associated with the known organ system failures that define the hematopoietic or gastrointestinal syndrome (2). NOS1−/− mice, distinct from NOS2−/− or NOS3−/− mice demonstrate fatal total irradiation-induced seizures (2) and cardiac conduction abnormalities (3). The neuro-anatomic developmental abnormalities, including esophageal dilation, in NOS1−/− mice led us to test for a radiotherapeutic effect of swallowed Manganese Superoxide Dismutase (MnSOD-PL) (4–6), and we reported that this treatment reduced irradiation-induced seizures and improved survival (2).
In the present studies, we evaluated differences in irradiation response of fetal brain neuron cultures and the role of the known esophageal-central nervous system neuro-anatomic pathways, specifically of the vagus nerve (7–10), in irradiation-induced seizures. We evaluated the effect of intraesophageal MnSOD-PL in ameliorating seizures in vagotomized NOS1−/− and C57BL/6NHsd wild-type (WT) mice.
Materials and Methods
NOS1−/−, NOS1+/− heterozygous and C57BL/6NHsd mouse total body irradiation (TBI)
All animal experiments and procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at University of Pittsburgh. NOS1−/− (Jackson Laboratories, Bar Harbor, ME, USA), NOS1+/− (Bred at the University of Pittsburgh, Pittsburgh, PA, USA), and control background strain C57BL/6NHsd WT mice (Harlan Sprague Dawley, Indianapolis, IN, USA), 6–10 weeks of age, were studied. The mice were females at approximately 20 to 23 gm with 10 mice per group. TBI to the lethal dose for 50% of mice at 30 days (LD50/30) of 9.5 Gy (2, 11) was delivered to mice by a 137Cs irradiator (Mark MKI-68, J. L. Shepherd and Associates, San Fernando, CA, USA) at a dose rate of 80 cGy per min. Mice were subsequently followed for survival.
Fetal neuronal brain cultures
Primary cortical glial and neuronal cells were prepared from embryonic day 16 C57BL/6NHsd and NOS1−/− mouse embryos and grown in 6 well plates at 500,000 cells/well. Cells were incubated at 37°C and 5% CO2, and grown in Dulbecco’s Modified Eagle’s medium (Mediatech, Inc., Manassas, VA) supplemented with 10% fetal bovine serum (FBS) for 8–10 days. Cells were then switched to FBS-free media for 9–18 days to limit glial cell proliferation. Mature mixed cortical culture cells were treated with either 1 μl/ml of MnSOD-PL (containing 1 μg of plasmid DNA) (8) 24 h before irradiation, or 1 μg/ml of the GS-nitroxide, JP4-039 (12), 30 min prior to 5 Gy or 10 Gy irradiation using a 137Cs gamma irradiator. Control groups consisted of 0 Gy, 5Gy and 10 Gy irradiation with neither antioxidant treatment. Following irradiation, cells were assayed for lipid peroxidation via Electron Paramagnetic Resonance (EPR) Spin Trapping with α-(4-pyridyl-1-oxide)-N-tert-butylnitrone (4-POBN) (Sigma Chemical Co, St. Louis, MO, USA) (13, 14).
Lipid hydroperoxide assay
Cell culture experiments were performed at indicated irradiation doses and assayed for lipid peroxidation using the lipid hydroperoxide assay (Cayman, USA) following the manufacturer’s instructions. Cells from single cell suspensions of E16 fetal brains were exposed to each irradiation dose and then 42 min post irradiation cells were assayed for lipid hydroperoxides (LOOH) (13, 14). Values are reported as nM LOOH/mg protein. Protein determinations were accomplished using the BCA assay (Pierce, USA).
EPR spin trapping
EPR spin trapping with 4-POBN was performed on NOS1−/− and C57BL/6NHsd neuronal cells as previously described (13–14). Briefly, cells were washed and medium was replaced with Chelex-treated Krebs-HEPES buffer (pH 7.4). Cells were then exposed to 4-POBN (10 mM) immediately before irradiation. To initiate lipid-derived free radical chain reactions via Fenton chemistry from irradiation-induced accumulation of cellular lipid hydroperoxides (LOOH + Fe2+ → LO• + OH− + Fe3+) at t=42 min post irradiation, cells were exposed to 20 μM ferrous sulfate (Fe2+) for 5 min. Cells were harvested by mechanical dissociation and the cells and buffer were collected and frozen in liquid N2 until analysis. The EPR data were derived by measuring the intensity of the low-field doublet from the 4-POBN-radical adduct spectrum following 10 additive scans over 2 min, as previously described (13, 14). Values represent arbitrary units and were thus normalized as a percentage of that at 0 Gy. Samples were analyzed on a Bruker eSCAN table-top EPR spectrometer (Bruker AXS, Inc., Madison, WI, USA) with instrument settings as follows: temperature 37°C, 10 G/21 s scan rate; 1.0 G modulation amplitude; 1.0×10 h receiver gain; 0.33 s time constant; and 40 mW nominal power.
Histopathologic analysis of tissues
Eight days after TBI to 9.5 Gy, NOS1−/− and C57BL/6NHsd mice were sacrificed and intravenously perfused with 50 ml of PBS prior to necropsy. Nonirradiated NOS1−/− and C57BL/6NHsd mice served as controls. The vagus nerve and esophagus were excised, fixed in 10% buffered formalin, sectioned (5 mm) (2), and stained with hematoxylin and eosin (H&E) for histological analysis.
