

Abstract
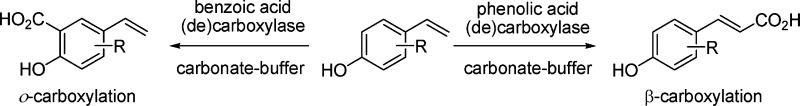
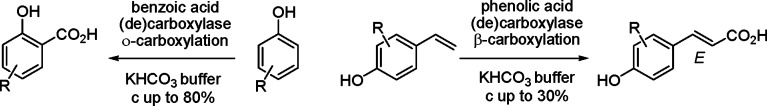
The enzymatic carboxylation of phenol and styrene derivatives using (de)carboxylases in carbonate buffer proceeded in a highly regioselective fashion: Benzoic acid (de)carboxylases selectively formed o-hydroxybenzoic acid derivatives, phenolic acid (de)carboxylases selectively acted at the β-carbon atom of styrenes forming (E)-cinnamic acids.
In order to alleviate the predominant dependence of the chemical industry from petroleum-based platform intermediates,1 the development of CO2-fixation reactions would allow conversion of a problematic waste gas into a useful carbon source for the production of chemicals.2 The biggest obstacle to surmount is the low energy level of CO2. Carboxylation is facilitated by using high-energy starting materials, such as H2, organometallics,3 unsaturated compounds (alkenes, alkynes, allenes4), or epoxides.5 Alternatively, carboxylation toward low-energy products containing carbon at a high oxidation state yields carbonates, carbamates, carboxylic acids, and esters or lactones. Although the number of processes based on chemical CO2-fixation is small, the volumes of production (e.g., urea, Kolbe–Schmitt reaction for phenol-carboxylation6) are impressive.7 Despite these isolated success stories, the use of CO2 as raw material for organic synthesis is still heavily underdeveloped.2,8 In this context, the biocatalytic CO2-fixation catalyzed by (de)carboxylases holds great potential. Analysis of biological CO2-fixation reveals that the major high-energy pathways9 are more substrate-specific and thus are less likely to be adapted to non-natural organic compounds.10 In contrast, detoxification pathways are more promising, because the removal of a multitude of toxins by a single broad-spectrum enzyme enhances the survival of the living system. Since carboxylation is a redox-independent process, it is ideally suited for the detoxification of phenolics in anaerobic organisms,10,11 which represents a biocatalytic equivalent to the Kolbe–Schmitt reaction,6 which requires pressurized CO2 and elevated temperatures (120–300 °C) and often suffers from incomplete regioselectivities.
Although the biodegradation of phenolic compounds via carboxylation by whole microbial cells is reasonably well understood,11b the respective enzymes were predominantly investigated for their (downhill) decarboxylation activities.12 In contrast, only limited data are available on the enzymatic carboxylation of (hetero)aromatics using (de)carboxylases running in the reverse (synthetic) direction:
-
(i)
p-Carboxylation of phenol yielding p-hydroxybenzoic acid is catalyzed by phenylphosphate carboxylase,11b,13 which requires activation of the substrate by (energy-consuming) phosphorylation with ATP prior to carboxylation. In contrast, 4-hydroxybenzoate decarboxylase was found to catalyze the direct (reverse) carboxylation of phenol at a slow rate14 (max. conversion 19%15).
-
(ii)
The regio-complementary o-carboxylation of phenol was catalyzed by salicylic acid decarboxylase with a respectable conversion of 27%.16 Most interestingly, the carboxylation of m-aminophenol selectively gave the antituberculostatic agent p-aminosalicylic acid with 70% conversion.17
-
(iii)
1,2-Dihydroxybenzene (catechol) was selectively carboxylated at the o-position by 3,4-dihydroxybenzoate decarboxylase in up to 28% conversion.18 The 1,3-analog (resorcinol) was carboxylated at the 2-position by 2,6-dihydroxybenzoate decarboxylase in up to 48% conversion.19 Although the enzyme was completely regioselective on its 'natural' substrate, it was also able to convert phenol, 1,2-dihydroxybenzene, and m-aminophenol at very low rates.20
-
(iv)
In contrast to the carboxylases mentioned above, which strictly depend on the presence of a phenolic functional group in the substrate, electron-excess heteroaromatic species (e.g., pyrrole, indole) were carboxylated at position 2 or 3, respectively, by pyrrole-2- carboxylate decarboxylase (max. conversion 80%21) and indole-3-carboxylase (max. conversion 34%22) (Scheme 1). Unfortunately, both enzymes appear to be highly substrate specific and only tolerate minimal structural variations.
