

Abstract
Patulin is a fungal mycotoxin of Aspergilus and Penicillium that is commonly found in rotting fruits and exerts its potential toxic effect mainly by reactive oxygen species (ROS) generation. However, the effect of patulin on cancer cells as well as its intracellular mechanism has been controversial and not clearly defined yet. In this study, patulin was found to induce G1/S accumulation and cell growth arrest accompanied by caspase-3 activation, PARP cleavage and ATF3 expression in human colon cancer cell line HCT116. Ser/Thr phosphorylation of a transcription factor, EGR-1, was increased while its expression did not change upon patulin treatment to the cells. Knockdown of ATF3 and EGR-1 using their respective siRNAs showed EGR-1 dependent ATF3 expression. Moreover, treatment of the cells with antioxidants N-acetylcysteine (NAC) and glutathione (GSH) revealed that patulin induced ATF3 expression and apoptosis were dependent on ROS generation. ATF3 expression was also increased by patulin in other colorectal cancer cell types, Caco2 and SW620. Collectively, our data present a new anti-cancer molecular mechanism of patulin, suggesting EGR-1 and ATF3 as critical targets for the development of anti-cancer chemotherapeutics. In this regard, patulin could be a candidate for the treatment of colorectal cancers.
Keywords: ATF3, EGR-1, ROS, Patulin
1. Introduction
Patulin, 4-hydroxy-4H-furo(3,2c)pyran-2(6H)-one, is a mycotoxin produced by molds including Penicillium expansum, Aspergillus, and Byssachlamys, also occurring world-wide in apple, apple products and sometimes in a number of foods including peaches, pears, and grain, or their products [1–6]. Patulin level of 50 μg/kg in apple juice was suggested to be sufficient for protection of human health [7]. Some in vivo works, however, reported that several organs or systems including kidney, liver, intestinal tissue and immune system were affected by the administration of patulin, possibly due to some teratogenic, genotoxic and carcinogenic effect of patulin in certain experimental animals [8–10]. Patulin exerts its toxic effect by covalently binding to reactive sulfhdryl groups in cellular proteins, as well as by glutathion depletion, resulting in oxidative damage and generation of reactive oxygen stress (ROS) [11–13]. Although several studies have shown patulin induced DNA strand breaks in mammalian cells [14,15], the mechanism of all these effects including apoptosis, however, is still poorly understood.
Activating transcription factor3 (ATF3), a member of the ATF/CREB family of bZIP transcription factors, is stress-responsive gene product [16]. Although ATF3 induction in astrocytes contributed to the cytoprotective function against oxidative insults [17], there have been other observations indicating that the expression of ATF3 was low in normal and quiescent cells but might be rapidly induced by a variety of physiological and pathological stimuli [18–20]. ATF3 expression plays an important role in berberine-induced apoptosis in human colorectal cancer cells [21]. Moreover, ATF3 is increased by nonsteroidal antiinflammatory drugs (NSAID), conjugated linoleic acid, LY294002, curcumin, and 3,3′-diindolylmethane, which are reported to have anti-tumor activity in human colorectal cancer cells [22–26]. These results showed that ATF3 could function as a pro-apoptotic protein and mediate cytotoxic agents induced apoptosis in various systems. However, the upstream regulations of ATF3 expression have not yet been clearly defined although activation of a few apoptosis associated proteins, including p53 and ERK1/2 was reported [10,27,28].
Although many mycotoxins including zearalenone, citrinin, gliotoxin, nivalenol and fuonisin B1 were shown to have cytotoxic effects [29–32], our study presents a new anticancer mechanism of patulin suggesting that EGR-1 phosphorylation by oxidative stress following patulin treatment might trigger EGR-1 binding to ATF3 promoter which leads to the apoptotic proteins expression and cell growth arrest of HCT-116 human colorectal cancer cells.
2. Materials and methods
2.1. Cells and materials
Cell lines were purchased from ATCC (Rockville,MD). HCT116, SW620 and Caco2 human colorectal cells were grown in McCoy’s 5A, L-15 and Eagle’s Minimum Essential Medium (MEM), respectively, supplemented with 10% FBS. Patulin, N-acetylcysteine (NAC) and glutathion (GSH) were obtained from Sigma (St Louis, MO). Antibodies against EGR-1, phospho-JNK, phospho-eIF2α, eIF2α and JNK were purchased from Cell Signaling (Beverly, MA). Antibodies against ATF3, poly (ADP-ribose) polymerase (PARP), HA and GAPDH were from Santa Cruz Biotechnology (Santa Cruz, CA). Antibody against caspase-3 was obtained from Calbiochem (Darmstadt, Germany).
2.2. Flow cytometric analysis
HCT116 was treated with vehicle or various concentrations of patulin for 24 h and then the cells were washed twice with cold PBS. The collected cells were resuspended in annexin-V binding Ca2+ buffer in annexin-V-FITC staining solution (1.0 μg/ml) and incubated for 15 min at room temperature in the dark. Flow cytometric analysis was performed using a FACS (Becton Dickinson, San Jose, CA).