Cervical vagotomy, sham surgery, and ionizing irradiation
NOS1−/− and C57BL/6NHsd mice were anesthetized using an intraperitoneal (i.p.) injection of Nembutal (1 μg in 0.2 ml). A cervical midline incision was made, the right vagus isolated and two incisions 0.2 cm. apart were made to the vagus nerve to ensure complete dissection and removal with minimal chance for spontaneous reanastomosis of the incised nerve. The incision was closed using staples. Staples were removed after 1 week, at which time the incision had healed. Sham operations consisted of i.p. injection of Nembutal, followed by a cervical midline incision, isolation of the vagus nerve without ligation, and then the incision was closed with staples. Staples were removed after 1 week, at which time the incision had healed. Mice were irradiated at day 30 after surgery to 9.5 Gy TBI using a Cesium irradiator (2). Irradiated subgroups received intraesophageal administration of MnSOD-PL 24 h prior to irradiation (2). A group of vagotomized mice did not receive TBI and were followed for survival to control for surgery-related mortality and irradiation plus surgery combined injury effect. A similar surgical procedure was used for repeat experiments performing incision of the left vagus nerve, or both right and left vagi.
Statistical methods
Data was analyzed using one-way analysis of variance followed by Tukey’s range test. For the comparison of survival between any two groups of mice, the two-sided log-rank test was used (2). A value of p<0.05 was determined to be statistically significant.
Results
NOS1+/− heterozygous mice show TBI response similar to that of C57BL/6NHsd mice
We previously reported that NOS1−/− mice were radiosensitive when compared to C57BL/6NHsd mice (2). NOS1−/−, NOS1+/−, and C57BL/6NHsd were irradiated to 9.5 Gy TBI (Figure 1). NOS1+/− mice were similar to irradiated C57BL/6NHsd mice (p=0.9269), and radioresistant compared to NOS1−/− mice (p=0.0245). C57BL/6NHsd mice were radioresistant compared to NOS1−/− mice (p=0.0286). Thus, deletion of one NOS1 gene copy did not duplicate the radiosensitive phenotype of homozygous deletion NOS1−/− mice.
Figure 1.
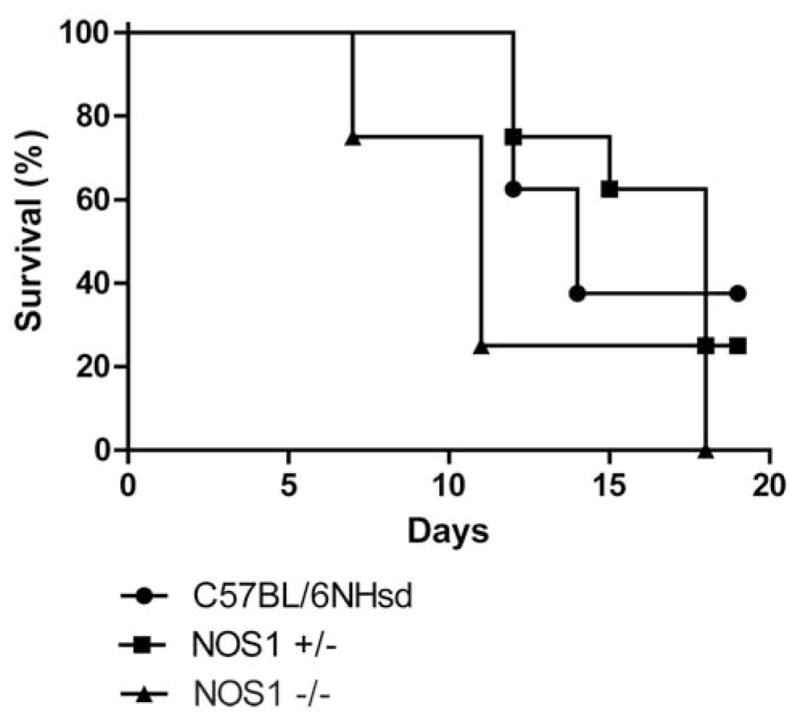
NOS1+/− mice are similar to C57BL/6NHsd controls in response to TBI. NOS1−/−, NOS1+/−, and C57BL/6NHsd mice (n=12) were irradiated to 9.5 Gy TBI. NOS1−/− mice were radiosensitive relative to C57BL/6NHsd and NOS1+/− mice (p=0.0286 and p=0.0245, respectively). There was no significant difference in survival between C57BL/6NHsd and NOS+/− mice.
Irradiation-induced lipid peroxidation in fetal brain, and isolated neuronal cultures from NOS1−/− mice, and its amelioration by MnSOD-PL
Exposure of fetal brain culture cells to 0–10 Gy induced dose-dependent cellular lipid peroxidation as detected by the formation of LOOH (Figure 2A). Similar results were obtained when lipid-derived free radical chain reactions were initiated by the addition of Fe2+ in the presence of the EPR spin trap, 4-POBN (Figure 2B). The 4-POBN radical adduct intensity increased with increasing irradiation dose. Shown in the inset is a representative spectrum of the 4-POBN radical adduct. Controls in the absence of added Fe2+ did not produce detectable differences between experimental groups, indicating endogenous lipid free radical formation was below the limit of detection under these experimental conditions (not shown).
