

Abstract
Kinesin spindle protein (KSP), a microtubule-associated motor protein essential for cell cycle progression, is overexpressed in many cancers and a potential anti-tumor target. We found that inhibition of KSP by a selective inhibitor, ARRY-520, blocked cell cycle progression, leading to apoptosis in acute myeloid leukemia cell lines which express high levels of KSP. Knockdown of p53, overexpression of XIAP, and mutation in caspase-8 did not significantly affect sensitivity to ARRY-520, suggesting that the response is independent of p53, XIAP, and the extrinsic apoptotic pathway. Although ARRY-520 induced mitotic arrest in both HL-60 and Bcl-2-overexpressing HL-60Bcl-2 cells, cell death was blunted in HL-60Bcl-2 cells, suggesting that the apoptotic program is executed through the mitochondrial pathway. Accordingly, inhibition of Bcl-2 by ABT-737 was synergistic with ARRY-520 in HL-60Bcl-2 cells. Furthermore, ARRY-520 increased Bim protein levels prior to caspase activation in HL-60 cells. ARRY-520 significantly inhibited tumor growth of xenografts in SCID mice and inhibited AML blast but not normal colony formation, supporting a critical role for KSP in proliferation of leukemic progenitor cells. These results demonstrate that ARRY-520 potently induces cell cycle block and subsequent death in leukemic cells via the mitochondrial pathway and has potential to eradicate AML progenitor cells.
Keywords: KSP inhibitor ARRY-520, Eg5, cell cycle, apoptosis, mitochondrial pathway, AML
Introduction
Microtubule-targeting agents such as taxanes, which stabilize microtubule polymers, and vinca alkaloids, which inhibit tubulin polymerization, are among the most effective drugs against a variety of cancers(1), including breast, ovarian, and lung carcinomas and leukemias. However, their use is hindered by the toxicity arising from disruption of overall cellular microtubule dynamics not associated with cellular proliferation. Therefore, selectively targeting microtubule components should constitute a novel therapeutic strategy to limit cancer cell proliferation.
Kinesin spindle protein (KSP, Eg5), a member of the kinesin-like protein family is a microtubule-associated motor protein, that hydrolyzes ATP, causing it to move toward the plus ends of microtubules (2;3). KSP plays an important role in maintaining spindle dynamics and is essential for chromosome positioning, centrosome separation, the establishment of a bipolar spindle, and separation of the spindle during mitosis (4;5). Its intrinsic biological functions suggest that KSP is an important target of anticancer therapy. Studies in mice have shown that Eg5 expression by retroviral insertion contributes to the development of mouse B-cell leukemia (6), suggesting that Eg5 plays a role in leukemogenesis. In pancreatic carcinoma cells, the kinesin-related protein HsEg5 has been identified as a central molecule involved in the antiproliferative action of all-trans-retinoic acid (7). In addition, KSP was shown to be highly expressed in transformed cells in culture, but less so in primary cells. Its expression is also higher in breast, colon, lung, ovary, and uterine carcinomas than in their adjacent tissues (8). Importantly, Eg5 was recently shown to be highly expressed in blast crisis CML (9). These findings indicate the potential importance of KSP as a target of anticancer therapy. Indeed, we observed that an Eg5 antisense oligonucleotide (ASO) was able to induce G2M cell cycle block and cell death in CML cells, independent of the cellular responses to imatinib (10).
Because KSP functions exclusively in mitosis, KSP inhibitors have recently been developed as a new generation of anti-mitotic agents for cancer therapy and some that have already been studied in phase 1/2 clinical trials have shown antiproliferative effects without causing significant neuropathy (11–15). ARRY-520, developed by Array BioPharma, is one such agent that has shown efficient KSP inhibition and pharmacodynamic activity in animal models of solid tumors (16;17). However, the effectiveness of these compounds in leukemia has not been tested, and their mechanisms of action are largely unknown.
In a search for more effective and improved therapies for patients with AML, an aggressive hematological malignancy associated with high relapse rates and a generally poor prognosis with chemotherapy as the current primary treatment, we examined the effect of ARRY-520 on various acute leukemia cells. We found that inhibition of KSP effectively induced cell cycle block and the death of these cells via the mitochondrion-mediated apoptotic pathway and that ARRY-520 potently inhibited tumor growth in xenografts and colony forming capacity of AML blasts.
