

Abstract
Background
Calcium transient triggered firing (CTTF) is induced by large intracellular calcium (Cai) transient and short action potential duration (APD). We hypothesized that CTTF underlies the mechanisms of early afterdepolarization (EAD) and spontaneous recurrent atrial fibrillation (AF) in transgenic (Tx) mice with overexpression of transforming growth factor β1 (TGF-β1).
Methods and Results
MHC-TGFcys33ser Tx mice develop atrial fibrosis because of elevated levels of TGF-β1. We studied membrane potential and Cai transients of isolated superfused atria from Tx and wild-type (Wt) littermates. Short APD and persistently elevated Cai transients promoted spontaneous repetitive EADs, triggered activity and spontaneous AF after cessation of burst pacing in Tx but not Wt atria (39% vs. 0%, P=0.008). We were able to map optically 4 episodes of spontaneous AF re-initiation. All first and second beats of spontaneous AF originated from the right atrium (4/4, 100%), which is more severely fibrotic than the left atrium. Ryanodine and thapsigargin inhibited spontaneous re-initiation of AF in all 7 Tx atria tested. Western blotting showed no significant changes of calsequestrin or sarco/endoplasmic reticulum Ca2+-ATPase 2a.
Conclusions
Spontaneous AF may occur in the Tx atrium because of CTTF, characterized by APD shortening, prolonged Cai transient, EAD and triggered activity. Inhibition of Ca2+ release from the sarcoplasmic reticulum suppressed spontaneous AF. Our results indicate that CTTF is an important arrhythmogenic mechanism in TGF-β1 Tx atria.
Keywords: Arrhythmia, Atrial fibrillation, Ca2+ triggers, Intracellular calcium, Optical mapping, Transgenic mice models
Fibrosis is a major substrate of atrial fibrillation (AF).1,2 A well-documented mechanism is that atrial fibrosis decreases the safety factor of propagation and promotes anisotropic reentry.3 However, in addition to causing conduction blocks, increased fibrosis also promotes early afterdepolarization (EAD) and triggered activity near the pulmonary veins of aged rat atria during glycolytic inhibition.4 The mechanisms may be related to intracellular calcium (Cai) accumulation, prolongation of the action potential duration (APD) and aging-induced remodeling. However, the vast majority of AF in ambulatory canine models occurs during simultaneous sympathovagal discharges.5 This is because APD shortening (induced by vagal activation), together with Cai elevation (induced by sympathetic activation), facilitates the development of EAD through activation of the inward Na+/Ca2+ exchanger current (INCX).6–10 This phenomenon has been named “late phase 3 EAD”.6,8 However, subsequent studies showed that these EADs can also occur much earlier during repolarization.7,11,12 Therefore, EAD and triggered activity associated with short APD and large Ca2+ transient is also known as “calcium transient triggered firing” (CTTF). This term is used to distinguish this EAD from the phase 2 and phase 3 EADs, which are used to describe EADs associated with APD prolongation, not shortening.13 This novel mechanism of AF was discovered in preparations of canine right atrium (RA) during acetylcholine infusion (which shortens APD) with a long pause that enhances Ca2+ transient.6
Transforming growth factor β1 (TGF-β1) plays a central role in the development of fibrosis and electroanatomical remodeling of the atria.14–16 Transgenic (Tx) mice with cardiac-restricted expression of a constitutively active form of TGF-β1 (MHC-TGFcys33ser mice) have increased atrial fibrosis17 and significantly increased vulnerability to AF by rapid pacing.18 However, it is unclear if fibrosis promotes spontaneous (non-paced) AF, and whether or not CTTF is important in spontaneous AF in this model. Here we hypothesize that CTTF is an important mechanism of spontaneous AF initiation in TGF-β1 Tx atria. The purpose of the present study was to perform simultaneous membrane potential (Vm) and Cai mapping in isolated atria from TGF-β1 Tx and wild-type (Wt) mice during carbachol superfusion to test this hypothesis.
Methods
Detailed methods are included in the Data S1. We used 18 Tx17 and 18 Wt mice for simultaneous optical mapping of Vm and Cai transient; 6 Tx and 6 Wt mice were used for the histological analyses; 6 Wt mice were used to determine the effects of extracellular Ca2+ concentration on APD. An additional 5 Tx and 5 Wt mice were used to determine the Ca-handling protein expressions. Atria were isolated and superfused with Tyrode’s solution containing 5 μmol/L of blebbistatin. We used 10 μmol/L blebbistatin in 6 experiments designed to test the effects of extracellular Ca2+ concentration on APD. After staining with both a Ca-sensitive dye (Rhod-2) and a voltage-sensitive dye (RH-237), the atria were illuminated with laser and optical signals acquired simultaneously with 2 cameras. Inducibility of atrial arrhythmias was tested with and without exposure to carbachol (carbamylcholine or CCH: 1–3 μmol/L, Sigma, St. Louis, MO, USA). Two series of 2-s electrical bursts of 20–40 ms pacing cycle length (PCL; burst pacing) were applied using an automated stimulator coupled with optical mapping acquisition software.18 Ryanodine (3 μmol/L) and thapsigargin (1 μmol/L) were then added to the superfusate for 30 min and the same pacing protocol was repeated. AF is defined as tachycardia with irregular intervals on the pseudo-ECG because of multiple foci of activation, alternating foci of activation or the coexistence of multiple reentrant wavefronts.