Figure 2.
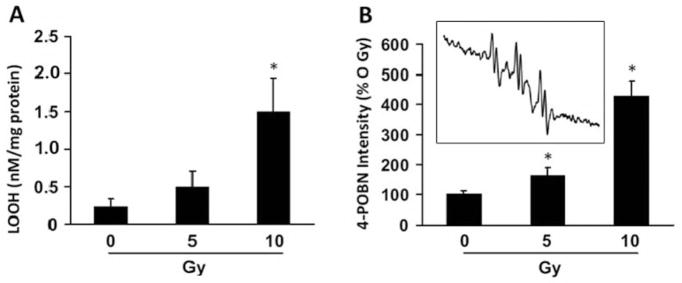
Irradiation-induced cellular lipid peroxidation in fetal brain cultures from NOS1−/− mice. A: Cell cultures prepared as described in the Materials and Methods were assayed for lipid hydroperoxide (LOOH) levels 42 min post irradiation with 0–10. B: Cells were treated as in A except cells were assessed for lipid-derived free radical formation by EPR spin trapping with 4-POBN. For the EPR experiments, cells were incubated with 4-POBN (10 mM) immediately before irradiation and at 42 min post irradiation were exposed to 20 μM Fe2+ for 5 min. The EPR data were derived by measuring the intensity of the up-field doublet of the 4-POBN-radical adduct spectrum following 10 additive scans over 2 min at 37°C. Values represent arbitrary units and were thus normalized as a percentage of 0 Gy. The inset is a representative 4-POBN radical adduct spectrum obtained from cells exposed to 10 Gy. Results are presented as means±standard error. Data for A and B represent the mean of at least three independent determinations. *Significant difference with Tukey’s range test for 10 Gy compared to 0 and 5 Gy in A and 10 Gy compared to 0 and 5 Gy, as well as 5 Gy compared to 0 Gy, for B.
In C57BL/6NHsd mouse neuronal cell cultures (Figure 3), lipid peroxidation was directly correlated with increasing radiation dose (EPR 0 Gy: 100±11, 5 Gy: 152±16, 10 Gy: 484±62). Exposure of fetal brain culture cells to 5 or 10 Gy increased cellular lipid peroxidation over controls (0 Gy) detected by the formation of 4-POBN radical adducts (Figure 3). Transfection of cells with MnSOD-PL did not significantly alter radical formation when cells were exposed to 5 Gy but did reduce 4-POBN radical intensity induced by 10 Gy by 32% (p<0.05) (Figure 3).
Figure 3.

MnSOD-PL inhibits irradiation-induced lipid peroxidation in C57BL/6NHsd mouse fetal brain neuronal cells. C57BL/6NHsd neuronal cells were exposed to 0, 5, or 10 Gy with or without treatment with JP4-039 or MnSOD-PL. Samples were analyzed for 4-POBN-radical adduct as described in Materials and Methods. Values represent the mean of 3 independent determinations and are presented in arbitrary units and were thus normalized as a percentage of 0 Gy presented as mean and standard error. *Significantly different at p<0.05 with Tukey’s range test.
In NOS1−/− mouse fetal brain cultures, lipid peroxidation was also directly increased with increased radiation dose (p>0.05) (Figure 4). Transfection of NOS1−/− cultures with MnSOD-PL reduced lipid peroxidation in response to 5 Gy (p<0.05) and 10 Gy (p<0.05) (Figure 4). No significant differences in the magnitude or pattern of increase in lipid peroxidation in response to irradiation were detected comparing C57BL/6NHsd to NOS1−/− neuronal cell cultures. Treatment of C57BL/6NHsd or NOS1−/− fetal brain cell cultures with JP4-039 did not significantly alter irradiation-induced lipid peroxidation by either 5 Gy or 10 Gy (Figures 3 and 4).
Figure 4.
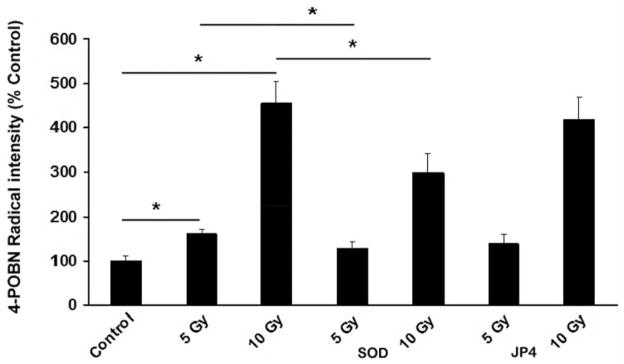
MnSOD-PL inhibits irradiation-induced lipid peroxidation by 5 or 10 Gy treatment of NOS1−/− mouse fetal brain neuronal cell cultures. NOS1−/− neuronal cells were exposed to 0, 5 or 10 Gy with or without treatment with JP4-039 or MnSOD. Samples were analyzed for 4-POBN-radical adduct as described in Materials and Methods. Values represent the mean of 3 independent determinations and are presented in arbitrary units and were thus normalized as a percentage of 0 Gy presented as mean and standard error. *Significantly different at p<0.05 with Tukey’s range test.