2.3. Western blot analysis
Cells were harvested and equal amount of cellular proteins was subjected to sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis as described previously [33]. Proteins were transferred to polyvinylidene difluoride (PVDF) membrane and the blots were probed with various primary antibodies. Enhanced chemiluminescence reagent was used to detect the proteins on the membranes.
2.4. Plasmid, transfections, and reporter assay
Chromosomal DNA was prepared from HCT116 cells using the DNAzol™ reagent (Gibco-BRL, Gaithersburg, MD). Human ATF3 promoter was amplified from chromosomal DNA with the following synthetic primers: 5′-CGACGCGTTCCCGCCACCCTGGCTTGAGG-3′ (sense), and 5′-CCCTCGAGAGCGTTGCATCACCCCTTTTATAGCCC-3′ (antisense). The PCR product was digested with Mlu and Xho and cloned upstream of the firefly luciferase gene in pGL3-basic vector (Promega). PCR products were confirmed by electrophoresis. For transfection, in brief, cells were plated onto 6-well plates at a density of 4×105 cells/well and grown overnight. Cells were co-transfected with the appropriate amount of plasmid and pCMV-β-galactosidase plasmid for 5 h using Lipofectamine. After transfection, cells were cultured in 10% FBS medium with or without compounds for 24 h. Luciferase and β-galactosidase activities were assayed according to the manufacturer’s protocol (Promega). Luciferase activity was normalized for β-galactosidase activity in cell lysate and expressed by an average of three independent experiments.
2.5. Cell proliferation
HCT116 was seeded in 96-well plates and treated with various concentrations of patulin, and cell viability was determined by MTT assays.
2.6. RNA isolation and reverse transcriptase-polymerase chain reaction (RT-PCR)
ATF3 mRNA expression was determined by RT-PCR. Total cellular RNA was extracted from cells using the TRIzol reagent (Life Technologies). cDNA was synthesized from 2 μg of total RNA using M-MLV reverse transcriptase (Gibco-BRL, Gaithersburg, MD). cDNA for ATF3 and GAPDH were amplified by PCR with specific primers. The sequences of the sense and anti-sense primer for ATF3 were 5′-GTTTGAGGATTTTGCTAACCTGAC-3′ and 5′-GCTGCAATCTTATTTCTTTCTCGT-3′, respectively. PCR products were analyzed by agarose gel electrophoresis and visualized by ethidium bromide.
2.7. Gel shift assay
In brief, cells (4×106 in 10 ml) grown in 100 mm dishes were lysed on ice for 15 min in a hypotonic solution containing 10 mM HEPES–KOH (pH 7.8), 10 mM KCl, 2 mM MgCl2, 0.1 mM EDTA (sodium salt), 0.2 mM NaF, 0.2 mM Na3VO4, 0.4 mM phenylmethylsulfonyl fluoride (PMSF), leupeptin (10 μg/ml), 1 mM dithiothreitol (DTT), and 0.15% NP-40. The lysate was centrifuged at 15,000 rpm for 1 min at 4 °C, and the resulting nuclear pellet was suspended in ice-cold extraction buffer (50 mM HEPES–KOH pH 7.8, 50 mM KCl, 300 mM NaCl, 0.1 mM EDTA, 0.2 mM NaF, 0.2 mM Na3VO4, 0.4 mM PMSF, 1 mM DTT and 10% glycerol) and incubated for 30 min at 4 °C with occasional vortex. The nuclear lysate was then centrifuged at 15,000 rpm for 30 min at 4 °C, and the resulting supernatant was stored at −70 °C or immediately subjected to EMSA analysis. Oligonucleotide containing GC-rich box (5′-CGACCGCGCCGCCAGCCGACCGCGCCGCCAGC-3′) (3.5 pmol) was incubated for 10 min at 37 °C in 10 μl containing 10 μCi of [γ-32P] ATP, 5 U of T4 polynucleotide kinase, and 1× kinase buffer (supplied with the kinase). The labeling reaction was terminated by the addition of 100 mM EDTA, after which the reaction mixture was centrifuged through a Sephadex G-25 column to remove unincorporated 32P. The 32P-labeled oligonucleotide was then stored at −70 °C until use. For EMSA assay, nuclear protein extract (10 μg) was incubated for 30 min at room temperature in a final volume of 10 μl containing 0.03 pmol of 32P-end-labeled oligonucleotide, 40 mM HEPES–KOH (pH 7.8), 10% glycerol, 1 mM MgCl2, 0.1 mM DTT and 1 μg of poly (dI–dC). The binding reaction was terminated by the addition of electrophoresis sample buffer, and the samples were fractionated on 5% non-denaturing polyacrylamide gels in 0.5× Tris–boric acid–EDTA (TBE) buffer. The gels were then subjected to autoradiography. For “supershift” analysis, the nuclear extract was incubated with specific antibodies for 30 min at room temperature before exposure to the 32P-labeled oligonucleotide. The reaction was stopped by the addition of gel loading dye, and the samples were subjected to electrophoresis on a non-denaturing 6% polyacrylamide gel in 0.5× Tris–borate–EDTA buffer followed by autoradiography.
