

Abstract
The AP-1 transcription factor modulates a wide range of cellular processes, including cellular proliferation, programmed cell death, and survival. JunD is a major component of the AP-1 complex following liver ischemia/reperfusion (I/R) injury; however, its precise function in this setting remains unclear. We investigated the functional significance of JunD in regulating AP-1 transcription following partial lobar I/R injury to the liver, as well as the downstream consequences for hepatocellular remodeling. Our findings demonstrate that JunD plays a protective role, reducing I/R injury to the liver by suppressing acute transcriptional activation of AP-1. In the absence of JunD, c-Jun phosphorylation and AP-1 activation in response to I/R injury were elevated, and this correlated with increased caspase activation, injury, and alterations in hepatocyte proliferation. The expression of dominant negative JNK1 inhibited c-Jun phosphorylation, AP-1 activation, and hepatic injury following I/R in JunD−/− mice but, paradoxically, led to an enhancement of AP-1 activation and liver injury in JunD+/− littermates. Enhanced JunD/JNK1-dependent liver injury correlated with the acute induction of diphenylene iodonium-sensitive NADPH-dependent superoxide production by the liver following I/R. In this context, dominant negative JNK1 expression elevated both Nox2 and Nox4 mRNA levels in the liver in a JunD-dependent manner. These findings suggest that JunD counterbalances JNK1 activation and the downstream redox-dependent hepatic injury that results from I/R, and may do so by regulating NADPH oxidases.
Transient tissue ischemia, caused by either surgical intervention or environmentally induced injury, can lead to a cascade of pathological events orchestrated by reoxygenation of the tissue. One example of this type of injury is liver ischemia/reperfusion (I/R)3 following transplantation and/or surgical resection, and this can significantly influence patient survival (1–4). The pathology of liver I/R injury is biphasic, involving an acute stage of reperfusion-initiated cellular changes that are mediated by increased oxygen tension to the ischemic organ and an ensuing stage characterized by an inflammatory response (5, 6). Both of these phases result in organ damage, and both appear to involve the production of abnormal levels of reactive oxygen species (ROS). Indeed, numerous studies suggest that the formation of oxygen free radicals following reoxygenation may initiate the cascade of hepatocellular injury, necrosis/apoptosis, and inflammatory cell infiltration characteristic of this pathology (7–10). Therefore, modulating oxidative stress responses in transplanted organs has traditionally been considered as a rational approach to decreasing the complications associated with I/R damage.
The AP-1 transcription complex is widely recognized as an important redox-activated factor involved in I/R liver injury. The family of AP-1 transcription factors consists of three main groups: the Jun proteins (c-Jun, JunB, and JunD), the Fos proteins (c-Fos, FosB, Fra1, and Fra2), and the activating transcription factors (ATF2, ATF3, and B-ATF) (11). Members of these families can form homodimers or heterodimers that make up the active AP-1 complex. In addition to the regulation of AP-1 complexes by their dimer protein composition, post-translational modifications that alter the DNA binding and transactivation abilities of these complexes play important roles in controlling their activity (12, 13). For example, c-Jun NH2-terminal kinase (JNK)-mediated phosphorylation of c-Jun on serines 63 and 73 of its NH2 terminus influences the transcriptional activity of the AP-1 complex (14). A second post-translational mechanism of AP-1 involves reduction-oxidation at a conserved cysteine residue within the DNA binding domains of Fos and Jun (15).
Previous studies have shown that I/R injury promotes the activation of JNK1, as well as the induction of AP-1 DNA binding complexes composed of Jun heterodimers, during the reperfusion phases of injury (16, 17). In this context, we have previously shown that hepatic activation of AP-1 following I/R injury primarily induces heterodimers composed of JunD and c-Jun (17). Although these two forms of Jun share a high degree of sequence homology, they have different transactivation properties and opposing effects on cellular proliferation, whereas c-Jun is a positive regulator of cell growth, JunD can function as a negative regulator of cell growth (18–22). In the context of liver I/R injury, hepatocytes undergo temporally regulated proliferation in a bimodal fashion, as evident from peaks in PCNA expression during the acute and subacute phases of reperfusion injury (23). However, it remains unclear whether these observed changes in cellular proliferation are coordinated by AP-1 transcriptional activity.
In this study, we sought to address the role of AP-1 transcriptional activation following hepatic I/R injury by monitoring the expression of an AP-1-responsive luciferase reporter in vivo. Additionally, we wanted to better understand the functional significance of c-Jun/JunD heterodimers in the liver following I/R injury. Results from these studies demonstrate that JunD deletion significantly enhances hepatic transcriptional activation of AP-1 following I/R, and these changes correlated with enhanced cellular proliferation and increased liver injury. The expression of dominant negative JNK1 (dnJNK1) in the liver prior to I/R significantly altered the temporal pattern of AP-1 transcriptional activation, hepatic injury, and NADPH-dependent superoxide production by the liver in a JunD-dependent manner. These findings suggest that JNK1 and JunD collaborate to control NADPH-regulated redox stress in the liver following I/R and that these changes may influence AP-1 transcriptional programs that determine hepatocellular fates.
EXPERIMENTAL PROCEDURES
Mouse Model of Partial Lobar I/R Injury
JunD knock-out mice used in this study were described previously (24). The JunD knock-out mice were bred onto a C57BL/6J background for 3–5 generations during the course of rederival into a specific pathogen-free barrier facility and maintained by breeding heterozygous JunD+/− males with knock-out JunD−/− females. Because these mice were not inbred, all experimental comparisons were performed with littermates from the colony (offspring from the mating scheme gave rise to litters composed of ~50% heterozygous and ~50% knock-out mice). Of note, male JunD−/− mice had reduced fertility and were not used for breeding. Male JunD+/− mice were compared with JunD−/− littermates in partial lobar liver I/R experiments as described previously (25). In brief, mice were anesthetized and injected with heparin (100 μg/kg) to prevent clotting of blood during lobar ischemia. The largest medial lobe of the liver was clamped at its base with a micro-aneurysm clamp, followed by placement of the liver and clamp back into the peritoneal cavity for 45 or 60 min. Following surgically implemented ischemia, the clamp was removed and the abdominal wall sutured, and the animals were returned to their cages. Removal of the clamp signified the start of reperfusion. The ischemic lobe of livers were harvested at 0, 6, 9, 12, 18, and/or 24 h after the initiation of reperfusion for experimental analysis.