Materials and Methods
Cells and cell cultures
U937, Jurkat, JurkatI9.2 (18), and HL-60 cells were purchased from the American Type Culture Collection (Manassas, VA) and Molm13 cells from Fujisaki Cell Center, Hayashibara Biochemical Labs, Inc. (Okayama, Japan). OCI-AML3 cells were kindly provided by Dr. M. Minden (Ontario Cancer Institute, Toronto, Ontario, Canada). OCI-AML3p53shRNA, p53 knockdown OCI-AML3 cells and OCI-AML3vec, the control cells were generated as described previously(19). XIAP overexpressing U937 (U937XIAP) and the control (U937neo) cells were kindly provided by Dr. D. Kufe (Dana-Farber Cancer Institute, Harvard Medical School, Boston, MA) (20). Bcl-2-overexpressing HL-60 cells (HL-60Bcl-2) were a gift of Dr. K. Bhalla (Medical College of Georgia Cancer Center, Augusta, GA). Fresh primary AML patient samples were acquired after informed consent following institutional guidelines. Mononuclear cells were purified by Ficoll-Hypaque (Sigma Chemical Co., St. Louis, MO) density-gradient centrifugation. Cells were cultured in RPMI 1640 medium containing 10% heat-inactivated fetal calf serum (FCS), 2 mM L-glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin.
Treatment of cells
Exponentially growing cells (0.4 × 106/mL) were treated with ARRY-520 (Array BioPharma, Inc., Boulder, CO) for up to 48 hours. For combination, HL-60 and HL-60Bcl-2 cells (0.4 × 106/mL) were incubated with ARRY-520, ABT-737, or both for up to 96 hours. ABT-737, a selective Bcl-2 inhibitor (21), was synthesized at M. D. Anderson Cancer Center based on the published structure. DMSO was used as the control agent.
To inhibit KSP expression, 3 × 106 exponentially growing HL-60 cells were transfected with 5 μg of either the KSP ASO or its control oligonucleotide (NSO) (both kindly supplied by ISIS Pharmaceuticals) using Nucleofector solution T and program K-17 following the manufacturer's instructions (Amaxa Biosystems, Cologne, Germany) and as previously described (10).
Cell viability assay
Apoptosis was estimated by flow cytometry measurements of phosphatidyl serine (22) with the Annexin-V-FLUOS Staining Kit (Roche Diagnostics, Indianapolis, IN). Membrane integrity was simultaneously assessed by 7-amino-actinomycin D (7-AAD). To measure changes in the mitochondrial membrane potential (MMP), cells were loaded with CMXRos (300 nM) and MitoTracker Green (500 nM) (both from Invitrogen, Carlsbad, CA) for 1 hour at 37°C. The loss of MMP was then assessed by measuring CMXRos retention while simultaneously adjusting for mitochondrial mass.
Cell cycle distribution
Cells were fixed with 70% ice-cold ethanol and stained with propidium iodide (PI) solution (25 μg/mL PI, 180 U/mL RNase, 0.1 % Triton X-100, and 30 mg/mL polyethylene glycol in 4 mM citrate buffer, pH 7.8). The DNA content was determined using a FACSCalibur flow cytometer (Becton Dickinson, San Jose, CA). The cell cycle distribution was analyzed using ModFit LT software (Verity Software House, Topsham, ME).
TUNEL assay
To determine the cell cycle stage of apoptotic cells, cells were fixed in 4% formaldehyde and permeabilized with 0.1% Triton X-100. TUNEL assay was performed using the Apo-Direct Kit following the manufacturer's instructions (BD Biosciences, San Jose, CA).
Western blot analysis
Western blot analysis was performed as described previously.(23)
Colony-formation assay
Colony-formation assay was performed as described previously (24) using 1 × 105 mononuclear cells from the bone marrow (BM) of AML patients and cells from normal blood obtained by apheresis treated with ARRY-520, 3.3 to 100 nM.
Xenograft studies in SCID mice
HL-60 or MV4-11 cells growing in IMDM supplemented with 20% or 10% FBS, Glutamax, and antibiotic-antimycotic were harvested when they reached approximately 106/mL. Female SCID-beige mice were implanted subcutaneously in the right flank with 107 HL-60 or MV4-11 cells/mouse in 100 μL PBS. Twenty-one days later for HL-60-injected mice and eighteen for MV4-11-injected mice, tumors were measured with calipers and tumor volume calculated: volume = (length × width2)/2. Mice were randomized into 5 or 8/group, with an average tumor volume of approximately 265 or 275 mm3 in each group for HL-60- or MV4-11-injected mice, respectively. Treatment began on the day of randomization (day 1). Mice injected with HL-60 were dosed with vehicle or ARRY-520 in 25% PEG400/10% EtOH/65% saline intraperitoneally, at 27 mg/kg, on days 1, 5 and 9. Mice injected with MV4-11 were dosed with vehicle or ARRY-520, at 20 mg/kg, on days 1, 5, 9, and 53, and the surviving vehicle-treated mice were later injected with ARRY-520 on days 28, 53, and 67. Tumor volume and animal weights were measured twice a week during the course of the study.