2.8. Small interfering RNA (siRNA)
ATF3 small interfering RNA (siRNA) (sc-29757) was purchased from Santa Cruz Biotechnology and EGR-1 small interfering RNA (siRNA) was designed by Dhamacon. Cells were transfected with 100 nM of siRNA using Lipofectamine 2000 transfection reagent (Invitrogen, Carlsbad, CA) in OPTI-MEMI-reduced serum medium (Invitrogen) for 6 h. The medium was removed and replaced with fresh McCoy’s 5A medium supplemented with 10% fetal calf serum. Cells were harvested 72 h after transfection for western blot analysis and FACS analysis.
2.9. Measurement of ROS
Intracellular ROS generation was measured by flow cytometry following staining with 10 μM of 2′,7′-dichlorofluorescein diacetate (DCFDA; Sigma), which has been shown to be somewhat specific for detection of H2O2. After cells were collected, the fluorescence was analyzed using FACSCalibur and a Leica DC300F fluorescence microscopy.
2.10. Immunoprecipitation
The total cell extracts were immunoprecipitated by incubating with antibody against EGR-1 overnight at 4 °C. Immune complexes were collected using protein A/G-Sepharose beads (Santa Cruz Biotechnology), washed and eluted in sample buffer. Samples were run on 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) gels, transferred to PVDF membranes, and probed with specific antibodies against phospho-Serine/threonine or EGR-1. Blots were developed using the ECL reagent.
2.11. Statistical analysis
Values are presented as the mean±S.E of three separate experiments. Statistical comparisons were made using the Student’s t-test when two groups were involved. A one way analysis of variance (ANOVA) with Dunnett post-test was used when more than two groups were compared. The P value of 0.05 or less was considered as statistically significant.
3. Results
3.1. Patulin induces apoptosis through G2/M arrest
To determine the anti-proliferative effect of patulin (Fig. 1A) on cell viability, HCT116 cells were treated with increasing concentrations of patulin for the various days and the cell viability was determined by MTT assay. Treatment of patulin dose-dependently decreased the cell viability (Fig. 1B), and increased the percentage of annexin-V+ stained apoptotic cells and G2/M phase cells (Fig. 1C). Accordingly, fluorescent microscopy analysis by differential staining with Hoechst 33342 and propidium iodide (PI) showed significantly increased staining of the cells when treated with patulin (50% of the cells PI-positive at 10 μM of patulin, Supplementary 1). Western blot analysis revealed cleavage of both caspase-3 and PARP by patulin treatment for 24 h (Fig. 1D). Thus, patulin induced apoptotic death of HCT116 cells.
Fig. 1.

Apoptotic effect of patulin in HCT116 cells. (A) Chemical structure of patulin. (B) Effect of patulin on cell proliferation measured by MTT assay. HCT116 cells were treated with vehicle or varying concentrations of patulin four days followed by lysis for MTT assay. Data are means±SE from representative triplicate experiments. (C) Flow cytometric analysis for DNA content and cell cycle distribution in response to patulin. HCT116 cells were treated with varying concentrations of patulin for 24 h and stained with annexin-V+ and PI. (D) Activation of apoptotic proteins by patulin. HCT116 cells were treated with the indicated concentrations of patulin for 24 h. After lysis, the level of cleaved caspase-3 and PARP was determined by western blot analysis.
3.2. Patulin-induced apoptosis is mediated by ATF3
In search for factors that influence the patulin-induced apoptosis, ATF3 was found to be highly expressed among some pro-apoptotic- or anti-apoptotic proteins (data not shown). Patulin induced ATF3 expression dose- and time-dependently. ATF3 mRNA expression was also increased by patulin (Fig. 2A). In accordance with this ATF expression, patulin dramatically increased ATF3 promotor activity in a dose-dependent manner (Fig. 2B). When ATF3 expression blocked by siRNA of ATF3, however, caspase-3 cleavage and the number of annexin-V stained cells were significantly reduced (Fig. 2C), demonstrating that ATF3 is required for patulin-induced apoptosis.
Fig. 2.

Effect of patulin on ATF3 expression in HCT116 cells. (A) ATF3 expression by patulin. Cells were treated with patuln at varying concentrations for 24 h, or at 5 μM for various times. Cell lysate and total RNA were subjected to western blot and RT-PCR analyses, respectively. (B) Reporter gene assay for ATF3 promoter. Cells were transiently transfected with 2 μg pATF3-luc plasmid or mock vector, and then incubated in the presence of patulin (10 μM) for 24 h. Luciferase activity was measured and expressed as means±SE from a representative triplicate experiments performed three times independently. Statistical significance was determined by Dunnett’s test (**p<0.01, ***p<0.001) followed by one-way ANOVA. (C) Effect of ATF3 knockdown on caspase-3 activation and apoptosis. Cells were transfected with ATF3 siRNA or scrambled siRNA for 48 h, and then treated with patulin at 5 μM for another 24 h. ATF3 expression and caspase-3 activation were determined by western blotting. Apoptotic cells were also determined by FACS analysis in the same condition and expressed as means±SE from a representative triplicate experiment. Statistical significance was determined by Student’s t-test **p<0.01.