Gene Delivery to the Liver Using Recombinant Adenovirus
Recombinant adenoviruse vectors Ad.LacZ (25) and Ad.dnJNK1 (26) were described previously. Ad.AP-1Luc virus was generated using a fragment from the pAP1(PMA)-TA-Luc plasmid (Clontech) containing the firefly luciferase gene, driven by six tandem copies of the AP1 enhancer linked to the minimal TATA box promoter from the herpes simplex virus thymidine kinase gene. An XhoI/NotI fragment from pAP1(PMA)-TA-Luc was inserted into a promoterless adeno-viral shuttle plasmid (pAd5mcspA), and Ad.AP-1Luc virus was generated by homologous recombination as described previously (25, 27). Viral infections with Ad.LacZ and Ad.dnJNK1 were performed 3 days before liver I/R surgery, by tail vein injection with 1011 particles of purified virus per 25 g of body weight in 200 μl of phosphate-buffered saline. Ad.AP-1Luc infections were performed at a lower dose of 5 × 1010 particles per 25 g of body weight.
Western Blot Analysis
Ischemic liver lobes were washed in phosphate-buffered saline and then homogenized in 2 ml of homogenization buffer (0.3 M sucrose, 10 mM HEPES (pH 7.6), 10 mM KCl, 0.74 mM spermidine, 0.15 mM spermine, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM dithiothreitol, 0.5 mM phenyl-methylsulfonyl fluoride, and 1 Complete protease inhibitor mixture tablet (Roche Applied Science) for 50 ml). 100 μg of protein was separated on a denaturing 10% SDS-PAGE and transferred to a nitrocellulose membrane. PCNA and p53 protein levels were determined by Western blot analysis using anti-PCNA and anti-p53 antibodies (Santa Cruz Biotechnology). Analysis of c-Jun phosphorylation was performed by immuno-precipitation of c-Jun from 1 mg of ischemic liver lobe lysate followed by Western blotting with a phosphoserine 63 c-Jun-specific antibody. Both the pan c-Jun (catalog number sc-45) and p-c-Jun(Ser-63) (catalog number sc-822) antibodies were from Santa Cruz Biotechnology. Westerns blots were quantified on a LI-COR Biosciences Odyssey Infrared Imaging System using infrared-labeled secondary antibody.
Luciferase Indicator Assays for AP-1 Transcriptional Activation
AP-1 transcriptional activity in the ischemic lobe of the liver was assessed using an Ad.AP-1Luc vector. 100 μg of total protein from each liver lysate was used to perform luciferase assays. Luciferase activity was measured using a kit from Promega (Madison, WI).
Electrophoretic Mobility Shift Assays for AP-1 DNA Binding
Nuclear extracts were generated from the ischemic lobe of the liver as described previously (25). Protein content was measured using a Bradford assay, and electrophoretic mobility shift assays (EMSAs) were performed as described previously with little modification (17). In brief, 6 μl of nuclear extract was incubated in EMSA buffer (250 mM KCl, 100 mM HEPES (pH 7.9), 25% glycerol, 5 mM EDTA, 5 mmol/liter dithiothreitol) with bovine serum albumin (1 μg/μl), poly(dI-dC) (1 μg/μl) and double-stranded, 32P-end-labeled oligonucleotide (200,000 cpm) in a total volume of 20 μl. This mixture was incubated for 30 min at room temperature before being separated on a 4% native polyacrylamide gel. The oligonucleotide sequence used for the AP-1 double-stranded probe was 5′-CGCTTGATGACTCAGCCGGAA-3′.
Serum GPT Assay for Liver Damage
Blood was collected from animals at the time of liver harvest at various times post-reperfusion. Blood samples were centrifuged for 2 min at 8,000 rpm to separate the serum from the cells. Serum transaminase (GPT) levels were measured using a microkinetic assay (2–6 μl of serum) as described previously (25).
Isolation of Liver Endomembranes and Chemiluminescence Assays for NADPH-dependent Superoxide ( ) Production
Liver tissue (ischemic lobe of the liver) was lysed in the homogenization buffer described above by nitrogen cavitation. 600 μg of crude lysate was then centrifuged at 3,000 × g to remove the heavy mitochondria and nuclei, and the post-nuclear supernatant was subsequently centrifuged at 100,000 × g for 1 h to pellet total endomembranes. The membranes were washed three times in homogenization buffer before being resuspended in 100 μl of homogenization buffer. NADPH oxidase activities were analyzed by measuring the rate of generation using a chemiluminescent, lucigenin-based system as described previously (28). In brief, 5 μM lucigenin in 50 μl of each endomembrane fraction was used to calculate the relative rate of production following the addition of β-NADPH at a final concentration of 100 μM, with or without 10 μM diphenylene iodonium (DPI).
Quantification of Nox Transcripts
JunD+/− and JunD−/− mice were infected with 1.0 × 1011 particles (intravenously) of either Ad.LacZ or Ad.dnJNK 3 days prior to harvesting the livers. Total RNA was purified from the liver using a standard guanidine thiocyanate solution and ultracentrifugation CsCl2 banding procedure. Poly(A) mRNA was purified from the total mRNA using the Micro-FastTract 2.0 mRNA mini kit (Invitrogen). Reverse transcription was performed on the mRNA samples using the High Capacity cDNA reverse transcription kit (Applied Biosystems) with random priming and a reaction volume of 100 μl. Reverse transcriptase negative samples were also carried along as a control for mRNA specificity in the PCR. Each sample was evaluated by real time PCR for Nox2, Nox4, and β-actin mRNA copies. For the three respective amplification reactions a Nox2, Nox4, and β-actin master mix was generated (per reaction: 10 μl of iQ SYBR Green Supermix, 375 nM primer 1, 375 nM primer 2, in a total volume of 16 μl). The primers used were as follows: Nox2, 5′-ACGCCCTTTGCCT-CCATTCTCAAGTC-3′ and 5′-ATGCGTGTCCCTGCACA-GCCAGTAG-3′; Nox4, 5′-TTCGAGGATCACAGAAGGTC-CCTAGCA-3′ and 5′-GGCGGCTACATGCACACCTGAG-AAA-3′; and β-actin, 5′-CCTGAACCCTAAGGCCAACC-GTGAAA-3′ and 5′-TACGACCAGAGGCATACAGGGAC-AGCA-3′. 16 μl of a given master mix and 4 μl of a cDNA sample were combined for each reaction. For each cDNA sample, three replicate real time reactions were prepared using each of the three sets of primers, for a total of nine reactions per liver sample. Additionally, to accurately quantify copy number, a series of standards were created using known copies numbers of Nox2, Nox4, and β-actin plasmids. Samples were run on a Bio-Rad MyIQ real time machine (step 1, heat at 95 °C for 2 min; step 2, 45 cycles alternating between 95 °C for 30 s and 71 °C for 15 s). Ct values for each reaction were used to calculate copy number of each cDNA transcript against the standard curves. Finally, the copy number of each Nox transcript was normalized to the copy number of the β-actin internal control. This normalization controlled for slight variations in the quality/quantity of each cDNA preparation used in each PCR.