Results
As compared with Wt atria, the atria of Tx mice were more white, consistent with increased fibrosis (Figure S1). There were significant differences in the electrophysiological characteristics and AF inducibility between the Wt and the Tx atria.
Reduced Conduction Velocity in Tx Atria
Figure 1A shows wavefront propagation during atrial pacing in Wt and Tx atria (Figure 1A-a,b, respectively). Figure 1B summarizes the means for APD, Cai transient duration (CaiTD) and conduction velocity (CV) in Tx and Wt atria with and without carbachol treatment. Carbachol shortened APD80 by 24.7% in the Tx-RA (P<0.001), 36.0% in the Wt-RA (P=0.002), 24.9% in the Tx left atrium (LA) (P=0.016) and 37.9% in the Wt LA (P=0.011; Figure 1B-a). However, CaiTD80 of the Tx and Wt atria did not change significantly during carbachol exposure (8.7% in Tx-RA, 10.6% in Wt-RA, 9.8% in Tx-LA and 7.6% in Wt-LA, all P>0.05; Figure 1B-b). The Tx RA and LA showed slower CV as compared with the Wt RA and LA, respectively (32.9±2.0 cm/s of Tx-RA vs. 41.1±2.8 cm/s of Wt RA; 33.1±1.6 cm/s of Tx-LA vs. 42.4±4.5 cm/s of Wt-LA, all P<0.001, Figure 1B-c). The lack of difference in APD between Tx and Wt atria, as well as the reduction of CV in Tx atria, is consistent with previous observations in MHC-TGF-cys33ser atria.18
Figure 1.
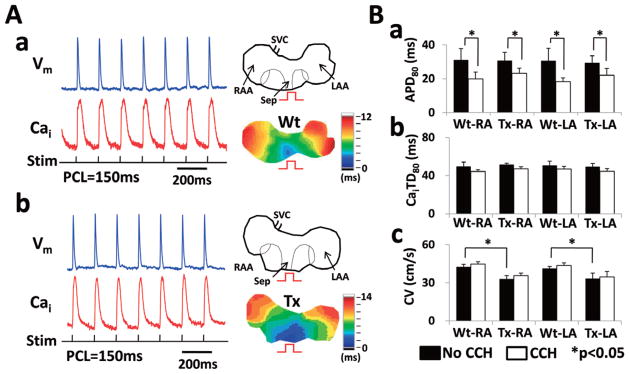
Atrial activation characteristics evaluated by dual optical mapping in wild-type (Wt) and transgenic (Tx) mice. (A) Both Wt and Tx mice showed atrial propagation during rapid atrial pacing (PCL 150 ms). (B) APD80, CaiTD80 and conduction velocity were compared between Wt and Tx mice. SVC, superior vena cava; RAA, right atrial appendage; Sep, septum; LAA, left atrial appendage; PCL, pacing cycle length; CV, conduction velocity; CCH, carbamylcholine; *P<0.05.
Increased Vulnerability to Pacing-Induced AF in Tx Atria
AF was induced by burst pacing at 20–40 ms in 11 of 18 (61%) Tx atria at baseline (in the absence of carbachol), but not in Wt atria (0 of 18, P<0.001). However, AF inducibility during burst pacing in the presence of carbachol was not significantly different between Tx atria (100%) and Wt atria (72%, P>0.05), suggesting that APD shortening similarly facilitated AF induction in both genotypes. A total of 229 AF episodes (43 episodes in Tx atria without carbachol, 107 episodes in Tx atria with carbachol; 79 episodes in Wt atria with carbachol) were analyzed. The combined duration of AF in Tx atria was significantly increased after carbachol superfusion (from 7.4±4.2 s to 17.5±8.4 s, P<0.001). The AF duration after carbachol superfusion was longer in Tx atria than in Wt atria with carbachol exposure (17.5±8.4 s vs. 11.0±4.9 s, P=0.031), whereas AF episodes in Tx atria before carbachol exposure were of similar duration as in Wt atria with carbachol exposure (P>0.05). The PCLs to induce AF in the Tx atria were not significantly different before (26.4±4.1 ms) or during (24.5± 3.2 ms) carbachol superfusion.
EAD and Triggered Activity in Tx But Not Wt Atria
A major difference between Tx and Wt atria is the development of EAD and triggered activity during rapid pacing (70–150 ms). We performed rapid atrial pacing 142 times (80 times without carbachol and 62 times with carbachol) in Tx (7.9±4.5 times per atria) and 143 times (77 times without carbachol and 66 times with carbachol) in Wt atria (7.9±2.3 times per atria); 38 episodes of ectopic beats were documented during rapid pacing in 10 (56%) Tx atria (4 atria without carbachol, 3 atria with carbachol and 3 atria in both conditions). In contrast, no ectopic beats occurred in the Wt atria. The ectopic beats occurred with similar frequency with (22 episodes, or 28%) and without (16 episodes, or 25%, P>0.05) carbachol exposure. Among the 38 episodes of ectopic beats, dual optical mapping was successfully performed in 12 episodes in 6 Tx atria (5 episodes without carbachol and 7 episodes with carbachol). Most of the episodes (10/12, 83%) originated from the RA, and 17% (2/12) were from the LA. Figure 2A shows Vm (blue) and Cai (red) tracings recorded in a typical preparation at the site marked with a blue dot in Figure 2B-a. There was Cai alternans during rapid pacing (Figure 2A-a). The first non-driven beat (Bc in Figure 2A-b) occurred during prolonged Cai transient. Isochronal voltage maps of this and subsequent ectopic beats (Figure 2B-b–e) show consecutive focal discharges from the RA that propagated centrifugally to the rest of the atria. These findings are consistent with triggered activity that occurred after >50% of repolarization of the preceding action potential. Figure 3 shows 6 additional examples of ectopic beats that occurred during prolonged Cai transients. These EADs occur because of a short APD and prolonged Cai transients. The take-off potential of triggered activity is mostly during the mid- to late phase 3.