Statistics
All experiments were performed in triplicate and results expressed as the mean±s.e., unless otherwise stated. The IC50 (the concentration inducing annexin V positivity in 50% of cells) was calculated using CalcuSyn software (Biosoft, Ferguson, MO). The combination index (CI) was determined by the Chou-Talalay method (25) using CalcuSyn software and was expressed as the average±s.e. of the CI values obtained at the ED50, ED75, and ED90. A CI<1 indicates a synergistic effect; CI=1, an additive effect; and CI>1, an antagonistic effect.
Results
Inhibition of KSP by ARRY-520 potently induces cell death in acute leukemic cells
We first showed by western blot that KSP, the target of ARRY-520, is highly expressed in HL-60, Jurkat, OCI-AML3, U937, and Molm13 cells and in most samples of AML blasts at various levels (Figure 1). We then treated these cell lines with ARRY-520 and found a decrease in cell viability with a concomitant increase in cell death in all cases. As shown in Figure 2A, ARRY-520, at nM concentrations, induced time- and dose-dependent cell death in these leukemic cells. Of the cell lines examined, OCI-AML3 and Molm13 cells were most sensitive (Figure 2A). To confirm that ARRY-520 acts by inhibiting KSP, we treated HL-60 cells with KSP ASO for 24 hours and then with ARRY-520 for an additional 48 hours. As shown in Figure 2B, downregulation of KSP sensitized HL-60 cells to ARRY-520 (IC50=2.0±0.4 versus IC50=6.1±1.3 in NSO treated cells). Of note, the IC50s of HL-60 cells were 11.3±3.3 nM in Figure 2A, in which cells were treated with ARRY-520 for 48 hours, versus 6.1±1.3 nM in Figure 2B (KSP-NSO), in which cells were electroporated with a NSO for 24 hours and then treated with ARRY-520 for 48 hours. Electroporated cells are normally more labile and therefore more sensitive to various agents.
Figure 1.
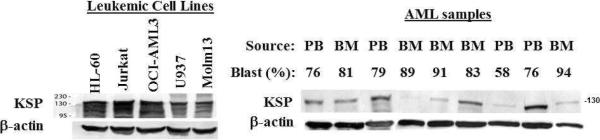
KSP is highly expressed in acute leukemia cell lines and in most samples of AML blasts. KSP levels were determined by western blot analysis. PB, peripheral blood.
Figure 2.
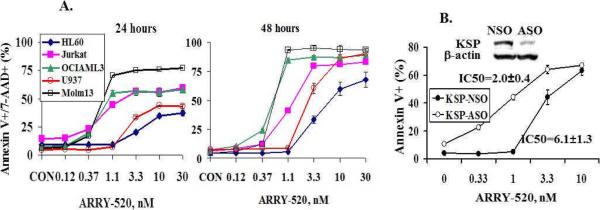
ARRY-520 induces dose- and time-dependent cell death in acute leukemic cells. Cells were treated with ARRY-520 and cell death was determined at 24 and 48 hours by annexin V/7-AAD staining (A). HL-60 cells were transfected with either KSP ASO or NSO for 24 hours and then treated with ARRY-520 for 48 hours. Cell death was determined by annexin V staining (B).
ARRY-520 impairs cell cycle progression and induces cell cycle block, leading to cell death
To determine its effect on cell cycle, we performed cell cycle analysis in OCI-AML3 cells treated with 1 nM ARRY-520. At 24 hours, a significant amount of G2M cells (24.1% versus 12.3% in DMSO control) and sub-G1 cells (26.7% versus 4.9 in DMSO control) were detected (Figure 3A). A time course analysis showed that cell cycle blockage was detected prior to cell death: G2M block was detectable at 6 hours and more prominent at 16 and 24 hours, while cell death was detectable at 16–24 hours and more pronounced at 48 hours (Figure 3B and 3C). TUNEL assay further demonstrated that dead cells were primarily derived from G2M cells (Figure 4). Similar results were obtained with U937 cells (not shown). These results suggest that KSP inhibition induces G2M cell cycle block, which leads to cell death.
Figure 3.

ARRY-520 induces G2M cell cycle block prior to cell death. ARRY-520 (1 nM) induced significant G2M cell cycle block in OCI-AML3 cells at 24 hours (A). Cell cycle distribution at different time points in OCI-AML3 cells treated with ARRY-520 (B). Annexin V positivity at different time points in OCI-AML3 cells treated with ARRY-520 (C).
Figure 4.

ARRY-520 induces primarily G2M cell death. OCI-AML3 cells were treated with 1.1 nM ARRY-520. Cell cycle distribution/cell death was assessed by TUNEL assay at 24 hours.
ARRY-520-induced cell death is independent of p53 status, XIAP levels, and activation of the extrinsic pathway
The finding that p53 wild-type OCI-AML3 and Molm13 cells are very sensitive to ARRY-520 prompted us to examine the role of p53 in ARRY-520-induced cell death. As shown in Figure 5A, ARRY-520 induced the expression of p53 in vector control OCI-AML3vec cells, but not in p53 knockdown OCI-AML3p53shRNA cells, confirming the p53 knockdown status of the cells. However, there were no apparent differences in the levels of apoptosis and cell cycle block between OCI-AML3p53shRNA cells and OCI-AML3vec cells determined by caspase-3 activation (Figure 5A), annexin V positivity (Figure 5B), or PI staining for DNA content (Figure 5C).