3.3. Patulin-induced ATF3 expression is associated with ROS generation
Patulin can induce an oxidative stress response in mammalian cells (10, 11). To determine whether ATF3 expression is related to oxidative stress after treatment of patulin, ROS generation was measured by determining the level of H2O2 using DCFDA staining in HCT116 cells. As shown in Fig. 3A, patulin treatment for 24 h highly increased ROS production. In determining the effect of patulin-induced ROS generation on ATF3 expression, it was found that ATF3 transcriptional activity was reduced by prior treatment of antioxidants, GSH and NAC for 1 h (Fig. 3B). Reduction of ATF3 expression by patulin also lowered caspase-3 activation (Fig. 3C). These results suggest that patulin-induced ATF3 expression might be associated with oxidative stress.
Fig. 3.

Association of ATF3 expression and apoptosis with ROS generation in response to patulin in HCT116 cells. (A) ROS accumulation by patulin. Cells were treated with varying concentrations of patulin followed by the addition of 5 μM H2DCFDA for 30 min at 37 °C. FACS analysis was performed to measure the intracellular accumulation of ROS. (B) Effect of antioxidants on patulin-induced ATF3 expression. Cells were transiently transfected with 2 μg pATF3-luc plasmid or mock vector, and then treated with glutathione (1 mM) or NAC (5 mM) for 1 h prior to patulin treatment at 5 μM for 24 h. Luciferase activity was measured and expressed as means±SE from representative triplicate experiments. Statistical significance was determined by Student’s test (***p<0.001). (C) Cells were pretreated with either GSH (5 mM) or NAC (10 mM) for 1 h prior to the exposure to patulin at 5 μM for 24 h. Cells were lysed and subjected to western blot analysis. The arrows represent cleaved caspase-3 fragments.
3.4. The EGR-1 is required for patulin-induced ATF3 expression
EGR-1 is a well known transcription factor that binds within the region of ATF3 promoter. In investigating the molecular mechanism of patulin-induced ATF3 transcriptional regulation, it was found that patulin did not affect EGR-1 expression (Fig. 4A). Electrophoretic mobility shift assay (EMSA) using an oligonucleotide containing the GC-rich sequence on the ATF3 promoter region, however, showed increased binding of nuclear protein(s) on the oligonucleotide (Fig. 4B). The specificity of this binding was confirmed by supershift analysis in which band retardation occurred by including anti-EGR-1 antibody into the reaction mixture while anti-SP1/SP3 antiobody had no effect. Next, to determine whether EGR-1 is responsible for patulin-induced ATF3 expression, HCT116 cells transfected with the scramble or EGR-1 siRNA were treated with patulin for 24 h. It was clearly found that EGR-1 knockdown noticeably reduced ATF3 expression in response to patulin (Fig. 4C). Reporter gene assay and annexin-V staining of the cells also showed that ATF3 expression and the number of apoptotic cells by patulin were reduced, respectively (Fig. 4D & E). On the other hand, EGR-1 phosphorylation at Ser/Thr was significantly increased by patulin treatment (Fig. 4F). These results suggest that EGR-1phosphorylation in patulin-treated cells at serine/threonine residues may be important for EGR-1-mediated ATF3 expression.
Fig. 4.

EGR-1 is required for patulin-induced ATF3 expression in HCT116 cells. (A) Cells were treated with various concentrations of patulin for 24 h, and then lysed for measurement of EGR-1 by western blot analysis. (B) EMSA analysis. Nuclear fraction (10 g) obtained from the cells treated with patulin at the indicated concentrations was applied to EMSA analysis using GC-rich oligonucleotide as described in Materials and methods. For supershift, EGR-1 or SP1/SP3 antibody (1 μg) was added into the EMSA reaction mixture. As a competitor, unlabeled cold oligonucleotide was added into the reaction mixture (lane 6). (C) EGR-1 knockdown abrogates patulin-induced ATF3 expression. Cells were transfected with EGR-1 siRNA or scrambled siRNA for 48 h followed by patulin treatment at 5 μM for 24 h. Proteins were detected by western blotting. (D) Effect of EGR-1 knockdown on patulin-induced ATF3 promoter activity. After transfection with EGR-1 siRNA or scrambled siRNA for 24 h, cells were transiently transfected 2 μg pATF3-luc plasmid prior to patulin treatment for further 24 h. Luciferase activity was measured and expressed as means±SE from a representative triplicate experiment performed three times independently. Statistical significance was determined by Dunnett’s test (*p<0.05, **p<0.01, ***p<0.001) followed by one-way ANOVA. (E) FACS analysis for the determination of the effect of EGR-1 knockdown on patulin-induced cell apoptosis. All the procedures were similar to Fig. 2C. Data are expressed as means±SE from a representative triplicate experiment. Statistical significance was determined by Dunnett’s test ((*p<0.05, **p<0.01, ***p<0.001) followed by one-way ANOVA. (F) Effect of patulin on EGR-1 phosphorylation. Cells were treated with varying concentrations of patulin for 24 h, and then lysed for immunoprecipitation with anti-EGR-1 antibody. The immunoprecipitates were subjected to SDS-PAGE and analyzed by immunoblotting with anti-p-Ser/Thr antibody. The membrane was then stripped and reprobed with anti-EGR-1 antibody.