Figure 2.

Focal discharges induced by rapid atrial pacing in transgenic (Tx) atria. Simultaneous membrane potential (Vm: blue) and intracellular calcium (Cai: red) tracings in Panel A shows apparent Cai alternans during rapid atrial pacing (PCL 70 ms). Vm tracing shows intermittent non-sustained tachycardia. Panel A-b shows the rapid activity in greater detail. The first beat (Bb) is a paced beat and the second (Bc) is a non-driven beat. Panel B-a is a schematic of the mapped region. Panels B-b–e shows the activation isochronal maps of beats Bb, Bc, Bd and Be respectively. Three different sites of focal discharge are depicted (blue, second beat; green, third beat, red, fourth beat; Ba). PCL, pacing cycle length.
Figure 3.

Timing of triggered activities (arrows) at the origin of focal activation. All 6 episodes occurred during large intracellular calcium (Cai) transient. Panels A-a,b show 2 examples of triggered activity during carbachol superfusion; Panel A-c shows triggered activity without carbachol superfusion. All 3 triggered beats occurred in late phase 3 (at 80–100% repolarization). Panel B shows 3 examples of triggered activity without carbachol infusion. The triggered activities occurred during mid-phase 3 of the action potential (50–80% of repolarization).
AF Induced by Sustained Triggered Activity During Rapid Atrial Pacing in Tx But Not Wt Atria
Another unique finding of the Tx atria was the development of triggered activities and AF during rapid atrial pacing (70–150 ms). WT atria showed inducible AF by burst pacing with extremely short PCL (20–40 ms) under carbachol exposure, but there was no AF induced when paced at longer PCLs. In contrast, atrial pacing at 70–150 ms PCL induced 38 episodes of ectopic beats in Tx atria. Most (30 of 38) episodes of ectopic beats were non-sustained (<30 s). However, 7 (18.4%) episodes of AF induced by rapid atrial pacing persisted more than 30 s. Figure 4A shows a representative example (PCL 150 ms). The boxed regions (B,C) are enlarged and shown in Panels 4B and 4C, respectively. There were repetitive ectopic beats with an increasing rate of occurrence (Figures 4B,C), leading to AF that was sustained for 1.0 s at the end of atrial pacing. After a 0.46 s pause, there was spontaneous recurrence of AF, which was sustained for 9.0 s. Membrane depolarization associated with ectopic activity on the ECG originated half-way during the repolarization of the preceding action potential, and were present both during pacing and at the onset of AF (Figure 4D). Figure 4E-a is a schematic of the mapped preparation and 4E-b shows the paced wavefront. Two different foci were mapped in Figures 4E-c,d. The 2nd, 4th, and 11–17th non-driven beats originated from the septum and the 3rd and 5–10th non-driven beats originated from the LA and initiated AF (Figure 4E-c). Note that beat Ec originated from 50% of the repolarization, in the presence of a large Cai transient.
Figure 4.

Ectopic beats and atrial fibrillation (AF) induced by rapid atrial pacing in transgenic (Tx) atria. (A) Pseudo-ECG during and after rapid atrial pacing (PCL 150 ms). The frequency of ectopic beats increased during pacing (B), and changed to spontaneous AF after pacing was halted (C). There were spontaneous recurrences of AF. Panel E-a is a schematic of the mapped region. Panels E-b–d shows the isochronal maps of beats labeled Eb, Ec and Ed in (D). Note that the non-driven beats came from 2 foci (mid-septum and left atrial appendage, respectively). PCL, pacing cycle length.