Figure 5.

ARRY-520 induces p53-independent cell cycle block and cell death. OCI-AML3vec and OCI-AML3p53shRNA cells were treated with ARRY-520. p53 and caspase-3 activation were determined by western blot at various time points (A), cell death by annexin V staining at 48 hours (B), and cell cycle distribution by PI staining at 24 hours (C).
To test whether XIAP, a potent caspase inhibitor that suppresses post-mitochondrial apoptosis, affects cell sensitivity and whether the activation of the extrinsic pathway is required for ARRY-520 action, we treated XIAP-overexpressing U937 cells (U937XIAP) and caspase-8-mutated Jurkat cells (JurkatI9.2) and their respective control cells (U937neo and Jurkat) with ARRY-520 and found that ARRY-520 had similar efficacy in U937neo and U937XIAP (Figure 6A) and in JurkatI9.2 and Jurkat cells (Figure 6B), regardless of the XIAP levels and caspase-8 status.
Figure 6.
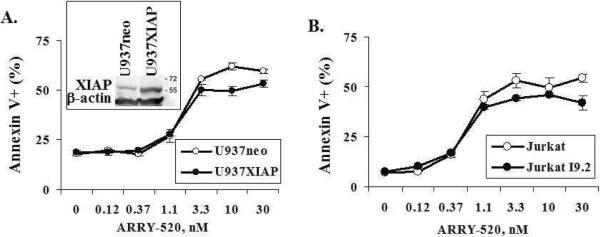
ARRY-520-induced cell death is independent of XIAP levels and activation of the extrinsic apoptotic pathway. XIAP-overexpressing U937 cells (U937XIAP) (A) and caspase-8-muated Jurkat cells (JurkatI9.2) (B) and their respective control cells (U937neo and Jurkat) were treated with ARRY-520, and cell death was determined by annexin V staining at 24 hours.
Activation of the intrinsic mitochondrial pathway is essential for cell death induced by KSP inhibition
Next, we examined the importance of the mitochondrial mediated intrinsic pathway to cell death induced by KSP inhibition. As shown in Figure 7A, ARRY-520 at 10 nM induced significant cell cycle block in both HL-60 and Bcl-2-overexpressing HL-60 (HL-60Bcl-2) cells at 24 hours. However, cell death was observed only in HL-60 cells under this condition, as shown by changes in MMP and annexin V/7-AAD positivity. Even with higher concentrations of ARRY-520 and prolonged treatment, HL-60Bcl-2 cells were resistant to ARRY-520-induced cell death (Figure 7A). These results not only further suggest that KSP inhibition induces cell cycle block leading to cell death but also indicate that KSP inhibition-induced cell death is mediated via the mitochondrial pathway and that overexpression of Bcl-2 abrogated this effect. We next treated HL-60 and HL-60Bcl-2 cells with ARRY-520, the Bcl-2 inhibitor ABT-737, or both. As shown in Figure 7B, at 24 hours, HL-60 cells were sensitive to both ARRY-520 and ABT-737. The combination only slightly increased the killing effect. In contrast, HL-60Bcl-2 cells were resistant to ARRY-520 or ABT-737 alone, but the combination significantly synergized their death (CI=0.011), confirming that Bcl-2 is a potent inhibitory factor of mitotic block-induced cell death. We then examined the protein levels of Bim, a BH3-only protein important in activating mitochondrial apoptotic pathway, in ARRY-520-treated HL-60 cells and found that the Bim level was increased in ARRY-520-treated HL-60 cells and that this increase occurred before caspase-3 activation (Figure 7C). Thus, induction of Bim by ARRY-520 provides a pro-apoptotic signal resulting in apoptosis induction.
Figure 7.

ARRY-520-induced cell death is mediated via the mitochondrial apoptotic pathway. HL-60 and HL-60Bcl-2 cells were treated with ARRY-520. Cell cycle distribution was determined at 24 hours by PI staining, changes in MMP at 24 hours by CMXRos and MitoTracker Green staining, and apoptosis by annexin V/7-AAD staining (A). HL-60 and HL-60Bcl-2 cells were treated with ARRY-520, ABT-737, or both and cell death was determined at 24 hours by annexin V/7-AAD staining (B). HL-60 cells were treated with 10 nM ARRY-520 for various time points, and Bim and caspase-3 levels were determined by western blot analysis (C). ABT, ABT-737; AR, ARRY-520.