RESULTS
JunD Modulates AP-1 Transcriptional Activation following Liver I/R Injury
Following I/R injury to the liver, increased nuclear DNA binding activity by AP-1 heterodimers composed of c-Jun and JunD occurs within the first 3 h of reperfusion (17, 25). Using a recombinant adenoviral vector encoding an AP-1 responsive luciferase gene, we sought to determine the transcriptional status of AP-1 following liver I/R. In light of these previous findings, we were surprised to find that maximal AP-1 transcriptional activation in JunD+/− animals occurred at 12 h after the initiation of hepatic reperfusion (Fig. 1A). Interestingly, JunD-deficient littermates demonstrated significantly earlier onset (6–9 h) and enhanced (more than 3-fold) AP-1 transcriptional induction (Fig. 1A), suggesting that JunD suppresses the transcriptional activity of the AP-1 complex during the early stages of reperfusion injury. Comparative analysis in these animals demonstrated an inverse relationship between DNA binding by AP-1 (Fig. 1B) and in vivo transcriptional activation mediated by AP-1. In both JunD+/− and JunD−/− animals, DNA binding by AP-1 complexes exhibited a bimodal distribution, peaking at 3–6 h, diminishing at intermediate time points and rising again by 18–24 h. Thus, maximal transcriptional activation by AP-1, which occurred at 12 h post-reperfusion, correlated with a decline in AP-1 binding activity. We interpret these results to suggest that AP-1-mediated transcription following I/R undergoes both activating and inhibitory stages. During the activating stages (3–6 h), similar levels of DNA binding were seen in JunD-deficient and control animals, supporting the notion that the AP-1 complex was more active for transcription in the absence of JunD. Furthermore, JunD ablation led to an earlier and accentuated reduction in nuclear AP-1 DNA binding during the refractory stage when DNA binding activity is inhibited (9–12 h).
FIGURE 1. JunD inhibits AP-1-mediated transcriptional activation, but not DNA binding, following liver I/R injury.

JunD−/− and JunD+/− mice were infected (intravenously) with Ad.AP-1Luc 3 days prior to I/R injury (45 min of partial lobar hepatic ischemia followed by the indicated times of reper-fusion). The 0-h time point represents non-I/R-injured control animals. A, AP-1 transcriptional activity was determined by measuring luciferase activity in liver lysates that were generated at the indicated post-reperfusion time points. Results depict the mean (±S.E.) for n = 4 animals tested at each experimental point. RLU, relative light units. B, nuclear extracts were also generated at each of the reperfusion time points and used in EMSAs to evaluate the DNA binding activity of AP-1. The positions of the AP-1-shifted and free probes are marked by arrows to the left of the gel. The samples on this gel are representative of those for four animals tested at each experimental point. Because of the limited number of lanes on a single gel, the JunD+/− zero time point is not shown but demonstrated no AP-1 shift as shown for JunD−/− mice.
JunD Protects the Liver from Injury following I/R
To determine the consequences of AP-1 activation in the liver following I/R injury and to investigate the role of JunD in this process, we evaluated the extent of liver injury by measuring serum transaminase (GPT) levels. These studies revealed significantly greater I/R liver injury in JunD-deficient mice as compared with heterozygous littermates, when the animals underwent 45 min of ischemia (Fig. 2A). Furthermore, when longer times of ischemia were used (1 h), lethality in JunD−/− mice within the first 90 min of reperfusion was significantly increased above that of JunD+/− littermates (Fig. 2B). This increased liver injury, as well as increased lethality, following liver I/R injury in the JunD−/− animals correlated with an elevation of caspase activity in liver homogenates during the early stages (6 h) of reperfusion (Fig. 2C). In JunD+/− littermates, by contrast, caspase activation in response to I/R treatment peaked much later (18 h post-reperfusion) and was attenuated in comparison with that seen in the JunD knock-out mice. In summary, these findings demonstrate that in the absence of JunD, enhanced AP-1-mediated transcriptional activation during the acute stage of reperfusion correlates with increased liver damage.
FIGURE 2. JunD protects against I/R-induced liver injury and alters hepatocellular remodeling.

JunD−/− and JunD+/− mice were subjected to I/R injury, with partial lobar hepatic ischemia taking place for 45 min (A, C, and D) or 60 min (B), followed by the indicated times of reperfusion. A, serum samples were collected by cardiac puncture at the indicated time points following the start of reperfusion and assayed for GPT. Results depict the mean (±S.E.) units/liter of GPT for n = 4 animals in each group. B, survival of mice following 60 min of ischemia was plotted using a Kaplan-Meier survival curve, p < 0.0004. C, caspase 3/7 activity in whole cell lysates from liver was determined using a caspase-Glo detection kit, for which a substrate emits light when cleaved by either caspase 3 or 7. Results depict the mean (±S.E.) relative light units (RLU). D, for each experimental point, pooled liver lysates from three animals were evaluated for PCNA and p53 expression by Western blotting (WB). The 0-h time point in A, C, and D represents the respective values (serum glutathione S-transferase, caspase 3/7 activity, or PCNA/p53 levels) for non-I/R injured control animals (i.e. noninjured base-line values).
JunD Alters Hepatocellular Responses following I/R Injury
JunD has been shown to slow cell cycle progression by inhibiting the G1-to-S phase transition (18, 19, 22). Previous studies on I/R injury to the liver have also demonstrated that PCNA expression (an index of cellular proliferation) is tightly regulated during the early stages of reperfusion injury, as indicated by its bi-modal expression pattern (23). We hypothesized that the lack of JunD expression might alter cell cycle progression during the acute stages of I/R injury and thereby lead to impaired hepatic repair (remodeling) mechanisms. Analysis of JunD+/− animals reproduced the previously described bi-modal expression of PCNA (23), with peaks at 3 and 18 h post-reperfusion (Fig. 2D). In contrast, this I/R-induced bi-modal response was dysregulated in mice lacking JunD, remaining elevated up to 12 h following reperfusion. Furthermore, JunD knock-out mouse livers expressed PCNA at elevated base-line levels in the absence of I/R injury (Fig. 2D, 0 h time points), indicating a pre-existing dysregulation of the cell cycle. These findings suggest that JunD does indeed alter cell cycle regulation in the liver, both prior to and following I/R injury. However, despite previous studies in fibroblasts demonstrating that JunD can act through p53 to modulate growth arrest and DNA repair (19), we did not observe significant differences in p53 levels between the two JunD genotypes (Fig. 2D).