Spontaneous Re-Initiation of AF in Tx But Not Wt Atria
Spontaneous re-initiation of AF, which is AF episodes that occur spontaneously after the termination of pacing-induced AF, was recorded in 7 of 18 Tx atria and in none of the Wt atria. In 4 of the 7 Tx atria, the re-initiated AF was sustained for >2 s. We recorded a total of 109 AF re-initiation events in these 4 atria after 15 episodes of AF or atrial tachycardia (AT) termination; 10 of 15 AF or AT termination episodes were followed by multiple spontaneous AF re-initiations (range 2–59 events, 6.8±14.3 per atria). Most of the initial AF episodes (n=14, 93%) were induced by 2-s burst atrial pacing during carbachol exposure (PCL 23.0±3.5 ms, range 20–30 ms), and 1 episode in the absence of carbachol exposure after pacing at 150 ms PCL (Figure 4). Most of the re-initiation events (n=13, 87%) were preceded by long episodes of AF (21.3±14.9 s, range 2.8–41.2 s), whereas 2 re-initiation events were preceded by short episodes of AF (0.9 and 1.1 s). The mean duration of re-initiated AF was 8.9±18.8 s (range 2–120 s). We were able to optically map 4 episodes of spontaneous AF re-initiation. All of the first and second beats of spontaneous AF originated from the RA (4/4, 100%). Figure 5 shows the latter episode of spontaneous AF re-initiation. Figure 5A shows the pseudo-ECG, and Figure 5B shows the optical signals at the earliest site of AF re-initiation. Figure 5C-a is a schematic of the mapped region. Figures 5C-b–e shows the isochronal voltage maps of the beats labeled in Figure 5B. Prolonged AT (25.2 s) was induced by 2-s burst atrial pacing (PCL 40 ms). After AT termination, spontaneous AF (3.1 s) occurred after a 0.4-s pause. The first beat (Cc) originated in the RA appendage. A second beat (Cd) occurred during the large Cai transient of the first beat. Isochronal maps show that the origin of activation of both beats was at the same site (Figure 5C-c,d). Figures 5C-e,f shows the patterns of activation of the beats labeled Ce and Cf, respectively, in Figure 5B. This episode was deemed to be AF because of the irregularities of activation on the pseudo-ECG and because more than 1 activation wavefront was present.
Figure 5.

Spontaneous re-initiation of atrial fibrillation (AF) in transgenic (Tx) atria. (A) Pseudo-ECG showing 2 episodes of spontaneous AF re-initiation. (B) Optical signals of intracellular calcium (Cai: red) and membrane voltage (Vm: blue) at the origin of spontaneous AF re-initiation (site 1) and from the origin of a competing activation wavefront from site 2. Note that after the first 2 beats of AF (Cc and Cd), site 2 activated faster than site 1. Panel C-a is a schematic of the mapped RA. An isochronal map during AT shows a single large reentrant wavefront with clockwise rotation (Panel C-b). After a pause, the first beat (Cc, blue) originated from the junction of the right atrial appendage (RAA) and the right atrium (RA) posterior wall, and the second beat (Cd, green) was triggered from the same site in parallel with elevated diastolic Cai. Beats 3–27 (Ce, red) originated from the same repetitive activation site in the septal area (site 2), with 3:2 conduction into site 1. Afterwards, 2 competing activation wavefronts were present (Cf), resulting in beats 28–31.
Effects of Ryanodine and Thapsigargin on Spontaneous Re-Initiation of AF
AF inducibility was not completely inhibited by ryanodine or thapsigargin; 6 of 7 (86%) Tx atria and 4 of 7 (57%) of Wt atria showed inducible AF by burst atrial pacing under carbachol, ryanodine or thapsigargin exposure. The duration of AF induced under carbachol, ryanodine and thapsigargin was significantly shorter than that with carbachol alone (22.5±9.9 vs. 8.8±3.4 s, P=0.037). The PCL to induce AF in Tx atria was not significantly different during carbachol and after ryanodine and thapsigargin superfusion (23.6±4.4 vs. 23.2±3.7 ms). However, ryanodine and thapsigargin effectively prevented all spontaneous AF re-initiation in Tx atria (Figure 6). There was no inducible AF in Wt atria without carbachol (Figure 6A). The Wt atria did not have spontaneous AF initiation (Figure 6C) despite a short APD and a large and persistent Cai transient during late phases 3 and 4 after carbachol exposure (Figures 6B,D). Without CCH, most of the Tx atria did not have spontaneous AF re-initiation (Figure 6E). Importantly, the difference between CaiTD and APD in the Wt vs. Tx atria was small (Figure 6F). Multiple episodes of recurrent AF occurred in a Tx atria during carbachol superfusion (Figure 6G), accompanied by short APD and large Cai transient (Figure 6H). Ryanodine and thapsigargin prevented the re-initiation of AF (Figure 6I). The CaiTD was 48.0±15.4% shorter than at baseline. The amplitude of Cai was significantly decreased, leading to a reduced signal to noise ratio of Cai transient (Figure 6J). The abbreviated CaiTD and the reduced Cai amplitude prevented the persistent Cai elevation during phase 3 or 4 of the action potential.
Figure 6.

Effects of carbachol, ryanodine and thapsigargin on atrial fibrillation (AF) inducibility. (A,C,E,G,I) Pseudo-ECG traces. (B,D,F,H,J) Respective Cai and Vm traces during 150 ms PCL. (A) No AF is inducible in the Wt atria without carbachol (CCH). (C) AF become inducible after CCH exposure, but no spontaneous AF occurs after AF termination. (E) In the absence of CCH, AF is inducible in Tx atria but no spontaneous AF re-initiation occurs after AF termination. (G) During CCH exposure, pacing-induced AF in the Tx atria is followed by recurrent spontaneous AF episodes. (I) Ryanodine and thapsigargin prevent AF recurrence. Although the Tx and Wt atria both show a short APD and large Cai transient after CCH exposure, multiple episodes of recurrent AF occur only in the Tx atria. Cai, intracellular calcium; CCH, carbamylcholine; Ry, ryanodine; Tg, thapsigargin; Tx, transgenic; Vm, membrane potential; Wt, wild-type.