ARRY-520 significantly inhibits tumor growth of xenografts in SCID mice
To evaluate its effect in vivo, we treated SCID mice implanted with HL-60 cells with ARRY-520. As shown in Figure 8A, ARRY-520 greatly decreased tumor volumes and all 5 mice showed complete responses (CR) on day 15. The drug was well tolerated with weight loss less than 20% over the course of the study in all animals and rapid recovery after completion of treatment. All the mice were sacrificed and the experiment was terminated on day 26 due to tumor sizes. It needs to be pointed out that although tumor growth was significantly inhibited during ARRY-520 treatment and became undetectable shortly after the treatment, tumors eventually outgrew suggesting that prolonged/repeated treatment is required to achieve better outcome. This is supported by our studies in MV4-11 xenograft in which we initially followed the same treatment schedule. Tumors shrank and became undetectable (Figure 8B) but started to grow back after day 39. We then retreated the mice with ARRY-520 on day 53. All 8 mice were followed through day 60 and 5 of them achieved CR. For the vehicle control group, 3 mice survived until day 28 (the other five were terminated due to excessive tumor burden) and they were then treated with three cycles of ARRY-520 (days 28, 53, and 67) and their tumors responded (Figure 8B). They were followed through day 74.
Figure 8.
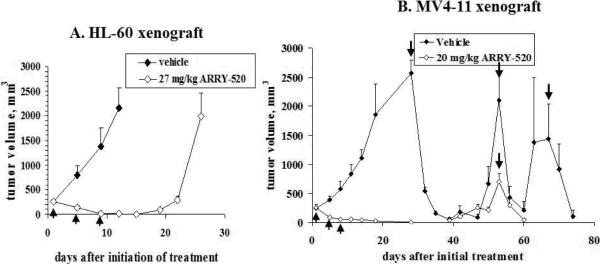
ARRY-520 significantly inhibits tumor growth in HL60 (A) and MV4-11 (B) xenografts of SCID mice. ↑, treatment days (days 1, 5, and 9). ↓, retreatment days (days 28, 53, and 67).
Inhibition of KSP by ARRY-520 significantly diminishes the blast colony-forming potential of AML samples
After demonstrating the effectiveness of KSP inhibition in inducing apoptosis in leukemic cell lines, we next examined the effects of ARRY-520 on primary blast cells from patients with AML. We found that ARRY-520 did not significantly affect the viability of blasts from these patients (results not shown), largely due to the fact that the blasts from AML patients do not proliferate under short-term culture conditions and would therefore not be susceptible to a selective anti-mitotic agent. We then examined the effect of ARRY-520 on the clonogenicity of AML blasts and normal blood cells. We treated BM samples from 5 AML patients (supplementary Table 1) and blood cells from 3 normal samples obtained by apheresis with ARRY-520. As shown in Figure 9, ARRY-520, at nM concentrations, strongly inhibited blast colony formation of BM samples from AML patients (9A), further supporting the antiproliferative role of KSP inhibition. At these concentrations, ARRY-520 did not affect the colony formation of cells from normal samples (9B).
Figure 9.
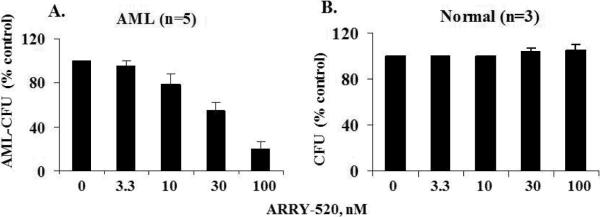
ARRY-520 greatly inhibits the colony formation capacity of BM from patients with AML. BM samples from 5 AML patients (A) and blood cells from 3 normal samples obtained by apheresis (B) were treated with ARRY-520, and CFU was determined. CFU, colony forming units.
Discussion
This study demonstrated that the inhibition of KSP by ARRY-520 impairs cell cycle progression, which leads to cell death in leukemic cell lines via the activation of the intrinsic pathway. This effect is independent of either p53 and XIAP levels or the extrinsic pathway. ARRY-520 strongly inhibited tumor growth of HL-60 xenograft and blocked growth and regrowth of MV4-11 xenograft without apparent toxicity in SCID mice. Further, while the bulk of AML blasts were not responsive to ARRY-520 in vitro, the capacity to form colonies of AML progenitor cells, but not the normal blood cells, was strongly inhibited by ARRY-520 supporting the critical role of KSP in leukemia progenitor cell proliferation.