Histopathologic evidence of greater inflammation in the esophagus of irradiated NOS1−/− mice
Previous studies demonstrated no significant change in radiation-induced increase in inflammatory cytokines, gross histopathologic, or microscopic morphology compared to results with esophagus from WT mice (2). To determine whether more extensive histopathologic studies would reveal evidence of a different irradiation response of the NOS1−/− mouse esophagus, transverse and longitudinal sections of esophagus were examined for histopathologic evidence of irradiation effects. Analysis of the inflammatory response included evaluation of a potential increase in morphologic parameters of infection, and other changes which might be associated with irradiation-induced changes that could affect the esophageal-central nervous system pathways leading to seizures.
Histopathological analysis was performed on the esophagus of unirradiated and 9.5 Gy TBI NOS1−/− and C57BL/6NHsd mice (n=3/group). Irradiated NOS1−/− esophagus had greater dilation and increased attenuation of tissue layers at day 8 after 9.5 Gy TBI (Figures 5 and 6). A greater inflammatory response was detectable in irradiated NOS1−/− esophagus compared to irradiated C57BL/6NHsd, with increased inflammatory cells and increased tissue necrosis (Figure 5 and 6). NOS1−/− mouse esophagus had increased numbers and size of areas of colonization with bacteria located at the stratified epithelial layer (Figure 5) compared to the C57BL/6NHsd esophagus. The NOS1−/− irradiated mouse esophagus also had greater ablation of the epithelial layer, necrosis of the outer longitudinal muscle layer, and greater infiltration with inflammatory cells (Figures 5 and 6).
Figure 5.

Cross sectional histopathology of esophagus from irradiated NOS1−/− and C57BL/6NHsd mice. Mice were irradiated to 9.5 Gy TBI as described in Materials and Methods and at eight days were sacrificed, perfused with PBS, and their esophagus excised, fixed in 10% buffered formalin, sectioned (5 mm), and stained with hematoxylin and eosin (H&E) for histological analysis. A: Nonirradiated C57BL/6NHsd mouse esophagus. Bar=100 μm. B: Nonirradiated NOS1−/− mouse esophagus showing relative dilation and attenuation compared to C57BL/6NHsd mice. C: Irradiated C57BL/6NHsd mouse esophagus showing an inflammatory response marked by thickening, swelling, necrosis and an infiltration of inflammatory cells. D: Irradiated NOS1−/− mouse esophagus showing relatively greater cellular inflammatory response (broad arrow) and necrosis (thin arrow) compared to irradiated C57BL/6NHsd mouse esophagus. (Bar=100 μM).
Figure 6.
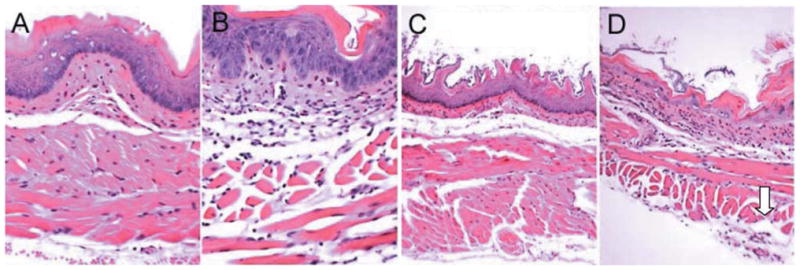
Longitudinal histopathology of esophagus of nonirradiated and irradiated NOS1−/− and C57BL/6NHsd mice. Once extracted, the esophagi were fixed in 10% buffered formalin, sectioned (5 mm), and subsequently stained with hematoxylin and eosin (H&E) for histological analysis. A: Nonirradiated C57BL/6NHsd mouse esophagus (×500). B: Nonirradiated NOS1−/− mouse esophagus (×500). C: C57BL/6NHsd mouse esophagus at day 8 following 9.5 Gy (TBI) (×500), showing moderate edema and atrophy of both inner circular and outer longitudinal muscle layers. D: NOS1−/− mouse esophagus at day 8 after 9.5 Gy TBI showing necrosis of outer circular muscle layer (arrow) (×100).
No evidence of neurodegeneration or increased apoptosis in abdominal vagus nerve of irradiated NOS1−/− mice
Histopathologic analysis of the vagus nerves was performed on fixed sections stained with H&E (n=3/group). After careful review of multiple sections of the nerve and nodose ganglion, no structural changes were noted between any of four control or irradiated groups at day 8 (Figure 7).
Figure 7.
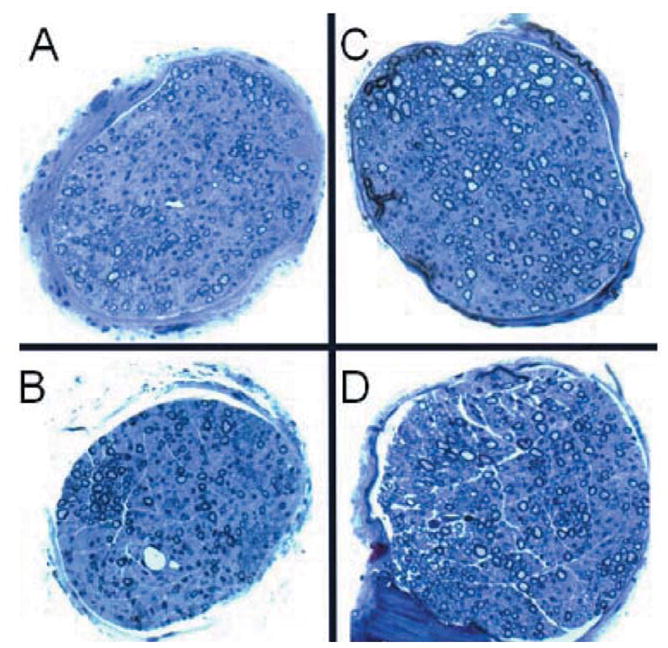
Histopathologic appearance of 9.5 Gy-irradiated NOS1−/− and C57BL/6NHsd mouse vagus nerve. Mice were irradiated to 9.5 Gy (TBI) and eight days later were sacrificed, perfused with PBS, and vagus nerves excised. A: Vagus nerve from nonirradiated C57BL/6NHsd mouse nerve. B: Nonirradiated NOS1−/− mouse vagus nerve. C: Irradiated C57BL/6NHsd mouse vagus nerve at day 8. D: Irradiated NOS1−/− mouse vagus nerve at day 8 (×1000).