The JNK Pathway Influences JunD-dependent AP-1 Transcriptional Activity following I/R Injury to the Liver
JNK plays a major role in regulating AP-1 activity by phosphorylating c-Jun on serine 63/73 (29). Additionally, JunD has been shown to be a substrate of JNK phosphorylation (30), but the functional consequences of this event remain largely unknown. Acute JNK1 activation occurs within the first 45–60 min following liver I/R and precedes the induction of nuclear AP-1 binding to DNA (17). Chemical inhibitors of JNK appear to suppress DNA binding by AP-1 following I/R injury, and they have been shown to protect the liver from apoptotically mediated damage (16). To this end, we hypothesized that expressing dominant negative JNK1 using a recombinant adenoviral vector (26) might suppress the elevation of AP-1 transcriptional activity in JunD knock-out mice, and thereby protect them from liver damage following I/R. To test this possibility, we infected JunD knock-out or heterozygous littermate mice with Ad.dnJNK1 or Ad.LacZ (as a control) virus prior to liver I/R. Consistent with this hypothesis, expression of dnJNK1 in JunD−/− mice significantly protected the liver from acute damage (Fig. 3, A versus B) and also significantly reduced the AP-1-mediated transcription (Fig. 3, C versus D). In contrast, control mice infected with Ad.LacZ virus demonstrated patterns of liver injury and AP-1 transcriptional induction similar to those seen in uninfected mice (Fig. 3A versus Fig. 2A and Fig. 3C versus Fig. 1A, respectively). Unexpectedly, dnJNK1 expression in JunD+/− mice led to a significant increase in liver injury and transcriptional induction of AP-1 following I/R (Fig. 3, B and D). AP-1 transcriptional activation during the acute phases of injury (6–9 h) was nearly completely inhibited by dnJNK1 expression, consistent with a previous report evaluating the effects of chemical inhibitors of JNK on AP-1 DNA binding (16). However, in both the JunD−/− and JunD+/− animals, the expression of dnJNK resulted in delayed transcriptional activation of AP-1, peaking 6 h later than in control LacZ-expressing animals (compare Fig. 3, C and D). Liver injury induced by dnJNK1 expression in JunD+/− animals also occurred 6 h later than in control Ad.LacZ-infected animals (Fig. 3, A versus B). These results suggest that in JunD+/− animals, inhibiting the JNK1 pathway following I/R retards the acute transcriptional activation of AP-1, but it also leads to compensatory transcriptional activation of AP-1 and consequent liver injury at later time points. Interestingly, this later phase of AP-1 transcriptional activation appears to be partially dependent on JunD, as such effects were significantly reduced in the JunD knock-out mice. Collectively, these results suggest that JNK1 and JunD collaborate to determine the temporal regulation of I/R-induced AP-1 transcriptional activation that is important in controlling liver injury.
FIGURE 3. Expression of dominant negative JNK1 alters AP-1 transcriptional activation and liver injury in a JunD-dependent manner following I/R.

JunD−/− and JunD+/− mice were infected (intravenously) with Ad.AP-1Luc and either Ad.LacZ or Ad.dnJNK1 viruses 3 days prior to I/R injury (partial lobar ischemia for 45 min followed by the indicated times of reperfusion). A and B, serum GPT levels; C and D, luciferase activities in liver lysates were assessed at the indicated time points, for each vector combination and JunD genotype, as indicated. Results depict the mean (± S.E.) for n = 3 animals at each experimental point. The 0-h time point represents non-I/R-injured control animals. E, 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside (X-gal)-stained liver section detecting β-galactosidase activity from uninfected and Ad.LacZ-infected mice.
The ability of dnJNK1 to suppress elevated levels of AP-1 activation selectively in JunD−/− mice suggested that targets of JNK1 (such as c-Jun) might have elevated levels of activation in the absence of JunD. To test this hypothesis, we quantified the time course of c-Jun phosphorylation in the liver following I/R injury of JunD−/− and JunD+/− animals infected with Ad.LacZ or Ad.dnJNK1. Indeed control Ad.LacZ-infected JunD−/− mice had significantly elevated levels of phosphoserine 63 c-Jun in the ischemic lobe of the liver at 6 and 9 h following I/R injury, as compared with Ad.LacZ-infected JunD+/− mice (Fig. 4A). Maximal c-Jun phosphorylation in Ad.LacZ-infected JunD−/− mice preceded maximal AP-1 transcriptional activation (Fig. 3C) by 3 h, suggesting that enhanced c-Jun phosphorylation in the setting of JunD deficiency elevates AP-1 transcriptional activity. To test whether JNK1 was responsible for enhanced c-Jun phosphorylation in JunD−/− mice, we evaluated the effects of dnJNK1 expression on c-Jun phosphorylation. dnJNK1 expression significantly reduced I/R-mediated activation of phospho-c-Jun in JunD−/− mouse liver (Fig. 4B), and this correlated with reduced AP-1 activation (Fig. 3, C versus D). Interestingly, dnJNK1 expression in JunD+/− mouse liver did not significantly alter the overall pattern of c-Jun serine 63 phosphorylation following liver I/R, in comparison with LacZ-expressing control JunD+/− mice. However, it was noted that there was a significantly earlier onset of c-Jun phosphorylation in Ad.dnJNK1-infected JunD+/− mice at 6 h post-I/R, in comparison with LacZ control animals (Fig. 4, A versus B).
FIGURE 4. JunD influences c-Jun phosphorylation following liver I/R in a JNK1-dependent manner.