It has been suggested that defects in the p53-dependent apoptotic pathway decrease the sensitivity of cells to some anti-tumor agents, constituting an obstacle to chemotherapy (26) and that an increase in p53 levels is required for maximal cell sensitivity to microtubule-targeting agents (27;28). In keeping with the notion that p53 expression is induced by DNA damage or DNA replication stress, we found that the inhibition of KSP by ARRY-520 increased p53 levels. However, we observed that ARRY-520-induced cell cycle block and cell death were independent of p53 status, supported by the finding that p53 knockdown cells were as sensitive to ARRY-520 as control cells. In addition, the effectiveness of ARRY-520 was essentially unchanged by XIAP overexpression or by lack of activation of the extrinsic pathway. The finding that caspase-8 mutation did not significantly change the effect of ARRY-520 is also in agreement with other reports that the extrinsic pathway is dispensable for apoptosis induced by microtubule-targeting agents (29;30). Thus, these agents are intriguing cancer therapeutics even in cells with XIAP overexpression or with a defect in p53 signaling or in the extrinsic pathway which is common in leukemic and other malignant cells.
Microtubule-targeting agents are known to induce mitochondrial membrane permeabilization and subsequent caspase activation by modulating Bcl-2 family proteins(31–34). KSP inhibitors are more selective microtubule-targeting agents that only affect spindle microtubules. The exact mechanisms by which these compounds induce cell death are less understood. The data present here demonstrated clearly that ARRY-520-induced cell death is mediated via the mitochondrial pathway. Cell death was significantly blunted in Bcl-2-overexpressing leukemic cells, which was overcome by Bcl-2 inhibition. Indeed, inhibition of Bcl-2 by ABT-737 synergized ARRY-520 in Bcl-2-overexpressing HL-60 cells, with the remarkable CI of 0.01 (Figure 7B).
Time course analysis demonstrated that the level of proapoptotic Bcl-2 protein Bim was increased by ARRY-520 before the activation of caspase-3 suggesting its causative effect on the activation of apoptosis. We observed a decrease in Bim levels in caspase-3-activated cells, which may result from its cleavage by caspase-3. The mechanism by which KSP inhibition induces Bim expression is unclear. Bim has been reported to be positively regulated by FOXO1 transcriptional factor (35) and CDK2-dependent phosphorylation of FOXO1 has been reported to be an apoptotic response to DNA damage and replication stress independent of p53 (36).
Because of their exquisite selectivity and the potential anti-tumor effect, various KSP inhibitors have been developed and their mechanisms of action studied. In accordance with our findings, Tao et al. reported in solid tumor cell lines that KSP-1A, a KSP inhibitor from Merck Research Lab, activates the mitochondrial apoptotic pathway in a p53-independent manner (37).
In the studies reported here, KSP inhibition by ARRY-520 exerted profound anti-proliferative and proapoptotic efficacy, independent of p53 status and XIAP overexpression, but dependent on Bcl-2. On the basis of these findings and comparatively reduced toxicity, ARRY-520 and related compounds warrant further investigation as agents for the treatment of leukemias and other cancers. Of note, a phase 1/2 study of ARRY-520 in patients with advanced myeloid leukemia is accruing patients at MD Anderson Cancer Center.
Supplementary Material
Acknowledgment
The authors thank Elena S. Vess and Betty Notzon for helping with the manuscript and Wenjing Chen for helping with collecting patient information.
Supported in part by grants from the National Institutes of Health (P01 CA49639, P01 CA55164, and CA16672) to M.A and Array BioPharma to B.Z.C.
Footnotes
Supplementary Information accompanies the paper on the Leukemia website http://www.nature.com/leu.
References
- (1).Jordan MA, Wilson L. Microtubules as a target for anticancer drugs. Nat Rev Cancer. 2004;4:253–265. doi: 10.1038/nrc1317. [DOI] [PubMed] [Google Scholar]
- (2).Goodson HV, Kang SJ, Endow SA. Molecular phylogeny of the kinesin family of microtubule motor proteins. J Cell Sci. 1994;107:1875–1884. doi: 10.1242/jcs.107.7.1875. [DOI] [PubMed] [Google Scholar]