Reentrant Wavefronts During AF in Tx and Wt Atria
We successfully mapped a total of 55 episodes of AF in 12 Tx atria both with (n=40) and without (n=15) carbachol infusion; 47 episodes of AF were mapped in 10 Wt atria during carbachol exposure. Among the mapped AF episodes in the Wt mice, 22 (47%) episodes were characterized by typical reentrant excitation. In contrast, the patterns of activation during AF in the Tx atria were accompanied by large reentrant wave fronts present at multiple locations. Figure 7A shows a representative AF episode induced directly by 2-s burst atrial pacing (PCL 22 ms) in Tx atria. Figure 7B shows the activation sequence of a reentrant wavefront in the RA. Figure 7C-a is a schematic of the preparation showing the pectinate muscle. In a cumulative phase-singularity map (Figure 7C-b), the highest density of phase singularities (indicated by white signal) occurred around the junction of right atrial appendage (RAA) and RA, where the pectinate muscular structures are located. Consistent with a previous report in the canine RA,19 the location of the phase singularity superimposed with the pectinate muscle. In addition to a dominant rotor in the RA, there were also phase singularities in the septum and LA. Typical large reentrant wavefronts, such as the ones shown in Figure 7C, were also present in 34 of 55 episodes of AF mapped in the Tx atria. Among them, 19 occurred with and 15 occurred without carbachol exposure.
Figure 7.

Reentrant wavefronts and atrial fibrillation (AF) in transgenic (Tx) atria. (A) Pseudo-ECG showing induction of AF by rapid pacing. (B) Vm and Cai tracings recorded during reentrant excitation at sites (1–5) marked in Panel C-c. Panel C-a is a schematic of the mapped region. Panel C-b shows the cumulative phase-singularity distribution. Panel C-c shows a typical phase map during AF. There was phase singularity (black arrow) in the RA (Panel C-c). However, additional phase singularities are also present in the septum and LA (blue arrows). All of them are represented in the cumulative phase map (Panel C-b). Cai, intracellular calcium; LA, left atrium; RA, right atrium; Vm, membrane potential.
Western Blot Analyses
There were no differences in calsequestrin or sarco/endoplasmic reticulum Ca2+-ATPase 2a expression between Tx and Wt in either the atria or the ventricles (Figure S6).
Discussion
Table summarizes the major results of the study. A major difference between Tx and Wt atria is that the Tx atria are prone to EAD, triggered activity and spontaneous AF during carbachol infusion. In contrast, reentrant activities may be induced in both types of atria by burst pacing. Inhibition of sarcoplasmic reticulum function by ryanodine and thapsigargin effectively suppressed spontaneous AF recurrence but not the initial induction of AF. These findings support the conclusion that EADs play an important role in spontaneous AF in TGF-β1 Tx atria when APD is abbreviated by cholinergic stimulation.
Table.
Summary of Results
| % (number of atria) | Wt atria
|
Tx atria
|
||
|---|---|---|---|---|
| No CCH | CCH | No CCH | CCH | |
| Ectopic beats during rapid atrial pacing* | 0% (0/18) | 0% (0/18) | 39% (7/18) | 33% (6/18) |
|
| ||||
| EAD and focal discharges (CTTF) | Absent | Absent | 5 episodes in 3 atria | 7 episodes in 3 atria |
|
| ||||
| AF inducibility† | 0% (0/18) | 72% (13/18) | 61% (11/18) | 100% (18/18) |
|
| ||||
| AF duration (mean ± SD, s) | 0 | 11.0 ± 4.9 | 7.4 ± 4.2 | 17.5 ± 8.4 |
|
| ||||
| Spontaneous AF without ryanodine/thapsigargin | 0% (0/18) | 0% (0/18) | 6% (1/18) | 39% (7/18) |
|
| ||||
| Spontaneous AF with ryanodine/thapsigargin | Not tested | 0% (0/7) | Not tested | 0% (0/7) |
Rapid atrial pacing with 70–50 ms PCL from the lower interatrial septum;
burst atrial pacing with 20–40 ms PCL for 2 s. CCH, carbamylcholine; EAD, early afterdepolarization; CTTF, calcium transient triggered firing; AF, atrial fibrillation: PCL, pacing cycle length.
Mechanisms of Afterdepolarization and Triggered Activity
It is postulated that rapid pacing, sympathetic stimulation and long pauses facilitate a large release of Ca2+ from the sarcoplasmic reticulum. If the APD is shortened by acetylcholine infusion or vagal stimulation, then INCX may be activated, leading to EAD and triggered activity originating from the late or mid-phase 3 of the action potential.6,7,11,12 This phenomenon is also known as CTTF.7,20 In the present study, we showed that Cai transient was unchanged while APD is significantly abbreviated in both Wt and Tx mice in the presence of CCH. Although CCH induced similar shortening of APD and no change of CaiTD in Tx and Wt mice, spontaneous recurrence of AF occurred only in Tx mice. These findings suggest that reduced CV and cell-to-cell coupling in TGF-β1 Tx atria play important roles in the induction of afterdepolarization and triggered activity in Tx mice.