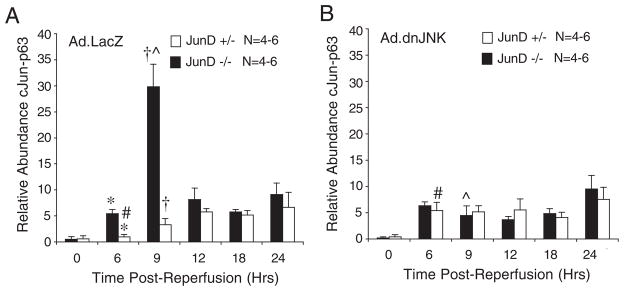
JunD−/− and JunD+/− mice were infected (intravenously) with Ad.LacZ (A) or Ad.dnJNK1 (B) viruses 3 days prior to I/R injury (partial lobar ischemia for 45 min followed by the indicated times of reperfusion). Hepatic lysates were generated from the ischemic lobe of the liver, and c-Jun was immunoprecipitated using a c-Jun antibody. Samples were then evaluated by Western blotting using a phosphoserine 63 c-Jun-specific antibody. Western blots were quantified using infrared dye-conjugated secondary antibody on a LI-COR Biosciences Odyssey Infrared Imaging System. Results depict the mean (±S.E.) for n = 4 – 6 animals at each experimental point. The 0-h time point represents non-I/R-injured control animals. *, †, #, and [caret] depict significant differences using the Student’s t test (*, †, and [caret], p < 0.01; #, p < 0.05).
JunD Collaborates with JNK1 to Modulate NADPH-dependent ROS Production following I/R Injury
Liver I/R-induced alterations in AP-1 DNA binding activity appear to be regulated in a redox-dependent fashion and are controlled by both cytoplasmic and mitochondrial superoxide production (17, 25). Given that NADPH oxidases (Nox) serve as major sources of cellular ROS, we hypothesized that JunD-mediated alterations in AP-1 transcriptional activity and cell cycle progression following liver I/R might be linked to changes in NADPH oxidase activity in this organ. To test this possibility, we assessed the extent of NADPH-dependent superoxide production in total endomembranes from livers harvested from JunD−/− and JunD+/− mice following I/R injury. Furthermore, we sought to determine whether the changes in AP-1 transcriptional activation resulting from dnJNK expression correlated with alterations in NADPH oxidase-derived ROS following I/R injury.
Results from these experiments demonstrated hyperactivation of NADPH-dependent superoxide production in the livers of Ad.LacZ-infected JunD−/− mice, with a 17-fold enhancement at 6 h of reperfusion as compared with Ad.LacZ-infected JunD+/− littermates (Fig. 5A, left panel). Furthermore, the time course of NADPH-dependent superoxide production was significantly affected by the presence of JunD, peaking at 6 and 12 h for JunD−/− and JunD+/− animals, respectively. Despite these differences, base-line levels of NADPH-dependent super-oxide in the absence of I/R (0-h time points) were not significantly different in JunD knock-out and heterozygous mice, suggesting that changes in JunD-dependent superoxide production were induced by I/R injury. Interestingly, hepatic dnJNK1 expression inhibited I/R-induced NADPH-dependent superoxide production in JunD−/− animals but promoted ROS production in JunD+/− littermates (Fig. 5A, right panel). These reversals in the Nox phenotype of dnJNK1-expressing animals were strikingly similar to the observed reversals in the liver injury and AP-1 transcription phenotypes also promoted by dnJNK expression (Fig. 3). Thus, our findings suggest that NADPH oxidase activation following I/R injury in the liver is influenced by both JunD and JNK1, and that these changes may be linked to JunD/JNK1-dependent changes in AP-1 transcriptional activation.
FIGURE 5. JunD modulates NADPH-dependent superoxide production in the liver following I/R injury.

JunD−/− and JunD+/− mice were infected (intravenously) with Ad.AP-1Luc and Ad.LacZ or Ad.dnJNK1 viruses 3 days prior to I/R injury (partial lobar ischemia for 45 min followed by the indicated times of reperfusion). The 0-h time point represents non-I/R-injured control animals. At each time point of reperfusion, the liver was harvested and used to generate total endomembranes for an assessment of NADPH-dependent superoxide production using lucigenin chemiluminescence. A, relative NADPH-dependent superoxide production in endomembranes from JunD−/− and JunD+/− mice infected with the indicated adenoviruses. Results depict the mean (±S.E.) for n = 4 animals at each experimental point. B and C, samples assessed in A were used to determine the fraction of DPI-sensitive NADPH-dependent superoxide for JunD−/− (C) and JunD+/− mice (D). In these studies, the endomembrane samples were treated with DPI (10 μm) or vehicle for 30 min prior to the addition of NADPH and measurements of lucigenin chemiluminescence. The fractions of non-DPI-sensitive and DPI-sensitive chemiluminescence are plotted for each sample (n = 4 animals in each group).
Although the observed changes in NADPH-dependent superoxide levels suggest that Nox activity may be involved in the response of the liver to I/R injury, other sources of superoxides may also account for the observed differences. We therefore evaluated the fraction of super-oxide that was dependent on NADPH oxidase, using the general NADPH oxidase inhibitor DPI (Fig. 5B and C). This led to several interesting findings. First, acute activation of DPI-sensitive NADPH-dependent superoxide following I/R injury correlated with enhanced AP-1 transcriptional activation for the various conditions analyzed; Ad.LacZ-infected JunD−/− mice (Fig. 5B, left panel) and Ad.dnJNK-infected JunD+/− mice (Fig. 5C, right panel) demonstrated the greatest rise in DPI-sensitive superoxide production; these same mice also demonstrated hyperactivated AP-1 transcriptional responses (Fig. 3, C and D). Under these conditions, superoxide levels rose during the acute phase (6 h), with the DPI-sensitive fraction accounting for the majority of NADPH-dependent superoxide in those samples. Second, the expression of dnJNK1 in JunD−/− mice inhibited DPI-sensitive superoxide production but elevated the overall levels of DPI-insensitive NADPH-dependent superoxide during the later stages of reperfusion (18 h) (Fig. 5B, right panel). Third, the majority of NADPH-dependent superoxide generated from JunD+/− mice infected with control Ad.LacZ virus was DPI-insensitive (Fig. 5C, left panel). These findings suggest that both NADPH oxidase and other sources of superoxide production in the liver following I/R are influenced by hepatic JunD and/or JNK1 activities. Furthermore, it appears that NADPH oxidase activation following liver I/R may have the greatest impact on the extent of AP-1 activation and liver injury.