Scheme 1. Regio-complementary Enzymatic Carboxylation of Phenols and Hydroxystyrene Derivatives.
In addition to the benzoic acid (de)carboxylases discussed above, which catalyze the carboxylation of an aromatic system, phenolic acid decarboxylases23 act on the side chain of hydroxycinnamic acids yielding styrenes. The reverse carboxylation activity of the latter enzymes is unknown. Based on the limited structural data available to date, both types of enzymes act through completely different mechanisms: Whereas benzoic acid decarboxylases are metal-dependent and require a catalytically active Zn2+ in the active site,24 the mechanism of phenolic acid decarboxylases proceeds via general (metal-independent) acid–base catalysis.25
In order to ensure the practical applicability of the enzymatic carboxylation, we avoided oxygen-sensitive enzymes13a,14a,14b,15 and thus selected (de)carboxylases, which are known to be oxygen-stable.
The following candidates were cloned and (over)expressed in E. coli BL21 (DE3) as host: 2,3-Dihydroxybenzoate decarboxylase from Aspergillus oryzae (2,3-DHBD_Ao),26 2,6-dihydroxybenzoate decarboxylase from Rhizobium sp. (2,6-DHBD_Rs),27 salicylic acid decarboxylase from Trichosporon moniliiforme (SAD_Tm),28 phenolic acid decarboxylases from Lactobacillus plantarum and Bacillus amyloliquefaciens (PAD_Lp,29 PAD_Ba).30 Carboxylation reactions were performed in glass vials using whole lyophilized E. coli cells (showing activities of 1.3 U/mg (PDC_Lp)) in phosphate buffer. After addition of substrate (10 mM) and KHCO3 (3 M, pH 8.5), the samples were shaken at 30 °C and 120 rpm. After extractive workup (24 h) reversed-phase HPLC was used to determine the conversion. Blank-experiments using empty E. coli host cells ensured the absence of competing (de)carboxylation activities (see Supporting Information).
For the mapping of the substrate tolerance, the candidate enzymes were tested using a representative set of phenols (1a–3a), as well as dihydroxybenzene (4a–6a) and styrene derivatives (7a–9a) bearing electron-donating or -withdrawing groups. Since the application of CO2-pressurized reaction conditions (up to 2 bar) did not have any significant effects on the conversion, it was assumed that the cosubstrate more likely is (bi)carbonate rather than CO2, which equilibrates rather slowly. Attempts to speed up CO2/(bi)carbonate equilibration using carbonic anhydrase failed, which was most likely caused by inhibition of carbonic anhydrase by the phenolic substrates.31 Variation of the (hydrogen) carbonate concentration within a range of 0.1–4.0 M revealed that a 3 M KHCO3 solution buffered at pH 8.5 gave an optimum in terms of reaction rate and conversion. All products were identified by comparison with authentic reference materials via coinjection on HPLC/UV and/or NMR.
The results depicted in Table 1 reveal the following trends: overall, all benzoic acid decarboxylases showed an exclusive regioselectivity for the carboxylation of the aromatic system in the o-position of the 'directing' phenolic group;32 no trace of regioisomeric p-carboxylation products, which are often formed in the Kolbe–Schmitt reaction, could be detected. In the absence of a free o-position (9a), no reaction occurred. Surprisingly, no clear preference for the corresponding 'natural' substrates (1a with SAD_Tm, 4a with 2,3-DHBD_Ao, 5a with 2,6-DHBD_Rs) could be detected, and several cross-activities within the same range of conversion (1a with 2,3-DHBD_Ao, 4a with 2,6-DHBD_Rs and SAD_Tm, 5a with 2,3-DHBD_Ao and SAD_Tm) suggest that the substrate tolerance of these enzymes was remarkably broad. Only 2,6-DHBD_Rs was inactive on phenol (1a). Fortunately, also non-natural substrates, such as 1-naphthol (2a) and m-aminophenol (3a), were regioselectively carboxylated to the corresponding o-products 2b and 3b in up to 62 and 80% conversion, respectively. Surprisingly, even olivetol (6a) bearing a sterically demanding C5-alkyl chain was regioselectively carboxylated to yield 6b in up to 56% conversion. In contrast, steric effects seem to play a major role for the o-carboxylation of styrene-type substrates: whereas 7a was carboxylated in up to 58% conversion, 8a gave 29% conversion at best with 2,3-DHBD_Ao and SAD_Tm; 2,6-DHBD_Rs was unreactive. In the absence of a free o-position (9a), no reaction took place, and the vinylic group always remained untouched. Overall, whereas 2,6-DHBD_Rs showed a clear preference for dihydroxybenzene derivatives, 2,3-DHBD_Ao also accepted simple phenols.