- (3).Walczak CE, Mitchison TJ. Kinesin-related proteins at mitotic spindle poles: function and regulation. Cell. 1996;85:943–946. doi: 10.1016/s0092-8674(00)81295-7. [DOI] [PubMed] [Google Scholar]
- (4).Blangy A, Lane HA, d'Herin P, Harper M, Kress M, Nigg EA. Phosphorylation by p34cdc2 regulates spindle association of human Eg5, a kinesin-related motor essential for bipolar spindle formation in vivo. Cell. 1995;83:1159–1169. doi: 10.1016/0092-8674(95)90142-6. [DOI] [PubMed] [Google Scholar]
- (5).Valentine MT, Fordyce PM, Block SM. Eg5 steps it up! Cell Div. 2006;1:31. doi: 10.1186/1747-1028-1-31. 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Hansen GM, Justice MJ. Activation of Hex and mEg5 by retroviral insertion may contribute to mouse B-cell leukemia. Oncogene. 1999;18:6531–6539. doi: 10.1038/sj.onc.1203023. [DOI] [PubMed] [Google Scholar]
- (7).Kaiser A, Brembeck FH, Nicke B, Wiedenmann B, Riecken EO, Rosewicz S. All-trans-retinoic acid-mediated growth inhibition involves inhibition of human kinesin-related protein HsEg5. J Biol Chem. 1999;274:18925–18931. doi: 10.1074/jbc.274.27.18925. [DOI] [PubMed] [Google Scholar]
- (8).Koller E, Propp SS, Hirsch S, Shepard PJ, Dean NM. Inhibition of Eg5 (Kinesin-like 1) with antisense oligonucleotide leads to arrest in G2/M of cell cycle and shows potent antitumor activity against a variety of tumors. Proc Am Assoc Cancer Res. 2003;44 Ref Type: Abstract. [Google Scholar]
- (9).Nowicki MO, Pawlowski P, Fischer T, Hess G, Pawlowski T, Skorski T. Chronic myelogenous leukemia molecular signature. Oncogene. 2003;22:3952–3963. doi: 10.1038/sj.onc.1206620. [DOI] [PubMed] [Google Scholar]
- (10).Carter BZ, Mak DH, Shi Y, Schober WD, Wang RY, Konopleva M, et al. Regulation and targeting of Eg5, a mitotic motor protein in blast crisis CML: overcoming imatinib resistance. Cell Cycle. 2006;5(19):2223–2229. doi: 10.4161/cc.5.19.3255. [DOI] [PubMed] [Google Scholar]
- (11).Burris H, LoRusso P, Jones S, McCormick J, Willcutt N, Hodge J, et al. A phase I study to determine the safety and pharmacokinetics of intravenous administration of SB715992 on a once weekly for three consecutive weeks schedule in patients with refractory solid tumors. Eur J Cancer. 2003;1(5):S172–S173. Ref Type: Abstract. [Google Scholar]
- (12).Williams D, Kathman S, Chu Q, Holen K, Rowinsky E, Wilding G, et al. A phase I study of SB-715992, a novel kinesin spindle protein (KSP) inhibitor: pharmacokinetic (PK)/pharmacodynamic (PD) modeling of absolute neutrophil counts (ANC) Eur J Cancer. 2008;2(8):20–21. Ref Type: Abstract. [Google Scholar]
- (13).Chu Q, Holen KD, Rowinsky EK, Alberti DB, Monroe P, Volkman JL, et al. A phase I study to determine the safety and pharmacokinetics of IV administered SB-715992, a novel kinesin spindle protein (KSP) inhibitor, in patients (pts) with solid tumors. Proc Am Soc Clin Oncol. 2003;22 Ref Type: Abstract. [Google Scholar]
- (14).Holen KD, Belani CP, Wilding G, Ramalingam S, Volkman JL, Ramanathan RK, et al. Phase I study to determine tolerability and pharmacokinetics (PK) of SB-743921, a novel kinesin spindle protein (KSP) inhibitor. J Clin Oncol. 2005 Jan 6;23(16S, Part I of II) Ref Type: Abstract. [Google Scholar]
- (15).Miglarese MR, Carlson RO. Development of new cancer therapeutic agents targeting mitosis. Expert Opin Investig Drugs. 2006;15(11):1411–1425. doi: 10.1517/13543784.15.11.1411. [DOI] [PubMed] [Google Scholar]
- (16).Lemieux C, DeWolf W, Voegtli W, Wallace E, Woessner R, Corrette C, et al. ARRY-520, a novel, highly selective KSP inhibitor with potent anti-proliferative activity. Proc Am Assoc Cancer Res. 2007 Ref Type: Abstract. [Google Scholar]
- (17).Woessner RD, Corrette C, Allen S, Hans J, Zhao Q, Aicher T, et al. ARRY-520, a KSP inhibitor with efficacy and pharmacodynamic activity in animal models of solid tumors. Proc Am Assoc Cancer Res. 2008 Ref Type: Abstract. [Google Scholar]
- (18).Juo P, Kuo CJ, Yuan J, Blenis J. Essential requirement for caspase-8/FLICE in the initiation of the Fas-induced apoptotic cascade. Curr Biol. 1998;8:1001–1008. doi: 10.1016/s0960-9822(07)00420-4. [DOI] [PubMed] [Google Scholar]