3.5. Patulin-induced ATF3 expression is not related with ER-stress or JNK
Endoplasmic reticulum stress (ER-stress) induces increased expression of chaperones including GRP78, which mediates the correct folding of proteins in the ER. Failure of misfolded protein removal could lead to an apoptotic death of cells through the activation of caspase-12 or c-Jun N-terminal kinase (JNK) downstream of ER-associated proteins PKR-like ER kinase (PERK) and eukaryotic translation initiation factor 2α (eIF2α) [34–38]. JNK could also be activated independently of ER-stress for cell apoptosis. Although HCT116 cells were treated with patulin, however, there was no obvious change in ER-stress. Moreover, patulin-induced ATF3 expression did not change even when the phosphorylation of JNK by patulin was significantly diminished by prior treatment of the cells with a JNK inhibitor (Fig. 5), which demonstrates that patulin-mediated ATF3 expression is regulated neither by ER-stress nor by JNK activity.
Fig. 5.
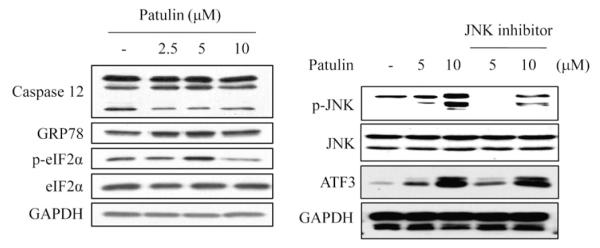
Effect of patulin on ER-stress and JNK activation in HCT116 cells. (A) Cells were treated with varying concentrations of patulin for 24 h, and then lysed for measurement of caspase-12, GRP78, p-eIF2α and eIF2α by western blot analysis. (B) Cells were pretreated with JNK inhibitor (25 μM) for 1 h before the exposure to patulin for further 24 h. After cell lysis, pJNK, JNK and ATF3 were measured by western blot analysis.
3.6. Effect of patulin in human colorectal cancer cell lines, Caco2 and SW620
To examine whether the effect of patulin on ATF3 protein expression and cell growth arrest could be generally occurring in other colorectal cancer cell types, SW480 and CaCo2 cells were treated with patulin for 24 h and the expression of ATF3 was measured. As revealed in Fig. 6, ATF3 expression was increased in the presence of patulin in all the cells dose- and time-dependently, accompanied by an increased cleavage of caspase-3 (Fig. 6A & B).
Fig. 6.

Effect of patulin on ATF3 expression and caspase 3 activation in human colorectal cancer cell lines, Caco2 and SW620. (A) Cells were treated with patulin at varying concentrations for 24 h, and lysed for the determination of ATF3 level and Caspase3 cleavage forms by western blot analysis. (B) Cells were treated with patulin at 5 μM for various times, and lysed for the determination of ATF3 level and Caspase3 cleavage forms as done in (A).
4. Discussion
Patulin is a secondary metabolite of fungi and has been extensively studied for its cytotoxic effects in various tumor models including in vitro and in vivo systems. Many studies revealed genotoxicity and mutagenicity by patulin [39]. The toxic effects of patulin may involve direct effects on cellular glutathione levels and mitochondrial function in addition direct effects on the plasma membrane [40]. It was also reported that glutathione depletion by patulin resulted in the generation of ROS, playing a critical role in initiation and execution of apoptosis, controlling mitotic cell division and senescence [41–44]. Although ERK1/2, p53 and p21/WAF1 signaling pathways were correlated with patulin-mediated ROS generation [10,27], our study showed more detailed apoptotic signaling by patulin. It was found that patulin arrested cell cycle at G2/M phase, leading to the increased ATF3 expression (Fig. 2) and apoptotic cell death (Fig. 1) which were dependent on ROS as revealed by the treatment of the cells with ROS scavengers, GSH and NAC (Fig. 3B & C). In exploiting the downstream signaling molecules of ROS, it was necessary to determine the expression of patulin-induced apoptotic proteins. Based upon the observations that ATF3 expression could be regulated by a variety of regulatory proteins, including NF-κB [45], ATF/CRE [46], MAPK [47], ER stress [48] and EGR-1/Sp1/Sp3 [49], we explored the relation of EGR-1 and ATF3 since ATF3 promoter region contains EGR-1 binding site. Although patulin did not affect the expression of EGR-1 in the cells (Fig. 4A), EMSA analysis, however, showed that EGR-1 binding to the ATF3 promoter region could be increased by patulin (Fig. 4B). In addition, EGR-1 knockdown by siRNA significantly repressed ATF3 expression by patulin (Fig. 4C and D). However, given the same binding site (GC-rich box) of EGR-1 and Sp1/Sp3 in the ATF3 promoter, it was interesting to note that only EGR-1 antibody retarded oligonucleotide-protein complex while there was no effect by Sp1/Sp3 antibody as revealed in the supershift assay (Fig. 4B, lanes 4 & 5). Additional study showed that patulin increased the levle of Ser/Thr phosphorylation of EGR-1 (Fig. 4F). Since EGR-1 has been reported to have several Ser/Thr phosphorylation sites, influencing the DNA binding and transcriptional activity of EGR-1 (24), it is presumable that patulin-induced phosphorylation of EGR-1 could contribute to the increased binding of EGR-1 to ATF3 promoter. Thus, this is the first report suggesting that patulin induced ROS generation and EGR-1 Ser/Thr phosphorylation might lead to the increased expression of ATF3 and apoptotic cell death. Although not confirmed yet, ROS generation is suggested to play a key role for the phosphorylation of EGR-1.