Table 1. Enzymatic Carboxylation of Phenols and Styrene Derivatives Using Benzoic and Phenolic Acid Decarboxylasesa.
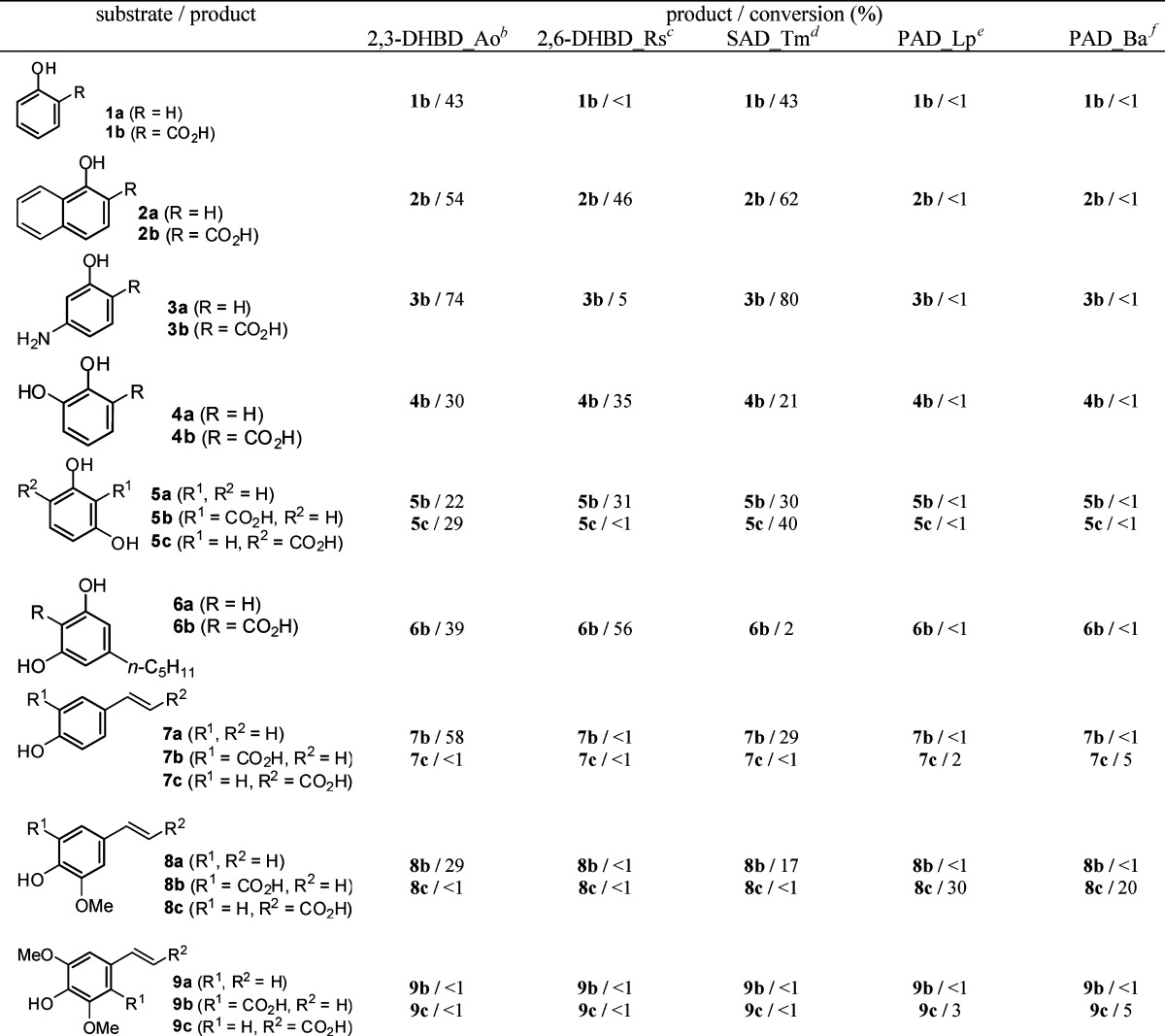
Reaction conditions: whole lyophilized E. coli cells containing overexpressed (de)carboxylase (30 mg), substrate (10 mM), KHCO3 (3 M), Pi buffer (pH 8.5, 100 mM), 30°C, 120 rpm, 24 h.