JunD Influences the Ability of dnJNK1 to Modulate Nox2 and Nox4 mRNA Levels
The mechanism by which JunD and JNK1 modulate NADPH-dependent ROS production in liver endo-membranes following I/R remains unclear. However, a previous study has suggested that JunD-deficient fibroblasts express 3-fold more Nox4 mRNA than do wild-type cells (31). This finding implicates a potential role for JunD in the regulation of Nox genes. To this end, we evaluated whether JunD- and dnJNK-mediated alterations in Nox activity in the liver following I/R might be the result of altered base-line changes in Nox gene expression caused by JunD deficiency and/or dnJNK expression.
We evaluated the abundance of Nox2 and Nox4 mRNA levels using quantitative RT-PCR and referenced these levels against β-actin mRNA levels as an internal control. Hepatic poly(A) mRNA was harvested from four groups of animals, including Ad.LacZ- or Ad.dnJNK1-infected JunD−/− and JunD+/− mice, and reverse-transcribed cDNA was used for quantitative PCR. Results from these experiments demonstrated that dnJNK1 expression significantly enhanced both Nox2 and Nox4 hepatic mRNA levels (~2.5-fold) only in JunD+/− mice (Fig. 6). Interestingly, JunD appeared to be required for this dnJNK1-dependent change in Nox mRNA expression, because no changes were seen in JunD-deficient animals expressing dnJNK1. The finding of increased base-line levels of Nox4 and Nox2 mRNA only in the livers of JunD+/− animals expressing dnJNK correlated with a rise in both DPI-sensitive NADPH-dependent superoxide production (Fig. 5B and C) and AP-1 transcription (Fig. 3) from this cohort of animals following I/R. These findings suggest that JunD and JNK may act in concert to control Nox gene expression in the liver, which influences ROS-dependent AP-1 responses.
FIGURE 6. The ability of dnJNK1 to modulate Nox2andNox4 mRNA levels in the liver is dependent on JunD.
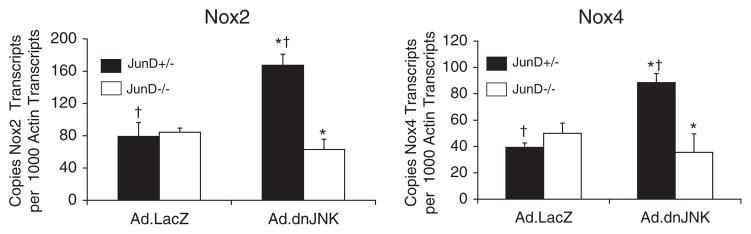
JunD−/− and JunD+/− mice were infected (intravenously) with Ad.LacZ or Ad.dnJNK1 viruses 3 days prior to harvesting the liver for generation of poly(A) mRNA. RT-PCR was performed for Nox2, Nox4, and β-actin, and Ct values for each reaction were used to calculate copy mRNA number by referencing a standard curve with plasmid controls for each gene. Results shown for both Nox2 and Nox4 are the mean ± S.E. for 3– 4 independent liver samples in each set. * and † depicts significant differences using the Student’s t test (* and †, p <0.01 for Nox2; †, p < 0.01; *, p < 0.05 for Nox4).
DISCUSSION
JNK1/AP-1 activation has traditionally been thought to play solely a deleterious role following liver I/R (16). Our current findings suggest that this view may be an oversimplification. In this regard, JNK1 and JunD dynamically controlled AP-1 transcriptional activation during both the acute and subacute stages of hepatic reperfusion. Altering the profile of the AP-1 transcriptional response by either dnJNK1 expression and/or JunD deletion significantly influenced I/R liver injury; an early peak activation of AP-1 was more deleterious and associated with higher levels of DPI-sensitive NADPH-dependent superoxide production by the liver, whereas a later peak in AP-1 activation was beneficial and associated with DPI-insensitive NADPH-dependent superoxide production. These findings demonstrate that JunD and JNK modulate both AP-1 transcriptional activation and NADPH-dependent ROS generation following liver I/R that may contribute to the extent of injury.
Alterations in JunD- and JNK-dependent hepatic ROS following I/R appear to be regulated by both Nox-dependent and Nox-independent mechanisms, given the diverse changes observed in DPI-sensitive and -insensitive ROS production. Control of NADPH-oxidases in the liver can theoretically occur by either altering transcription of these genes or by influencing the activation of co-factors that are required for Nox complex function. Indeed, dnJNK1 expression in the liver enhanced both Nox2 and Nox4 mRNA expression in a JunD-dependent manner, and this appeared to predispose the liver to enhance ROS production, AP-1 activation, and liver injury following I/R. This finding of enhanced liver injury in the setting of elevated Nox2 expression is consistent with the previous finding that liver I/R-induced injury and mortality is reduced in mice lacking the Nox2 isoform of NADPH oxidase (32). However, given the fact that uninjured livers demonstrated no increase in base-line NADPH-dependent ROS production, I/R-induced pathways responsible for activation of the Nox2/4 complexes must also be involved. Interestingly, we did not observe previously reported enhancement of Nox4 gene expression seen in JunD-deficient fibroblasts (31), suggesting that cell-specific differences (potentially involving JNK activity) may be at play in these two models.
In contrast to JunD+/− mice, JunD−/− mice did not demonstrate changes in Nox gene expression in the setting of hepatic dnJNK1 expression, despite elevation in DPI-sensitive Nox activity following I/R injury. These findings suggest that alternative JunD-dependent mechanisms of post-transcriptional Nox regulation must also exist. In this context, it is interesting that the onset of DPI-sensitive Nox hyperactivation following liver I/R coincided with enhanced c-Jun phosphorylation at 6 h; this was true for both Ad.dnJNK1-infected JunD+/− mice and Ad.LacZ-infected JunD−/− mice. The ability of dnJNK1 to reverse both c-Jun hyperphosphorylation and Nox hyperactivation in JunD−/− mice while promoting these same effects in JunD+/− mice remains unclear. However, the common finding of c-Jun hyperphosphorylation during the acute phase of the I/R response (6–9 h) in both cases of Nox hyperactivation suggests that a functional relationship exists between JunD-dependent JNK pathways and post-transcriptional regulation of Nox complexes.
The ability of JunD to retard the cell cycle and prevent the transformation of fibroblasts has been well documented (18). JunD could potentially regulate the cell cycle through cyclin D1, an important cell cycle check point regulator with an AP-1-binding site in its promoter. Cyclin D1 transcription has been linked to JNK1/2 activation during liver regeneration (33). This study demonstrated that pharmacologic inhibition of JNK1/2 following partial hepatectomy of rats results in reduced PCNA expression, c-Jun phosphorylation, and AP-1 DNA binding leading to increased liver injury (33). Our studies demonstrating an enhancement of c-Jun phosphorylation, AP-1-mediated transcription, and elevated cellular proliferation (as indexed by PCNA expression) in JunD-deficient mice are consistent with the conclusions drawn from these previous studies.