EGR-1 plays a pivotal role in the regulation of cell growth, differentiation and apoptosis, and its pro-apoptotic activity may depend on the cell type and the nature of the stimulus. EGR-1 expression was suppressed or absent in a variety of human cancer cell lines [50,51] and tumors including breast carcinoma [50], glioblastoma [52] and lung cancer [53]. Our results also showed an important role of EGR-1 in ATF3 expression and apoptosis. On the other hand, another apoptosis-associated pathway, endoplasmic reticulum stress (ER-stress), does not seem to be involved in patulin-induced apoptosis since there was little change in the level of ER-stress associated proteins, caspase-12, GRP78 and eIF2α, in response to patulin in the cells (Fig. 5). Interestingly, however, JNK phosphorylation was increased by patulin but was not relevant with ATF3 expression since JNK inhibitor could not affect the expression of ATF3 by patulin (Fig. 5). Although the involvement of upstream regulators including ERK1/2, p53 and p21 for EGR-1 phosphorylation has not yet been defined, our data clearly demonstrate that ROS-mediated EGR-1 phosphorylation and ATF3 expression are critical in patulin-induced apoptosis. Moreover, two other human colorectal cancer cells showed similar pattern of ATF3 expression by patulin (Fig. 6), supporting further that ROS generation, EGR-1 phosphorylation and ATF3 expression might mainly function for patulin-induced apoptosis of many colorectal cancer cells.
We have tried several times to identify the direct binding target protein of patulin by attaching biotinylated links to the compound. However, due to the instability of patulin, it was not easy to perform the proteomic analysis further. Thus, we are currently chemically modifying patulin to obtain more stable patulin derivatives. This will help find a new therapeutic target against cancer.
In summary, patulin, through increased oxidative stress, induces phosphorylation of a transcription factor EGR-1 for elevated expression of ATF3, which in turn results in the cell cycle arrest and activation of apoptotic protein cascades (Fig. 7). Our study provides more detailed mechanism of patulin for colorectal cancer cell death. When the direct target protein to which patulin binds is identified, both the target and patulin could be applied to anti-cancer therapeutics development.
Fig. 7.
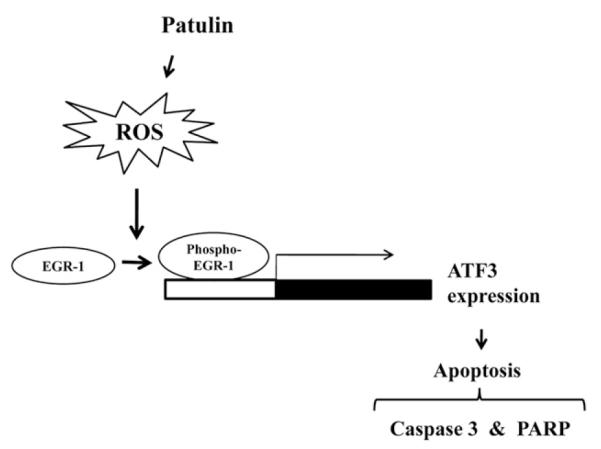
A simplified proposed model for patulin-induced ATF3 activation and apoptosis. Patulin-induced ROS generation triggers EGR-1 phosphorylation at Ser/Thr residue increased binding of the phosphorylated EGR-1 to the GC-rich box region of ATF3 promoter, consequently leading to the activation of apoptotic proteins and cancer cell death.
Acknowledgements
This work was supported by the World Class Institute (WCI) Program, Global R&D Center (GRDC) Program of the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (MEST), and also supported by KRIBB Research Initiative Program.
Footnotes
Supplementary materials related to this article can be found online at doi:10.1016/j.cellsig.2011.12.017.