2,3-DHBD_Ao = 2,3-dihydroxybenzoate decarboxylase (Aspergillus oryzae).26
2,6-DHBD_Rs = 2,6-dihydroxybenzoate decarboxylase (Rhizobium sp.).27
SAD_Tm = salicylic acid decarboxylase (Trichosporon moniliiforme).28
PAD_Lp = phenolic acid decarboxylase (Lactobacillus plantarum).29
PAD_Ba = phenolic acid decarboxylase (Bacillus amyloliquefaciens).30
In contrast to benzoic acid decarboxylases, both phenolic acid decarboxylases PAD_Lp and PAD_Ba showed complete regio-complementary behavior: The aromatic system remained completely unaffected, and carboxylation selectively occurred at the β-carbon atom of the side chain by forming the corresponding (E)-cinnamic acid derivatives (7c–9c) in low to modest conversion (cmax. 30%). This biocatalytic transformation is remarkable, because (to the best of our knowledge) it does not have any direct counterpart in the arsenal of chemical catalysis, the closest analogue being the Ni-catalyzed hydrocarboxylation of styrenes,33 which forms 2-arylpropionic acids, rather than cinnamic acid derivatives. Detailed studies on the exploitation of the regio-complementary carboxylation of styrene derivatives are underway.
Acknowledgments
This study was performed in cooperation within the strategic research programme of the Austrian Centre of Industrial Biotechnology (ACIB, funded through the FFG-COMET-Program) and the DK Molecular Enzymology (Project W9), and support by the FWF, BMWFJ, BMVIT, SFG, Standortagentur Tirol and ZIT is gratefully acknowledged. Klaus Zangger (University of Graz, Department of Chemistry) is thanked for his great support in NMR spectroscopy. Byung-Gee Kim (School of Chemical and Biological Engineering, Seoul National University, Seoul, South Korea) is cordially thanked for the generous donation of phenolic acid decarboxylase plasmids.
Supporting Information Available
Gene sequences, cloning and overexpression of (de)carboxylases, SDS-PAGE analysis, general carboxylation procedure, blank experiments using empty host cells, synthesis of substrates and reference materials, analytical procedures, optimization of carbonate concentration, CO2 pressure experiments, and NMR spectra are given in the electronic Supporting Information. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- The major intermediates derived from crude oil encompass methanol, alkenes (ethene, propene, butene, butadiene), and aromatics (benzene, toluene, xylenes).
- a Behr A.Angewandte Homogene Katalyse; Wiley-VCH: Weinheim, 2008; Chapter 28, pp 441–464. [Google Scholar]; b Aresta M.; Dibenedetto A. Dalton Trans. 2007, 2975–2992. [DOI] [PubMed] [Google Scholar]; c Huang K.; Sun C.-L.; Shi Z.-J. Chem. Soc. Rev. 2011, 40, 2435–2452. [DOI] [PubMed] [Google Scholar]; d Baiker A. Appl. Organomet. Chem. 2000, 14, 751–762. [Google Scholar]; e Omae I. Catal. Today 2006, 115, 33–52. [Google Scholar]; f Zevenhoven R.; Eloneva S.; Teir S. Catal. Today 2006, 115, 73–79. [Google Scholar]; g Aresta M., Ed. Carbon Dioxide as Chemical Feedstock; Wiley-VCH: Weinheim, 2010. [Google Scholar]
- a Zhang L.; Cheng J.; Ohishi T.; Hou Z. Angew. Chem., Int. Ed. 2010, 49, 8670–8673. [DOI] [PubMed] [Google Scholar]; b Bernhardt S.; Metzger A.; Knochel P. Synthesis 2010, 3802–3810. [Google Scholar]
- Zhang Y.-G.; Riduan S. N. Angew. Chem., Int. Ed. 2011, 50, 6210–6212. [DOI] [PubMed] [Google Scholar]
- Decortes A.; Castilla A. M.; Kleij A. W. Angew. Chem., Int. Ed. 2010, 49, 9822–9837. [DOI] [PubMed] [Google Scholar]
- a Ijima T.; Yamaguchi T. Tetrahedron Lett. 2007, 48, 5309–5311. [Google Scholar]; b Kosugi Y.; Rahim M. A.; Takahashi K.; Imaoka Y.; Kitayama M. Appl. Organomet. Chem. 2000, 14, 841–843. [Google Scholar]; c Kolbe H.; Lautermann E. Ann. 1860, 115, 157–206. [Google Scholar]; d Schmitt R. J. Prakt. Chem. 1885, 31, 397–411. [Google Scholar]; e Lindsey A. S.; Feskey H. Chem. Rev. 1957, 57, 583–614. [Google Scholar]
- It has been estimated that ca. 56.1 Mt of CO2 are “chemically recycled” by these processes to date.