Right-sided cervical vagotomy prior to TBI does not alter response of NOS1−/− or C57BL/6HNsd mice to TBI
C57BL/6NHsd and NOS1−/− mice were irradiated to 9.5 Gy TBI 30 days after right vagotomy to allow sufficient time for healing of surgical wounds (Figure 8 and Figure 9, respectively). Right vagotomized, sham operated and subgroups receiving MnSOD-PL prior to irradiation were studied. Survival of C57BL/6NHsd vagotomized, and sham-operated groups of mice was similar to that of non-vagotomized C57BL/6NHsd mice (p=0.1464 and 0.1864, respectively) (Figure 8). Thus, vagotomy did not induce rapid death in irradiated WT mice. Right vagotomized NOS1−/− mice were not relatively radioresistant and were in fact more radiosensitive than non-vagotomized NOS1−/− mice (p=0.0390) (Figure 9). Thus, NOS1−/− mice were protected by right vagotomy. NOS1−/− vagotomized mice, administered intraesophageal MnSOD-PL were not significantly protected compared to right vagotomized NOS1−/− mice and were more radioresistant (p=0.0060). All control vagotomized mice receiving no TBI survived, confirming that the surgery alone did not cause mortality. Our results with left vagotomized NOS1−/− and C57BL/6NHsd mice did not show any effect of the procedure on the intrinsic strain differences in response to TBI. Bilateral vagotomized mice could not be studied as the surgery produced fatal respiratory arrest.
Figure 8.
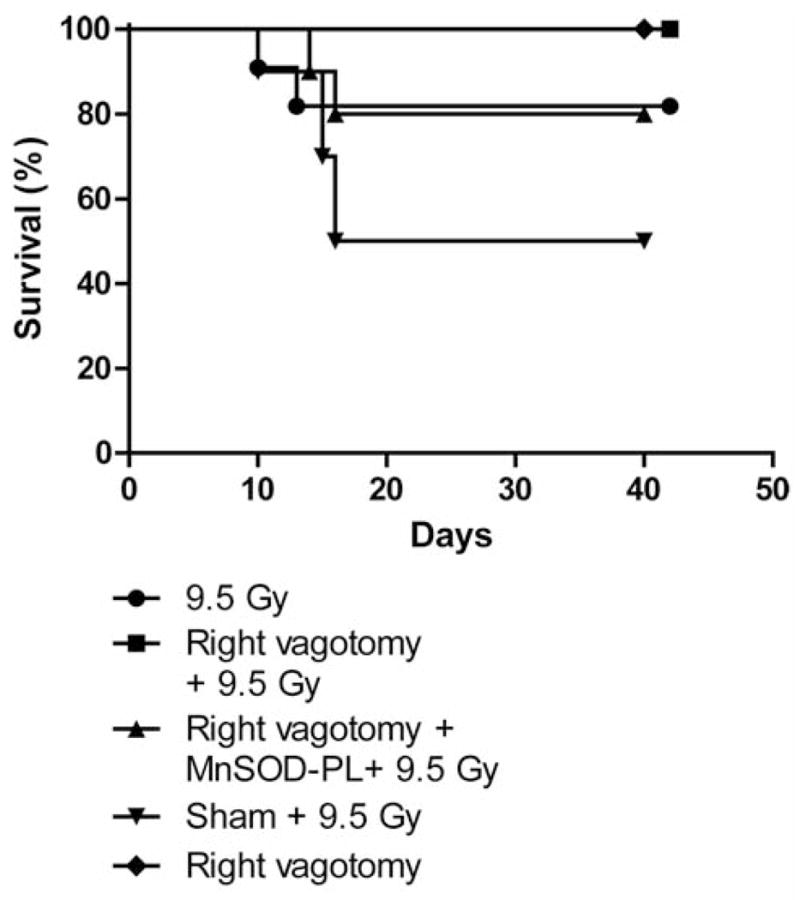
Right-vagotomized C57BL/6NHsd mice show no significant alteration in response to 9.5 Gy TBI. Groups of mice (n=12) included: right-vagotomized, non-vagotomized, vagotomized then intraesophageal MnSOD-PL-treated, or sham-operated C57BL/6NHsd mice. Mice were irradiated to 9.5 TBI 30 days after surgery. MnSOD-PL was given by swallow 24 hr prior to irradiaiton as described in Materials and Methods and in (2). All mice were followed for survival. No statistically significant difference in survival was detectable between any of the groups of vagotomized compared to non-vagotomized mice.