References
- [1].Sommer NF, Buchanan JR, Fortlage RJ. Appl. Microbiol. 1974;28(4):589–593. doi: 10.1128/am.28.4.589-593.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Frank HK, Orth R, Figge A. Z. Lebensmitt. Unters. Forsch. 1977;163(2):111–114. doi: 10.1007/BF01126030. [DOI] [PubMed] [Google Scholar]
- [3].Scott PM, Fuleki T, Harwig J. J. Agric. Food Chem. 1977;25(2):434–437. [PubMed] [Google Scholar]
- [4].Kwon O, Jeong SJ, Kim SO, He L, Lee HG, Jang KL, Osada H, Jung M, Kim BY, Ahn JS. Carcinogenesis. 2010;31(7):1194–1201. doi: 10.1093/carcin/bgq071. [DOI] [PubMed] [Google Scholar]
- [5].Morales H, Sanchis V, Coromines J, Ramos AJ, Marin S. Food Microbiol. 2008;25(2):378–385. doi: 10.1016/j.fm.2007.09.008. [DOI] [PubMed] [Google Scholar]
- [6].Chan AO, Peng JZ, Lam SK, Lai KC, Yuen MF, Cheung HK, Kwong YL, Rashid A, Chan CK, Wong BC. Gut. 2006;55(4):463–468. doi: 10.1136/gut.2005.077776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Kwon GY, Yoo BC, Koh KC, Cho JW, Park WS, Park CK. J. Korean Med. Sci. 2005;20(2):242–247. doi: 10.3346/jkms.2005.20.2.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Becci PJ, Hess FG, Johnson WD, Gallo MA, Babish JG, Dailey RE, Parent RA. J. Appl. Toxicol. 1981;1(5):256–261. doi: 10.1002/jat.2550010504. [DOI] [PubMed] [Google Scholar]
- [9].Liu BH, Yu FY, Wu TS, Li SY, Su MC, Wang MC, Shih SM. Toxicol. Appl. Pharmacol. 2003;191(3):255–263. doi: 10.1016/s0041-008x(03)00254-0. [DOI] [PubMed] [Google Scholar]
- [10].Liu BH, Wu TS, Yu FY, Su CC. Toxicol. Sci. 2007;95(2):340–347. doi: 10.1093/toxsci/kfl156. [DOI] [PubMed] [Google Scholar]
- [11].Wu TS, Liao YC, Yu FY, Chang CH, Liu BH. Toxicol. Lett. 2008;183(1–3):105–111. doi: 10.1016/j.toxlet.2008.09.018. [DOI] [PubMed] [Google Scholar]
- [12].Heussner AH, Dietrich DR, O’Brien E. Toxicol In Vitro. 2006;20(3):332–341. doi: 10.1016/j.tiv.2005.08.003. [DOI] [PubMed] [Google Scholar]
- [13].Barhoumi R, Burghardt RC. Fundam. Appl. Toxicol. 1996;30(2):290–297. doi: 10.1006/faat.1996.0067. [DOI] [PubMed] [Google Scholar]
- [14].Zhou SM, Jiang LP, Geng CY, Cao J, Zhong LF. Toxicon. 2009;53(5):584–586. doi: 10.1016/j.toxicon.2009.01.030. [DOI] [PubMed] [Google Scholar]
- [15].Zhou SM, Jiang LP, Geng CY, Cao J, Zhong LF. Toxicon. 2010;55(2–3):390–395. doi: 10.1016/j.toxicon.2009.08.019. [DOI] [PubMed] [Google Scholar]
- [16].Liang G, Wolfgang CD, Chen BP, Chen TH, Hai T. J. Biol. Chem. 1996;271(3):1695–1701. doi: 10.1074/jbc.271.3.1695. [DOI] [PubMed] [Google Scholar]
- [17].Das S, Eisenberg LD, House JW, Lee KJ, Lusk RP, Nielsen DR, Patel MM, Steckowych JM, Ermini EB. Otolaryngol. Head Neck Surg. 2011;144(2):135–141. doi: 10.1177/0194599810393441. [DOI] [PubMed] [Google Scholar]
- [18].Syed V, Mukherjee K, Lyons-Weiler J, Lau KM, Mashima T, Tsuruo T, Ho SM. Oncogene. 2005;24(10):1774–1787. doi: 10.1038/sj.onc.1207991. [DOI] [PubMed] [Google Scholar]
- [19].Mashima T, Udagawa S, Tsuruo T. J. Cell. Physiol. 2001;188(3):352–358. doi: 10.1002/jcp.1130. [DOI] [PubMed] [Google Scholar]
- [20].Hartman MG, Lu D, Kim ML, Kociba GJ, Shukri T, Buteau J, Wang X, Frankel WL, Guttridge D, Prentki M, Grey ST, Ron D, Hai T. Mol. Cell. Biol. 2004;24(13):5721–5732. doi: 10.1128/MCB.24.13.5721-5732.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Rondon C, Romero JJ, Lopez S, Antunez C, Martin-Casanez E, Torres MJ, Mayorga C, RP R, Blanca M. J Allergy Clin Immunol. 2007;119(4):899–905. doi: 10.1016/j.jaci.2007.01.006. [DOI] [PubMed] [Google Scholar]
- [22].Hataoka Y, Zhang L, Yukimasa T, Baba Y. Anal. Sci. 2005;21(1):53–56. doi: 10.2116/analsci.21.53. [DOI] [PubMed] [Google Scholar]
- [23].Lee SH, Yamaguchi K, Kim JS, Eling TE, Safe S, Park Y, Baek SJ. Carcinogenesis. 2006;27(5):972–981. doi: 10.1093/carcin/bgi268. [DOI] [PubMed] [Google Scholar]