Importance of Fibrosis in Afterdepolarization and Triggered Activity
A plausible explanation for the increased arrhythmogenesis in Tx mice is the electrotonic inhibition of pacemaking cells by non-pacemaking cells. Wilders et al21 used computer simulation to study the importance of intercellular coupling conductance (Gc) on arrhythmogenesis and found that at high Gc, automaticity is suppressed by the loading effects of the surrounding cells. At intermediate Gc, the ectopic activity may propagate into the surrounding structure. Increased fibrosis might reduce Gc among myocytes and facilitate the development of triggered activity. In agreement with a previous study,22 fibrosis is more severe in the RA than in the LA of MHC-TGFcys33ser mice. In addition, we demonstrated that most of the triggered activities and all of the first and second beats after spontaneous AF re-initiation originated from the RA. These findings indicate that the severity of fibrosis is important in the initiation of AF. TGF-β1 is also a potent trigger of myofibroblast activation.23 Myofibroblasts are known to form heterocellular gap junctions with cardiomyocytes24 and promote arrhythmogenesis by altering CV and promoting reentry of cultured cells in vitro.25 Indeed, decreased conduction velocity was observed in the Tx atria via optical mapping or multi-electrode array recordings.18 However, whether or not effective myofibroblast–myocyte coupling occurs in intact atria remains unclear.
Variations of APD in Isolated Mouse Atrial Preparations
The APD values in Wt mice have varied considerably in different studies. Nygren et al26 reported APD70 of 14–15 ms and Chelu et al27 reported APD80 of 24±1 ms in superfused Wt mouse atria using superfused atrial preparations. Bagwe et al reported APD70 of 20.7±5.7 ms and APD90 of 34±11 ms in a Langendorff-perfused preparation.28 The APD distribution in mouse atria is highly heterogeneous, with the RA having a longer APD than the LA.26 Multiple mechanisms could contribute to the variations of APD, including superfusion vs. perfusion, pacing rate, APD measurements to 70%, 80% or 90% of repolarization, effects of optical dyes and electromechanical uncoupler, and serum electrolyte concentrations. In the Data S1, we report that the APD80 measured in 1.8 mmol/L of Ca2+ is much longer than that measured in 0.9 mmol/L Ca2+, consistent with the fact that single L-type Ca2+ channel conductance increases steeply over the range of 0.9–1.8 mmol/L in rodents.29 In addition, because of the limitation of oxygen perfusion, the center of superfused atrial tissues is most likely ischemic. The ischemic cells may have IKATP activation, which through electrotonic coupling could shorten the APD of the non-ischemic cells on the surface. The APD measured with optical mapping techniques in superfused tissues is therefore shorter than that measured with glass microelectrodes, which usually sample from the top layer of well-perfused cells. In the present study, it is likely that the use of 0.9 mmol/L of Ca2+, carbachol infusion and superfused preparations all contributed to the short APD and the development of CTTF. However, because recurrent AF occurred only in the Tx atria and not the Wt atria under the same experimental conditions, the results support the hypothesis that fibrosis contributes to the development of CTTF and spontaneous AF.
Study Limitations
TGF-β1 has many downstream signaling targets that may also contribute to the arrhythmogenesis in this model. We proposed that focal discharges are consistent with activation originated by triggered activity. However, as shown in Figure S3, the thickness of the atrial wall is probably insufficient to accommodate a persistent transmural reentrant wavefront. It is unlikely that these focal discharges were wavefronts initiated by epicardial breakthrough of a transmural rotor. Finally, the mouse atria have abundant apamin-sensitive K currents (IKAS),30 which may be activated and shorten the APD during rapid pacing. However, we do not know if there is differential expression of IKAS in Tx and Wt atria to explain the increased incidence of spontaneous AF in Tx atria.
Conclusions
TGF-β1 overexpression was accompanied by increased atrial fibrosis, vulnerability to EADs, triggered activity and spontaneous recurrence of AF. Pharmacological inhibition of sarcoplasmic reticulum Ca2+ release effectively suppressed spontaneous AF recurrence. Our results support a role of Ca2+-dependent triggered activity in spontaneous AF recurrence in the atria of mice with TGF-β1 overexpression.
Supplementary Material
Figure S1. Photograph of a typical preparation of isolated mouse atrium, showing the positioning of the electrogram and stimulus.
Figure S2. Dominant frequency (DF) map during atrial fibrillation (AF) in wild-type (Wt) and transgenic (Tx) mice.
Figure S3. Histological findings.
Figure S4. Histological evaluation of transgenic (Tx) and wild-type (Wt) atria.
Figure S5. Effects of pacing cycle length (PCL) on Ca transient duration (CaTD80).
Figure S6. Western blot analyses.
Acknowledgments
We thank Dorothy Field, Lei Lin and Jian Tan for their assistance and Drs Sufen Wang and Miguel Valderrabano for helping us with mapping the isolated atrial preparation.
Funding Sources
This work was supported by National Institutes of Health grants P01 s HL78931, R01 HL78932, R01 HL71140 to P.-S.C; and P01 HL85098, R01 HL83126, R01 HL75165 and R21 HL091189 to LJF; a Korea Research Foundation Grant (KRF-2008-357-E00028, 2011-0026089) funded by the Korean Government (E.C.), a Nihon Kohden/St Jude Medical electrophysiology fellowship (M.M.); an American Heart Association Established Investigator Award (S.L.), a Piansky Family Trust (M.C.F.) and a Medtronic-Zipes endowments (P.-S.C.).
Footnotes
Disclosures
None.