- (19).Carter BZ, Mak DH, Schober WD, Dietrich MF, Pinilla C, Vassilev LT, et al. Triptolide sensitizes AML cells to TRAIL-induced apoptosis via decrease of XIAP and p53-mediated increase of DR5. Blood. 2008;111(7):3742–3750. doi: 10.1182/blood-2007-05-091504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Datta R, Oki E, Endo K, Biedermann V, Ren J, Kufe D. XIAP regulates DNA damage-induced apoptosis downstream of caspase-9 cleavage. J Biol Chem. 2000;275:31733–31738. doi: 10.1074/jbc.M910231199. [DOI] [PubMed] [Google Scholar]
- (21).Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- (22).Martin S, Reutelingsperger C, McGahon A, Rader J, van Schie R, LaFace D, et al. Early redistribution of plasma membrane phosphatidylserine is a general feature of apoptosis regardless of the initiating stimulus: inhibition by overexpression of Bcl-2 and ABL. J Exp Med. 1995;182:1545–1556. doi: 10.1084/jem.182.5.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Carter BZ, Milella M, Tsao T, McQueen T, Schober WD, Hu W, et al. Regulation and targeting of antiapoptotic XIAP in acute myeloid leukemia. Leukemia. 2003;17:2081–2089. doi: 10.1038/sj.leu.2403113. [DOI] [PubMed] [Google Scholar]
- (24).Estrov Z, Black RA, Sleath PR, Harris D, Van Q, LaPushin R, et al. Effect of interleukin-1 beta converting enzyme inhibitor on acute myelogenous leukemia progenitor proliferation. Blood. 1995;86:4594–4602. [PubMed] [Google Scholar]
- (25).Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- (26).Lowe SW, Bodis S, McClatchey A, Remington L, Ruley HE, Fisher DE, et al. p53 status and the efficacy of cancer therapy in vivo. Science. 1994;266(5186):807–810. doi: 10.1126/science.7973635. [DOI] [PubMed] [Google Scholar]
- (27).Tan G, Heqing L, Jiangbo C, Ming J, Yanhong M, Xianghe L, et al. Apoptosis induced by low-dose paclitaxel is associated with p53 upregulation in nasopharyngeal carcinoma cells. Int J Cancer. 2002;97(2):168–172. doi: 10.1002/ijc.1591. [DOI] [PubMed] [Google Scholar]
- (28).Esteve MA, Carre M, Braguer D. Microtubules in apoptosis induction: are they necessary? Curr Cancer Drug Targets. 2007;7(8):713–729. doi: 10.2174/156800907783220480. [DOI] [PubMed] [Google Scholar]
- (29).Goncalves A, Braguer D, Carles G, Andre N, Prevot C, Briand C. Caspase-8 activation independent of CD95/CD95-L interaction during paclitaxel-induced apoptosis in human colon cancer cells (HT29-D4) Biochem Pharmacol. 2000;60(11):1579–1584. doi: 10.1016/s0006-2952(00)00481-0. [DOI] [PubMed] [Google Scholar]
- (30).Kim R, Tanabe K, Emi M, Uchida Y, Toge T. Death receptor-dependent and -independent pathways in anticancer drug-induced apoptosis of breast cancer cells. Oncol Rep. 2003;10(6):1925–1930. [PubMed] [Google Scholar]
- (31).Yamaguchi H, Chen J, Bhalla K, Wang HG. Regulation of Bax activation and apoptotic response to microtubule-damaging agents by p53 transcription-dependent and -independent pathways. J Biol Chem. 2004;279(38):39431–39437. doi: 10.1074/jbc.M401530200. [DOI] [PubMed] [Google Scholar]
- (32).Tudor G, Aguilera A, Halverson DO, Laing ND, Sausville EA. Susceptibility to drug-induced apoptosis correlates with differential modulation of Bad, Bcl-2 and Bcl-xL protein levels. Cell Death Differ. 2000;7(6):574–586. doi: 10.1038/sj.cdd.4400688. [DOI] [PubMed] [Google Scholar]
- (33).Li R, Moudgil T, Ross HJ, Hu HM. Apoptosis of non-small-cell lung cancer cell lines after paclitaxel treatment involves the BH3-only proapoptotic protein Bim. Cell Death Differ. 2005;12(3):292–303. doi: 10.1038/sj.cdd.4401554. [DOI] [PubMed] [Google Scholar]
- (34).Esteve MA, Carre M, Braguer D. Microtubules in apoptosis induction: are they necessary? Curr Cancer Drug Targets. 2007;7(8):713–729. doi: 10.2174/156800907783220480. [DOI] [PubMed] [Google Scholar]
- (35).Gilley J, Coffer PJ, Ham J. FOXO transcription factors directly activate bim gene expression and promote apoptosis in sympathetic neurons. J Cell Biol. 2003;162(4):613–622. doi: 10.1083/jcb.200303026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Huang H, Regan KM, Lou Z, Chen J, Tindall DJ. CDK2-dependent phosphorylation of FOXO1 as an apoptotic response to DNA damage. Science. 2006;314(5797):294–297. doi: 10.1126/science.1130512. [DOI] [PubMed] [Google Scholar]
- (37).Tao W, South VJ, Diehl RE, Davide JP, Sepp-Lorenzino L, Fraley ME, et al. An inhibitor of the kinesin spindle protein activates the intrinsic apoptotic pathway independently of p53 and de novo protein synthesis. Mol Cell Biol. 2007;27(2):689–698. doi: 10.1128/MCB.01505-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.