- a Behr A. Angew. Chem., Int. Ed. Engl. 1988, 27, 661–678. [Google Scholar]; b Marks T. J. Chem. Rev. 2001, 101, 953–996. [DOI] [PubMed] [Google Scholar]; c Louie J. Curr. Org. Chem. 2005, 9, 605–623. [Google Scholar]; d Quadrelli E. A.; Centi G.; Duplan J.-L.; Perathoner S. ChemSusChem 2011, 4, 1194–1215. [DOI] [PubMed] [Google Scholar]; e Martin R.; Kleij A. W. ChemSusChem 2011, 4, 1259–1263. [DOI] [PubMed] [Google Scholar]
- Calvin–Benson–Bassham cycle, Arnon–Buchanan cycle, Wood–Ljungdahl pathway, acetyl-CoA carboxylase pathways.
- Glueck S. M.; Gümüs S.; Fabian W. M. F.; Faber K. Chem. Soc. Rev. 2010, 39, 313–328. [DOI] [PubMed] [Google Scholar]
- a Brackmann R.; Fuchs G. Eur. J. Biochem. 1993, 213, 563–571. [DOI] [PubMed] [Google Scholar]; b Boll M.; Fuchs G. Biol. Chem. 2005, 386, 989–997. [DOI] [PubMed] [Google Scholar]; c Gobson J.; Harwood C. S. Annu. Rev. Microbiol. 2002, 56, 345–369. [DOI] [PubMed] [Google Scholar]; d Ackermann L. Angew. Chem., Int. Ed. 2011, 50, 3842–3844. [DOI] [PubMed] [Google Scholar]
- Liu A.; Zhang H. Biochemistry 2006, 45, 10407–10411. [DOI] [PubMed] [Google Scholar]
- a Aresta M.; Quaranta E.; Liberio R.; Dileo C.; Tommasi I. Tetrahedron 1998, 54, 8841–8846. [Google Scholar]; b Dibenedetto A.; Lo Noce R.; Pastore C.; Aresta M.; Fragale C. Environ. Chem. Lett. 2006, 3, 145–148. [Google Scholar]; c Schühle K.; Fuchs G. J. Bacteriol. 2004, 186, 4556–4567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Liu J.; Zhang X.; Zhou S.; Tao P.; Liu J. Curr. Microbiol. 2007, 54, 102–107. [DOI] [PubMed] [Google Scholar]; b He Z.; Wiegel J. Eur. J. Biochem. 1995, 229, 77–82. [DOI] [PubMed] [Google Scholar]; c Lupa B.; Lyon D.; Shaw L. N.; Sieprawska-Lupa M.; Wiegel J. Can. J. Microbiol. 2008, 54, 75–81. [DOI] [PubMed] [Google Scholar]
- Matsui T.; Yoshida T.; Hayashi T.; Nagasawa T. Arch. Microbiol. 2006, 186, 21–29. [DOI] [PubMed] [Google Scholar]
- Kirimura K.; Gunji H.; Wakayama R.; Hattori T.; Ishii Y. Biochem. Biophys. Res. Commun. 2010, 394, 279–284. [DOI] [PubMed] [Google Scholar]
- Kirimura K.; Yanaso S.; Kosaka S.; Koyama K.; Hattori T.; Ishii Y. Chem. Lett. 2011, 40, 206–208. [Google Scholar]
- a He Z.; Wiegel J. J. Bacteriol. 1996, 178, 3539–3543. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Yoshida T.; Inami Y.; Matsui T.; Nagasawa T. Biotechnol. Lett. 2010, 32, 701–705. [DOI] [PubMed] [Google Scholar]
- This enzyme is also termed γ-resorcylate decarboxylase. ; a Yoshida M.; Fukuhara N.; Oikawa T. J. Bacteriol. 2004, 186, 6855–6863. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Ishii Y.; Narimatsu Y.; Iwasaki Y.; Arai N.; Kino K.; Kirimura K. Biochem. Biophys. Res. Commun. 2004, 324, 611–620. [DOI] [PubMed] [Google Scholar]; c Yoshida T.; Hayakawa Y.; Matsui T.; Nagasawa T. Arch. Microbiol. 2004, 181, 391–397. [DOI] [PubMed] [Google Scholar]
- a Iwasaki Y.; Kino K.; Nishide H.; Kirimura K. Biotechnol. Lett. 2007, 29, 819–822. [DOI] [PubMed] [Google Scholar]; b Matsui T.; Yoshida T.; Yoshimura T.; Nagasawa T. Appl. Microbiol. Biotechnol. 2006, 73, 95–102. [DOI] [PubMed] [Google Scholar]