References
- 1.Benjamin EJ, Chen PS, Bild DE, Mascette AM, Albert CM, Alonso A, et al. Prevention of atrial fibrillation: Report from a National Heart, Lung, and Blood Institute workshop. Circulation. 2009;119:606–618. doi: 10.1161/CIRCULATIONAHA.108.825380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tanaka K, Zlochiver S, Vikstrom KL, Yamazaki M, Moreno J, Klos M, et al. Spatial distribution of fibrosis governs fibrillation wave dynamics in the posterior left atrium during heart failure. Circ Res. 2007;101:839–847. doi: 10.1161/CIRCRESAHA.107.153858. [DOI] [PubMed] [Google Scholar]
- 3.Spach MS, Dolber PC. Relating extracellular potentials and their derivatives to anisotropic propagation at a microscopic level in human cardiac muscle: Evidence for electrical uncoupling of side-to-side fiber connections with increasing age. Circ Res. 1986;58:356–371. doi: 10.1161/01.res.58.3.356. [DOI] [PubMed] [Google Scholar]
- 4.Ono N, Hayashi H, Kawase A, Lin SF, Li H, Weiss JN, et al. Spontaneous atrial fibrillation initiated by triggered activity near the pulmonary veins in aged rats subjected to glycolytic inhibition. Am J Physiol Heart Circ Physiol. 2007;292:H639–H648. doi: 10.1152/ajpheart.00445.2006. [DOI] [PubMed] [Google Scholar]
- 5.Tan AY, Zhou S, Ogawa M, Song J, Chu M, Li H, et al. Neural mechanisms of paroxysmal atrial fibrillation and paroxysmal atrial tachycardia in ambulatory canines. Circulation. 2008;118:916–925. doi: 10.1161/CIRCULATIONAHA.108.776203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burashnikov A, Antzelevitch C. Reinduction of atrial fibrillation immediately after termination of the arrhythmia is mediated by late phase 3 early afterdepolarization-induced triggered activity. Circulation. 2003;107:2355–2360. doi: 10.1161/01.CIR.0000065578.00869.7C. [DOI] [PubMed] [Google Scholar]
- 7.Patterson E, Lazzara R, Szabo B, Liu H, Tang D, Li YH, et al. Sodium-calcium exchange initiated by the Ca2+ transient: An arrhythmia trigger within pulmonary veins. J Am Coll Cardiol. 2006;47:1196–1206. doi: 10.1016/j.jacc.2005.12.023. [DOI] [PubMed] [Google Scholar]
- 8.Burashnikov A, Antzelevitch C. Late-phase 3 EAD. A unique mechanism contributing to initiation of atrial fibrillation. Pacing Clin Electrophysiol. 2006;29:290–295. doi: 10.1111/j.1540-8159.2006.00336.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Watanabe I, Okumura Y, Nagashima K, Ohkubo K, Ashino S, Kofune M, et al. Electrical remodeling in fibrillating canine atrium: Action potential alternans during rapid atrial pacing and late phase 3 early afterdepolarization after cessation of rapid atrial pacing. Int Heart J. 2010;51:354–358. doi: 10.1536/ihj.51.354. [DOI] [PubMed] [Google Scholar]
- 10.Chen PS, Joung B, Shinohara T, Das M, Chen Z, Lin SF. The initiation of the heart beat. Circ J. 2010;74:221–225. doi: 10.1253/circj.cj-09-0712. [DOI] [PubMed] [Google Scholar]
- 11.Patterson E, Po SS, Scherlag BJ, Lazzara R. Triggered firing in pulmonary veins initiated by in vitro autonomic nerve stimulation. Heart Rhythm. 2005;2:624–631. doi: 10.1016/j.hrthm.2005.02.012. [DOI] [PubMed] [Google Scholar]
- 12.Patterson E, Jackman WM, Beckman KJ, Lazzara R, Lockwood D, Scherlag BJ, et al. Spontaneous pulmonary vein firing in man: Relationship to tachycardia-pause early afterdepolarizations and triggered arrhythmia in canine pulmonary veins in vitro. J Cardiovasc Electrophysiol. 2007;18:1067–1075. doi: 10.1111/j.1540-8167.2007.00909.x. [DOI] [PubMed] [Google Scholar]
- 13.Maruyama M, Lin SF, Xie Y, Chua SK, Joung B, Han S, et al. Genesis of phase 3 early afterdepolarizations and triggered activity in acquired long-QT syndrome. Circ Arrhythm Electrophysiol. 2011;4:103–111. doi: 10.1161/CIRCEP.110.959064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baudino TA, Carver W, Giles W, Borg TK. Cardiac fibroblasts: Friend or foe? Am J Physiol Heart Circ Physiol. 2006;291:H1015–H1026. doi: 10.1152/ajpheart.00023.2006. [DOI] [PubMed] [Google Scholar]
- 15.Katoh Y, Nakazato Y. Can we predict electroanatomical remodeling of left atrium in patients with non-valvular atrial fibrillation by transforming growth factor-β and tissue inhibitor of metalloproteinase-1? Circ J. 2011;75:536–537. doi: 10.1253/circj.cj-11-0071. [DOI] [PubMed] [Google Scholar]
- 16.Kim SK, Park JH, Kim JY, Choi JI, Joung B, Lee MH, et al. High plasma concentrations of transforming growth factor-beta and tissue inhibitor of metalloproteinase-1: Potential non-invasive predictors for electroanatomical remodeling of atrium in patients with non-valvular atrial fibrillation. Circ J. 2011;75:557–564. doi: 10.1253/circj.cj-10-0758. [DOI] [PubMed] [Google Scholar]