Figure 9.
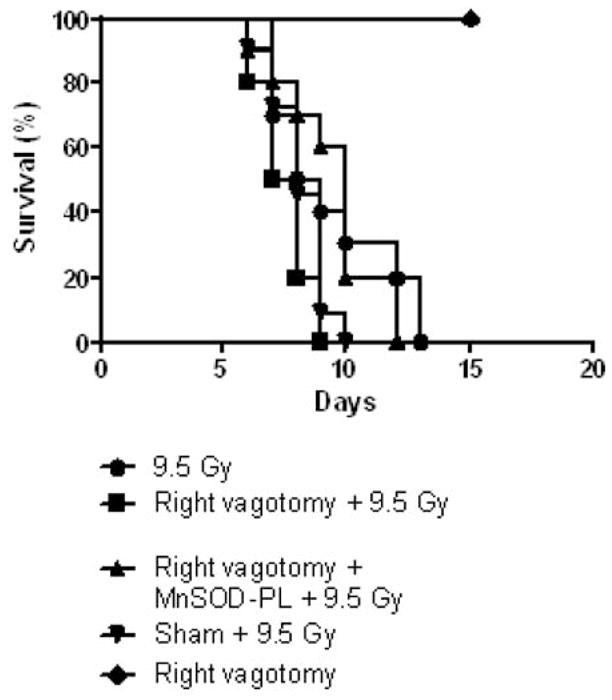
Right-vagotomized NOS1−/− mice show no alteration in radiosensitivity in response to 9.5 Gy (TBI). Right-vagotomized, non-vagotomized, vagotomized then MnSOD-PL-treated and sham-operated NOS1−/− mice (n=12) were irradiated to 9.5 TBI 30 days after surgery. Right-vagotomized mice without irradiation were also followed for survival. Right-vagotomized NOS1−/− mice were not protected and were in fact more radiosensitive than non-vagotomized, irradiated mice (p=0.039). MnSOD-PL-treated right-vagotomized NOS1−/− mice were not protected and were in fact more radioresistant than the non-MnSOD-PL-treated vagotomized irradiated mice (p=0.0060).
Discussion
Homozygous deletion recombinant negative NOS1−/− mice have been shown to be radiosensitive to TBI compared to control C57BL/6NHsd mice and die of grand mal seizures as opposed to listlessness and fatigue seen in the controls (2). NOS1 is involved in production of peroxynitrite, a potent reactive oxygen species which leads to apoptosis through activation of the caspase system, and NOS1 localizes to the mitochondria (15–17). In the present studies, we sought to determine if expression of low levels of NOS1 in NOS1+/− mice altered TBI radiosensitivity. Heterozygous NOS1+/− mice were similar in TBI response to control C57BL/6NHsd mice and were radioresistant compared to NOS1−/− mice. These data are similar to those with homozygous but not heterozygous deletion of p53, since only the former leads to an increased radiosensitivity (18, 19). Thus, complete absence but not reduced expression of NOS1, as with p53, is required for TBI radiosensitivity.
Through activation of mitochondrial death pathways, radiation leads to the production of reactive oxygen species (15–17, 20), and lipid peroxidation (11). Epilepsy and seizures have been linked to increased oxidative stress and to increased lipid peroxidation (21–22). We measured brain lipid peroxidation in irradiated NOS1−/− compared to C57BL/6NHsd mouse fetal brain cultures. Neuronal cell lipid peroxidation in response to increasing doses of irradiation and the effect of JP4-039 and the antioxidant MnSOD-PL was also tested in NOS1−/− and C57BL/6NHsd fetal brain cultures. In both NOS1−/− and C57BL/6NHsd neuronal cell cultures, increasing irradiation doses increased lipid peroxidation. Treatment with MnSOD-PL significantly lowered lipid peroxidation, at 10 Gy in both C57BL/6NHsd and NOS1−/− neuronal cultures. These results confirm that oxidative stress in response to irradiation is reduced by the antioxidant MnSOD (3–6). No significant differences were detected in the patterns of lipid peroxidation between NOS1−/− and control C57BL/6NHsd neuronal cultures. Thus, absence of the NOS1 gene product does not alter irradiation-induced peroxynitrate formation in whole-brain cultures, perhaps attributable to the low relative number of NOS1 neurons in the fetal brain and in brain-derived cell cultures (11, 15–16).
In prior studies, gross and histopathological analysis of tissues, including esophagus, peripheral blood and serum chemistry analysis, and RT-PCR measurement of inflammatory cytokines revealed no cause of the increased radiosensitivity or grand mal seizures in NOS1−/− mice. Both NOS1−/− and C57BL/6NHsd mice were protected by intraesophageal administration of MnSOD-PL (2). To determine the role of the esophagus and swallowed MnSOD-PL in TBI-induced seizure activity, the vagus nerve was examined after TBI.