Our study also suggests that JunD can directly influence the context in which JNK1 functions following I/R injury in the liver. This conclusion may be viewed as contradicting the findings from chemical inhibitor studies in which JNK is functionally blocked prior to I/R injury (16, 34). These studies demonstrated that chemical inhibition of JNK1/2, in both liver and lung models of I/R injury, led to a decrease in tissue damage following I/R injury. However, these earlier studies used chemical inhibitors that either selectively inhibited JNK activity by competing for binding between c-Jun and JNK or specifically inhibited JNK by directly blocking induction of its activity, and none was specific to particular isoforms of JNK. Recent studies also suggest that JNK1 and JNK2 may have distinct roles in c-Jun-dependent regulation of cellular proliferation (35). However, these results conflict with other studies demonstrating that both JNK1 and JNK2 are positive regulators of cellular proliferation (36, 37). Interestingly, one of these studies demonstrated that expression of dnJNK1 (same construct used in our studies) in JNK2−/− cells enhances cellular proliferation (36). This may be one explanation for our observation of detrimental effects of expressing dnJNK1 in JunD+/− mice. Additionally, JunD-dependent regulation of both JNK1 and JNK2 might account for the observations that dnJNK1 protects from liver injury, reduces AP-1 activation, and attenuates Nox-dependent ROS production by the liver only in JunD−/− mice following I/R. Although the potential for JunD regulation of JNK2 remains speculative, our findings demonstrating that dnJNK1 inhibits I/R-induced c-Jun hyperphosphorylation in JunD−/− mice suggests a potential role for JunD in JNK1 regulation. Given that JNK2 has also been shown to negatively regulate cellular proliferation (38), compensatory changes in JNK2 activation in response to dnJNK1 expression may act in a JunD-specific manner to alter hepatic responses following I/R injury.
In conclusion, our study demonstrates that JunD plays an important role in repressing AP-1 transcriptional activation following I/R injury to the liver and that this process is beneficial to liver remodeling. Alterations in AP-1 transcriptional activation appeared to be linked to both hepatic proliferative responses and NADPH-dependent ROS production by the liver following I/R injury. Whether enhanced NADPH-dependent ROS production by the liver is a consequence of enhanced AP-1-mediated damage, or instead acts as an effector thereof, remains unclear. However, our findings support the hypothesis that JunD suppresses AP-1-mediated transcriptional activation of cell cycle regulatory genes, thereby giving the liver time to repair cellular damage prior to proliferation. In the absence of JunD, enhanced proliferation following I/R injury leads to increased caspase-mediated damage.
Acknowledgments
We gratefully acknowledge Dr. Christine Blaumueller for editorial assistance and Dr. Ziying Yan and Sarah Desprat for technical assistance. The core facilities of The Center for Gene Therapy was recipient of National Institutes of Health Grant P30 DK54759.
Footnotes
The abbreviations used are: I/R, ischemia/reperfusion; JNK, c-Jun NH2-terminal kinase; PCNA, proliferating cell nuclear antigen; ROS, reactive oxygen species; EMSA, electrophoretic mobility shift assay; GPT, glutamic pyruvate transaminase; DPI, diphenylene iodonium; dn, dominant negative; Nox, NADPH oxidases.
This work was supported in part by NIDDK Grants DK067928 and DK51315 from the National Institutes of Health (to J. F. E.).
References
- 1.Kretzschmar M, Kruger A, Schirrmeister W. Exp Toxicol Pathol. 2003;54:423–431. doi: 10.1078/0940-2993-00291. [DOI] [PubMed] [Google Scholar]
- 2.Smyrniotis V, Kostopanagiotou G, Lolis E, Theodoraki K, Farantos C, Andreadou I, Polymeneas G, Genatas C, Contis J. J Am Coll Surg. 2003;197:949–954. doi: 10.1016/j.jamcollsurg.2003.07.009. [DOI] [PubMed] [Google Scholar]
- 3.Kim YI. J Hepatobiliary Pancreat Surg. 2003;10:195–199. doi: 10.1007/s00534-002-0730-x. [DOI] [PubMed] [Google Scholar]
- 4.Kupiec-Weglinski JW, Busuttil RW. Transplant Proc. 2005;37:1653–1656. doi: 10.1016/j.transproceed.2005.03.134. [DOI] [PubMed] [Google Scholar]
- 5.Okaya T, Lentsch AB. J Investig Surg. 2003;16:141–147. [PubMed] [Google Scholar]
- 6.Colletti LM, Kunkel SL, Walz A, Burdick MD, Kunkel RG, Wilke CA, Strieter RM. Hepatology. 1996;23:506–514. doi: 10.1002/hep.510230315. [DOI] [PubMed] [Google Scholar]
- 7.Arthur MJ, Bentley IS, Tanner AR, Saunders PK, Millward-Sadler GH, Wright R. Gastroenterology. 1985;89:1114–1122. doi: 10.1016/0016-5085(85)90218-5. [DOI] [PubMed] [Google Scholar]
- 8.Atalla SL, Toledo-Pereyra LH, MacKenzie GH, Cederna JP. Transplantation. 1985;40:584–590. doi: 10.1097/00007890-198512000-00002. [DOI] [PubMed] [Google Scholar]