- 17.Nakajima H, Nakajima HO, Salcher O, Dittie AS, Dembowsky K, Jing S, et al. Atrial but not ventricular fibrosis in mice expressing a mutant transforming growth factor-β1 transgene in the heart. Circ Res. 2000;86:571–579. doi: 10.1161/01.res.86.5.571. [DOI] [PubMed] [Google Scholar]
- 18.Verheule S, Sato T, Everett T, Engle SK, Otten D, Rubart-von der LM, et al. Increased vulnerability to atrial fibrillation in transgenic mice with selective atrial fibrosis caused by overexpression of TGF-beta1. Circ Res. 2004;94:1458–1465. doi: 10.1161/01.RES.0000129579.59664.9d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu TJ, Yashima M, Xie F, Athill CA, Kim YH, Fishbein MC, et al. Role of pectinate muscle bundles in the generation and maintenance of intra-atrial reentry: Potential implications for the mechanism of conversion between atrial fibrillation and atrial flutter. Circ Res. 1998;83:448–462. doi: 10.1161/01.res.83.4.448. [DOI] [PubMed] [Google Scholar]
- 20.Nakagawa H, Scherlag BJ, Patterson E, Ikeda A, Lockwood D, Jackman WM. Pathophysiologic basis of autonomic ganglionated plexus ablation in patients with atrial fibrillation. Heart Rhythm. 2009;6:S26–S34. doi: 10.1016/j.hrthm.2009.07.029. [DOI] [PubMed] [Google Scholar]
- 21.Wilders R, Wagner MB, Golod DA, Kumar R, Wang YG, Goolsby WN, et al. Effects of anisotropy on the development of cardiac arrhythmias associated with focal activity. Pflugers Arch. 2000;441:301–312. doi: 10.1007/s004240000413. [DOI] [PubMed] [Google Scholar]
- 22.Nakajima H, Nakajima HO, Dembowsky K, Pasumarthi KB, Field LJ. Cardiomyocyte cell cycle activation ameliorates fibrosis in the atrium. Circ Res. 2006;98:141–148. doi: 10.1161/01.RES.0000197783.70106.4a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cucoranu I, Clempus R, Dikalova A, Phelan PJ, Ariyan S, Dikalov S, et al. NAD(P)H oxidase 4 mediates transforming growth factor-β1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circ Res. 2005;97:900–907. doi: 10.1161/01.RES.0000187457.24338.3D. [DOI] [PubMed] [Google Scholar]
- 24.Miragoli M, Gaudesius G, Rohr S. Electrotonic modulation of cardiac impulse conduction by myofibroblasts. Circ Res. 2006;98:801–810. doi: 10.1161/01.RES.0000214537.44195.a3. [DOI] [PubMed] [Google Scholar]
- 25.Zlochiver S, Munoz V, Vikstrom KL, Taffet SM, Berenfeld O, Jalife J. Electrotonic myofibroblast-to-myocyte coupling increases propensity to reentrant arrhythmias in two-dimensional cardiac monolayers. Biophys J. 2008;95:4469–4480. doi: 10.1529/biophysj.108.136473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nygren A, Lomax AE, Giles WR. Heterogeneity of action potential durations in isolated mouse left and right atria recorded using voltage-sensitive dye mapping. Am J Physiol Heart Circ Physiol. 2004;287:H2634–H2643. doi: 10.1152/ajpheart.00380.2004. [DOI] [PubMed] [Google Scholar]
- 27.Chelu MG, Sarma S, Sood S, Wang S, van Oort RJ, Skapura DG, et al. Calmodulin kinase II-mediated sarcoplasmic reticulum Ca2+ leak promotes atrial fibrillation in mice. J Clin Invest. 2009;119:1940–1951. doi: 10.1172/JCI37059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bagwe S, Berenfeld O, Vaidya D, Morley GE, Jalife J. Altered right atrial excitation and propagation in connexin40 knockout mice. Circulation. 2005;112:2245–2253. doi: 10.1161/CIRCULATIONAHA.104.527325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rubart M, Patlak JB, Nelson MT. Ca2+ currents in cerebral artery smooth muscle cells of rat at physiological Ca2+ concentrations. J Gen Physiol. 1996;107:459–472. doi: 10.1085/jgp.107.4.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu Y, Tuteja D, Zhang Z, Xu D, Zhang Y, Rodriguez J, et al. Molecular identification and functional roles of a Ca2+-activated K+ channel in human and mouse hearts. J Biol Chem. 2003;278:49085–49094. doi: 10.1074/jbc.M307508200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Photograph of a typical preparation of isolated mouse atrium, showing the positioning of the electrogram and stimulus.
Figure S2. Dominant frequency (DF) map during atrial fibrillation (AF) in wild-type (Wt) and transgenic (Tx) mice.
Figure S3. Histological findings.
Figure S4. Histological evaluation of transgenic (Tx) and wild-type (Wt) atria.
Figure S5. Effects of pacing cycle length (PCL) on Ca transient duration (CaTD80).
Figure S6. Western blot analyses.