Histopathological analysis of the vagus nerve and esophagus in 9.5 Gy TBI-treated NOS1−/− and control mice was carried out. Irradiated NOS1−/− mouse esophagus showed increased necrosis and inflammatory cell infiltration compared to that of C57BL/6NHsd mice. Since prior data showed no differences in mRNA expression for inflammatory cytokines NF-κβ (nuclear factor kappa-light-chain-enhancer of activated B cells), TNF-ά (tumor necrosis factor-alpha), or IFN-γ (interferon-gamma) (2), the present result detecting an increase in histopathologic markers of inflammation in the NOS1−/− esophagus may have been the result of infection or another parameter of organ damage. No difference was detectable between control or irradiated groups in the morphologic appearance of the vagus nerve. The vagus nerve has been associated with seizure activity, and vagal nerve stimulation has been used to control epilepsy (7, 8). Stimulation of the vagus nerve has also been shown to be a treatment option for seizures in patients refractory to antiepileptic drugs (7, 8). Through its afferent connection of the esophagus to the brain (23, 24), the vagus nerve has been reported to be associated with up-regulation of inflammatory responses (9, 10), and also has anti-inflammatory functions through reducing the level of inflammatory mediators (23, 24).
We tested the physiologic effect of vagotomy on TBI-induced seizures and early death. Right vagotomized NOS1−/− mice had no restoration of resistance to TBI compared to non-vagotomized mice. Furthermore, no change in relative resistance to TBI was detected in right vagotomized C57BL/6NHsd mice. Sham-operated mice were similar to non-operated control mice. All nonirradiated vagotomized mice survived in both strains. Thus, an intact right vagus nerve does not explain the phenotype of either radiosensitivity in NOS1−/− mice or the relative radioresistance in WT C57BL/6NHsd mice. If the vagus nerve has a potent anti-inflammatory function (9, 10), it may suppress irradiation-induced inflammation and increase survival similar to MnSOD-PL treatment (2, 4–5, 11, 25). NOS1−/− mice may be more sensitive to TBI due to lack of the anti-inflammatory function of the vagus nerve. C57BL/6NHsd mice might have a compensatory function of the left vagus nerve meditated through NOS1, a function absent in NOS1−/− mice. In more recent studies with groups of 10 NOS1−/− and control C57Bl/HNsd mice irradiated to 9.25 Gy TBI at 30 days after left vagotomy, there was also no effect of this surgical procedure on the intrinsic radiosensitivity (survival) of either strain. These data establish that the mechanism of radioprotection of NOS1−/− mice by swallowed MnSOD-PL cannot be attributed to the vagus nerve pathway and direct attention to either the esophageal spinal afferent nerves or another developmental abnormality in the neuro-esophageal pathway in these mice. Bilateral cervical vagotomized mice could not be tested as they expired from respiratory failure. Further studies will be required to define the mechanism of TBI-induced early death from seizures in NOS1−/− mice.
Acknowledgments
This project was supported by NIH/NCI Grant RO1 CA83876 and NIA/NIH grant T32-AG21885.
References
- 1.Huang PL, Dawson TM, Bredt DS, Snyder SH, Fishman MC. Targeted disruption of the neuronal nitric oxide synthase gene. Cell. 1993;75:1273–1286. doi: 10.1016/0092-8674(93)90615-w. [DOI] [PubMed] [Google Scholar]
- 2.Rajagopalan MS, Stone B, Rwigema JC, Salimi U, Epperly MW, Goff J, Franicola D, Dixon T, Cao S, Zhang X, Buchholz BM, Bauer AJ, Choi S, Bakkenist C, Wang H, Greenberger JS. Intraesophageal manganese superoxide dismutase-plasmid liposomes ameliorates novel total-body and thoracic radiation sensitivity of NOS1−/− mice. Radiat Res. 2010;174(3):297–312. doi: 10.1667/RR2019.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zahid I, Epperly MW, Haider M, Greenberger JS, London B. Cardiac conduction defect may contribute to total body irradiation sensitivity of NOS1 knockout mice. The International Society of Heart Research, North American Section Meeting; Philadelphia. 22–25th May, 2001. [Google Scholar]
- 4.Epperly MW, Gretton JA, DeFilippi SJ, Sikora CA, Liggitt D, Koe G, Greenberger JS. Modulation of radiation-induced cytokine elevation associated with esophagitis and esophageal stricture by manganese superoxide dismutase-plasmid/liposome (SOD-PL) gene therapy. Radiat Res. 2001;155:2–14. doi: 10.1667/0033-7587(2001)155[0002:morice]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 5.Epperly MW, Kagan VE, Sikora CA, Gretton JE, DeFilippi SJ, Bar-Sagi D, Greenberger JS. Manganese superoxide dismutase-plasmid/liposome (MnSOD-PL) administration protects mice from esophagitis associated with fractionated irradiation. Int J Cancer (Radiat Oncol Invest) 2001;96(4):221–233. doi: 10.1002/ijc.1023. [DOI] [PubMed] [Google Scholar]