- a Wieser M.; Yoshida T.; Nagasawa T. J. Mol. Catal. B: Enzym. 2001, 11, 179–184. [Google Scholar]; b Wieser M.; Fujii N.; Yoshida T.; Nagasawa T. Eur. J. Biochem. 1998, 257, 495–499. [DOI] [PubMed] [Google Scholar]; c Wieser M.; Yoshida T.; Nagasawa T. Tetrahedron Lett. 1998, 39, 4309–4310. [Google Scholar]
- Yoshida T.; Fujita K.; Nagasawa T. Biosci. Biotechnol. Biochem. 2002, 66, 2388–2394. [DOI] [PubMed] [Google Scholar]
- These enzymes are also denoted as hydroxycinnamic acid decarboxylase, ferulic acid decarboxylase, or p-coumaric acid decarboxylase. ; a Rodriguez H.; Landete J. M.; Curiel J. A.; de las Rivas B.; Mancheno J. M.; Munoz R. J. Agric. Food Chem. 2008, 56, 3068–3072. [DOI] [PubMed] [Google Scholar]; b Zago A.; Degrassi G.; Bruschi C. V. Appl. Environ. Microbiol. 1995, 61, 4484–4486. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Cavin J.-F.; Barthelmebs L.; Divies C. Appl. Environ. Microbiol. 1997, 63, 1939–1944. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Gu W.; Li X.; Huang J.; Duan Y.; Meng Z.; Zhang K.-Q.; Yang J. Appl. Microbiol. Biotechnol. 2011, 89, 1797–1805. [DOI] [PubMed] [Google Scholar]; e Prim N.; Pastor F. I. J.; Diaz P. Appl. Microbiol. Biotechnol. 2003, 63, 51–56. [DOI] [PubMed] [Google Scholar]
- Goto M.; Hayashi H.; Miyahara I.; Hirotsu K.; Yoshida M.; Oikawa T.. Crystal structures of nonoxidative Zn-dependent 2,6-dihydroxybenzoate (γ-resorcylate) decarboxylase from Rhizobium sp. strain Mtp-10005, pdb accession number entry 2DVT_A. [DOI] [PubMed]
- a Matte A.; Grosse S.; Bergeron H.; Abokitse K.; Lau P. C. K. Acta Crystallogr., Sect. F 2010, F66, 1407–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Gu W.; Yang J.; Lou Z.; Liang L.; Sun Y.; Huang J.; Li X.; Cao Y.; Meng Z.; Zhang K.-Q. PLoS ONE 2011, 6, e16262, DOI: 10.1371/journal.pone.0016262. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Rodriguez H.; Angulo I.; de las Rivas B.; Campillo N.; Paez J. A.; Munoz R.; Mancheno J. M. Proteins 2010, 78, 1662–1676. [DOI] [PubMed] [Google Scholar]
- NCBI Reference GI: 94730373. Santha R.; Appaji Rao N.; Vaidyanathan C. S. Biochim. Biophys. Acta 1996, 1293, 191–200. [DOI] [PubMed] [Google Scholar]
- NCBI Reference GI: 116667102; see ref (19a).
- NCBI Reference GI: 225887918; see ref (16).
- NCBI Reference GI: 300769086; see ref (25c).
- NCBI Reference GI: 308175189.
- Lindskog S. Pharmacol. Ther. 1997, 74, 1–20. [DOI] [PubMed] [Google Scholar]
- Preliminary data indicate that the phenolic moiety seems to play an important role in catalysis, since the nonphenolic substrates tested were unreactive (data not shown).
- a Williams C. M.; Johnson J. B.; Rovis T. J. Am. Chem. Soc. 2008, 130, 14936–14937. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zhang Y.; Riduan S. N. Angew. Chem., Int. Ed. 2011, 50, 6210–6212. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.