- 9.Koo A, Komatsu H, Tao G, Inoue M, Guth PH, Kaplowitz N. Hepatology. 1992;15:507–514. doi: 10.1002/hep.1840150325. [DOI] [PubMed] [Google Scholar]
- 10.Mathews WR, Guido DM, Fisher MA, Jaeschke H. Free Radic Biol Med. 1994;16:763–770. doi: 10.1016/0891-5849(94)90191-0. [DOI] [PubMed] [Google Scholar]
- 11.Chinenov Y, Kerppola TK. Oncogene. 2001;20:2438–2452. doi: 10.1038/sj.onc.1204385. [DOI] [PubMed] [Google Scholar]
- 12.Sheerin A, Thompson KS, Goyns MH. Mech Ageing Dev. 2001;122:1813–1824. doi: 10.1016/s0047-6374(01)00319-0. [DOI] [PubMed] [Google Scholar]
- 13.Troen G, Nygaard V, Jenssen TK, Ikonomou IM, Tierens A, Matutes E, Gruszka-Westwood A, Catovsky D, Myklebost O, Lauritzsen G, Hovig E, Delabie J. J Mol Diagn. 2004;6:297–307. doi: 10.1016/S1525-1578(10)60525-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ronai Z. Mol Cell. 2004;15:843–844. doi: 10.1016/j.molcel.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 15.Abate C, Patel L, Rauscher FJ, III, Curran T. Science. 1990;249:1157–1161. doi: 10.1126/science.2118682. [DOI] [PubMed] [Google Scholar]
- 16.Uehara T, Bennett B, Sakata ST, Satoh Y, Bilter GK, Westwick JK, Brenner DA. J Hepatol. 2005;42:850–859. doi: 10.1016/j.jhep.2005.01.030. [DOI] [PubMed] [Google Scholar]
- 17.Zhou W, Zhang Y, Hosch MS, Lang A, Zwacka RM, Engelhardt JF. Hepatology. 2001;33:902–914. doi: 10.1053/jhep.2001.23073. [DOI] [PubMed] [Google Scholar]
- 18.Pfarr CM, Mechta F, Spyrou G, Lallemand D, Carillo S, Yaniv M. Cell. 1994;76:747–760. doi: 10.1016/0092-8674(94)90513-4. [DOI] [PubMed] [Google Scholar]
- 19.Weitzman J, Fiette L, Matsuo K, Yaniv M. Mol Cell. 2000;6:1109–1119. doi: 10.1016/s1097-2765(00)00109-x. [DOI] [PubMed] [Google Scholar]
- 20.Bakiri L, Lallemand D, Bossy-Wetzel E, Yaniv M. EMBO J. 2000;19:2056–2068. doi: 10.1093/emboj/19.9.2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ameyar-Zazoua M, Wisniewska MB, Bakiri L, Wagner EF, Yaniv M, Weitzman JB. Oncogene. 2005;24:2298–2306. doi: 10.1038/sj.onc.1208424. [DOI] [PubMed] [Google Scholar]
- 22.Xiao L, Rao JN, Zou T, Liu L, Marasa BS, Chen J, Turner DJ, Passaniti A, Wang JY. Biochem J. 2007;403:573–581. doi: 10.1042/BJ20061436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schlossberg H, Zhang Y, Dudus L, Engelhardt JF. Hepatology. 1996;23:1546–1555. doi: 10.1002/hep.510230635. [DOI] [PubMed] [Google Scholar]
- 24.Thepot D, Weitzman JB, Barra J, Segretain D, Stinnakre MG, Babinet C, Yaniv M. Development (Camb) 2000;127:143–153. doi: 10.1242/dev.127.1.143. [DOI] [PubMed] [Google Scholar]
- 25.Zwacka RM, Zhou W, Zhang Y, Darby CJ, Dudus L, Halldorson J, Oberley L, Engelhardt JF. Nat Med. 1998;4:698–704. doi: 10.1038/nm0698-698. [DOI] [PubMed] [Google Scholar]
- 26.Qiao L, Han SI, Fang Y, Park JS, Gupta S, Gilfor D, Amorino G, Valerie K, Sealy L, Engelhardt JF, Grant S, Hylemon PB, Dent P. Mol Cell Biol. 2003;23:3052–3066. doi: 10.1128/MCB.23.9.3052-3066.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Anderson RD, Haskell RE, Xia H, Roessler BJ, Davidson BL. Gene Ther. 2000;7:1034–1038. doi: 10.1038/sj.gt.3301197. [DOI] [PubMed] [Google Scholar]
- 28.Li Q, Harraz MM, Zhou W, Zhang LN, Ding W, Zhang Y, Eggleston T, Yeaman C, Banfi B, Engelhardt JF. Mol Cell Biol. 2006;26:140–154. doi: 10.1128/MCB.26.1.140-154.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Davis RJ. Biochem Soc Symp. 1999;64:1–12. doi: 10.1515/9781400865048.1. [DOI] [PubMed] [Google Scholar]
- 30.Yazgan O, Pfarr CM. J Biol Chem. 2002;277:29710–29718. doi: 10.1074/jbc.M204552200. [DOI] [PubMed] [Google Scholar]
- 31.Gerald D, Berra E, Frapart YM, Chan DA, Giaccia AJ, Mansuy D, Pouyssegur J, Yaniv M, Mechta-Grigoriou F. Cell. 2004;118:781–794. doi: 10.1016/j.cell.2004.08.025. [DOI] [PubMed] [Google Scholar]
- 32.Harada H, Hines IN, Flores S, Gao B, McCord J, Scheerens H, Grisham MB. Arch Biochem Biophys. 2004;423:103–108. doi: 10.1016/j.abb.2003.08.035. [DOI] [PubMed] [Google Scholar]
- 33.Schwabe RF, Bradham CA, Uehara T, Hatano E, Bennett BL, Schoonhoven R, Brenner DA. Hepatology. 2003;37:824–832. doi: 10.1053/jhep.2003.50135. [DOI] [PubMed] [Google Scholar]
- 34.Ishii M, Suzuki Y, Takeshita K, Miyao N, Kudo H, Hiraoka R, Nishio K, Sato N, Naoki K, Aoki T, Yamaguchi K. J Immunol. 2004;172:2569–2577. doi: 10.4049/jimmunol.172.4.2569. [DOI] [PubMed] [Google Scholar]
- 35.Sabapathy K, Hochedlinger K, Nam SY, Bauer A, Karin M, Wagner EF. Mol Cell. 2004;15:713–725. doi: 10.1016/j.molcel.2004.08.028. [DOI] [PubMed] [Google Scholar]
- 36.Dong C, Yang DD, Tournier C, Whitmarsh AJ, Xu J, Davis RJ, Flavell RA. Nature. 2000;405:91–94. doi: 10.1038/35011091. [DOI] [PubMed] [Google Scholar]
- 37.Jaeschke A, Karasarides M, Ventura JJ, Ehrhardt A, Zhang C, Flavell RA, Shokat KM, Davis RJ. Mol Cell. 2006;23:899–911. doi: 10.1016/j.molcel.2006.07.028. [DOI] [PubMed] [Google Scholar]
- 38.Sabapathy K, Wagner EF. Cell Cycle. 2004;3:1520–1523. doi: 10.4161/cc.3.12.1315. [DOI] [PubMed] [Google Scholar]