- 6.Tarhini AA, Belani C, Luketich JD, Ramalingam SS, Argiris A, Gooding W, Petro D, Kane K, Liggitt D, Championsmith T, Zhang X, Epperly MW, Greenberger JS. A phase I study of concurrent chemotherapy (Paclitaxel and Carboplatin) and thoracic radiotherapy with swallowed manganese superoxide dismutase (MnSOD) plasmid liposome (PL) protection in patients with locally advanced stage III non-small cell lung cancer. Human Gene Therapy. 2011;22(3):336–343. doi: 10.1089/hum.2010.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schachter SC, Saper CB. Vagus nerve stimulation. Epilepsia. 1998;39(7):677–686. doi: 10.1111/j.1528-1157.1998.tb01151.x. [DOI] [PubMed] [Google Scholar]
- 8.Handforth A, DeGiorgio CM, Schachter SC, Uthman BM, Naritoku DK, Tecoma ES, Henry TR, Collins SD, Vaughn BV, Gilmartin RC, Labar DR, Morris GL, 3rd, Salinsky MC, Osorio I, Ristanovic RK, Labiner DM, Jones JC, Murphy JV, Ney GC, Wheless JW. Vagus nerve stimulation therapy for partial-onset seizures: a randomized active-control trial. Neurology. 1998;51(1):48–55. doi: 10.1212/wnl.51.1.48. [DOI] [PubMed] [Google Scholar]
- 9.Borovikova LV, Ivanova S, Zhang M, Yang H, Botchkina GI, Watkins LR, Wang H, Abumrad N, Eaton JW, Tracey KJ. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature. 2000;405(6785):458–462. doi: 10.1038/35013070. [DOI] [PubMed] [Google Scholar]
- 10.Borovikova LV, Ivanova S, Nardi D, Zhang M, Yang H, Ombrellino M, Tracey KJ. Role of vagus nerve signaling in CNI-1493-mediated suppression of acute inflammation. Auton Neurosci. 2000;85(1–3):141–147. doi: 10.1016/S1566-0702(00)00233-2. [DOI] [PubMed] [Google Scholar]
- 11.Epperly MW, Dixon T, Wang H, Schlesselman J, Franicola D, Greenberger JS. Modulation of radiation-induced life shortening by systemic intravenous MnSOD-plasmid liposome gene therapy. Radiat Res. 2008;170(4):437–443. doi: 10.1667/rr1286.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rajagopalan MS, Gupta K, Epperly MW, Franicola D, Zhang X, Wang H, Zhao H, Tyurin VA, Kagan VE, Wipf P, Kanai A, Greenberger JS. The mitochondria-targeted nitroxide JP4-039 augments potentially lethal irradiation damage repair. In Vivo. 2009;23:717–726. [PMC free article] [PubMed] [Google Scholar]
- 13.Kelley EE, Domann FE, Buettner GR, Oberley LW, Burns CP. Increased efficacy of in vitro photofrin photosensitization of human oral squamous cell carcinoma by iron and ascorbate. J Photochem Photobiol B: Biol. 1997;40:273–277. doi: 10.1016/s1011-1344(97)00068-7. [DOI] [PubMed] [Google Scholar]
- 14.Kelley EE, Buettner GR, Burns CP. Production of lipid-derived free radicals in L1210 murine leukemia cells is an early oxidative event in the photodynamic action of photofrin. Photochem Photobiol. 1997;65:576–580. doi: 10.1111/j.1751-1097.1997.tb08608.x. [DOI] [PubMed] [Google Scholar]
- 15.Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281(5381):1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- 16.Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86(1):147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- 17.Wang ZB, Liu YQ, Cui YF. Pathways to caspase activation. Cell Biol Int. 2005;29(7):489–496. doi: 10.1016/j.cellbi.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 18.Pulkkinen JO, Servomaa K, Pekkola K, Kulmala J, Grenman R. The effect of irradiation on mitotic and apoptotic frequency in head and neck cancer cell lines, the correlation to p53 mutations and clonogenic survival. Anticancer Res. 2000;20(3A):1503–1511. [PubMed] [Google Scholar]
- 19.Liang L, Shao C, Deng L, Mendonca MS, Stambrook PJ, Tischfield JA. Radiation-induced genetic instability in vivo depends on p53 status. Mutat Res. 2002;502(1–2):69–80. doi: 10.1016/s0027-5107(02)00029-5. [DOI] [PubMed] [Google Scholar]
- 20.Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. 2004;116(2):205–219. doi: 10.1016/s0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- 21.Sathisha TG, Shetty BV, Rao GM, Sudha K. Effect of grandmal seizures on oxidative stress and antioxidants in epileptics. Biomedical Research. 2007;18(3):167–169. [Google Scholar]
- 22.Júnior HV, de França Fonteles MM, Mendes de Freitas R. Acute seizure activity promotes lipid peroxidation, increased nitrite levels and adaptive pathways against oxidative stress in the frontal cortex and striatum. Oxid Med Cell Longev. 2009;2(3):130–137. doi: 10.4161/oxim.2.3.8488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Picker O, Scheeren TW, Arndt JO. Nitric oxide synthases in vagal neurons are crucial for the regulation of heart rate in awake dogs. Basic Res Cardiol. 2001;96(4):395–404. doi: 10.1007/s003950170048. [DOI] [PubMed] [Google Scholar]
- 24.Choate JK, Danson EJ, Morris JF, Paterson DJ. Peripheral vagal control of heart rate is impaired in neuronal NOS knockout mice. Am J Physiol Heart Circ Physiol. 2001;281(6):H2310–2317. doi: 10.1152/ajpheart.2001.281.6.H2310. [DOI] [PubMed] [Google Scholar]
- 25.Greenberger JS, Epperly MW, Gretton J, Jefferson M, Nie S, Bernarding M, et al. Radioprotective gene therapy. Curr Gene Ther. 2003;3(3):183–195. doi: 10.2174/1566523034578384. [DOI] [PubMed] [Google Scholar]