- [24].Yamaguchi K, Lee SH, Kim JS, Wimalasena J, Kitajima S, Baek SJ. Cancer Res. 2006;66(4):2376–2384. doi: 10.1158/0008-5472.CAN-05-1987. [DOI] [PubMed] [Google Scholar]
- [25].Lee SH, Kim JS, Yamaguchi K, Eling TE, Baek SJ. Biochem. Biophys. Res. Commun. 2005;328(1):63–69. doi: 10.1016/j.bbrc.2004.12.138. [DOI] [PubMed] [Google Scholar]
- [26].Yan C, Jamaluddin MS, Aggarwal B, Myers J, Boyd DD. Mol. Cancer Ther. 2005;4(2):233–241. [PubMed] [Google Scholar]
- [27].Saxena N, Ansari KM, Kumar R, Dhawan A, Dwivedi PD, Das M. Toxicol. Appl. Pharmacol. 2009;234(2):192–201. doi: 10.1016/j.taap.2008.09.033. [DOI] [PubMed] [Google Scholar]
- [28].Zhou SM, Jiang LP, Geng CY, Cao J, Zhong LF. Toxicon. 55(2-3):390–395. doi: 10.1016/j.toxicon.2009.08.019. [DOI] [PubMed] [Google Scholar]
- [29].Ooi EM, Janus ED, Grant SJ, Sinclair LM, RB PH. J. Lipid Res. 2010;51(8):2413–2421. doi: 10.1194/jlr.M004705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Johannessen LN, Nilsen AM, Lovik M. Clin. Exp. Allergy. 2005;35(6):782–789. doi: 10.1111/j.1365-2222.2005.02249.x. [DOI] [PubMed] [Google Scholar]
- [31].Ben-Ami R, Lewis RE, Leventakos K, Kontoyiannis DP. Blood. 2009;114(26):5393–5399. doi: 10.1182/blood-2009-07-231209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Minervini F, Fornelli F, Flynn KM. Toxicol. In Vitro. 2004;18(1):21–28. doi: 10.1016/s0887-2333(03)00130-9. [DOI] [PubMed] [Google Scholar]
- [33].Kwon O, Kim KA, Kim SO, Ha R, Oh WK, Kim MS, Kim HS, Kim GD, Kim JW, Jung M, Kim CH, Ahn JS, Kim BY. Carcinogenesis. 2006;27(11):2258–2268. doi: 10.1093/carcin/bgl097. [DOI] [PubMed] [Google Scholar]
- [34].Li J, Lee B, Lee AS. J. Biol. Chem. 2006;281(11):7260–7270. doi: 10.1074/jbc.M509868200. [DOI] [PubMed] [Google Scholar]
- [35].Srinivasan S, Ohsugi M, Liu Z, Fatrai S, Bernal-Mizrachi E, Permutt MA. Diabetes. 2005;54(4):968–975. doi: 10.2337/diabetes.54.4.968. [DOI] [PubMed] [Google Scholar]
- [36].Galehdar Z, Swan P, Fuerth B, Callaghan SM, Park DS, Cregan SP. J. Neurosci. 2010;30(50):16938–16948. doi: 10.1523/JNEUROSCI.1598-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Kim DM, Won M, Chung CS, Kim S, Yim HJ, Jung SH, Jeong S. Apoptosis. 2010;15(12):1540–1548. doi: 10.1007/s10495-010-0531-7. [DOI] [PubMed] [Google Scholar]
- [38].Kristiansen M, Hughes R, Patel P, Jacques TS, Clark AR, Ham J. J. Neurosci. 2010;30(32):10820–10832. doi: 10.1523/JNEUROSCI.2824-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Mahfoud R, Maresca M, Garmy N, Fantini J. Toxicol. Appl. Pharmacol. 2002;181(3):209–218. doi: 10.1006/taap.2002.9417. [DOI] [PubMed] [Google Scholar]
- [40].Burghardt RC, Barhoumi R, Lewis EH, Bailey RH, Pyle KA, Clement BA, Phillips TD. Toxicol. Appl. Pharmacol. 1992;112(2):235–244. doi: 10.1016/0041-008x(92)90193-v. [DOI] [PubMed] [Google Scholar]
- [41].Simon HU, Haj-Yehia A, Levi-Schaffer F. Apoptosis. 2000;5(5):415–418. doi: 10.1023/a:1009616228304. [DOI] [PubMed] [Google Scholar]
- [42].Herrera B, Alvarez AM, Sanchez A, Fernandez M, Roncero C, Benito M, Fabregat I. FASEB J. 2001;15(3):741–751. doi: 10.1096/fj.00-0267com. [DOI] [PubMed] [Google Scholar]
- [43].Moustafa MH, Sharma RK, Thornton J, Mascha E, Abdel-Hafez MA, Thomas AJ, Jr., Agarwal A. Hum. Reprod. 2004;19(1):129–138. doi: 10.1093/humrep/deh024. [DOI] [PubMed] [Google Scholar]
- [44].Juan ME, Wenzel U, Daniel H, Planas JM. J. Agric. Food Chem. 2008;56(12):4813–4818. doi: 10.1021/jf800175a. [DOI] [PubMed] [Google Scholar]
- [45].Kaszubska W, Hooft van Huijsduijnen R, Ghersa P, DeRaemy-Schenk AM, Chen BP, Hai T, DeLamarter JF, Whelan J. Mol. Cell. Biol. 1993;13(11):7180–7190. doi: 10.1128/mcb.13.11.7180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Tamura K, Hua B, Adachi S, Guney I, Kawauchi J, Morioka M, Tamamori-Adachi M, Tanaka Y, Nakabeppu Y, Sunamori M, Sedivy JM, Kitajima S. EMBO J. 2005;24(14):2590–2601. doi: 10.1038/sj.emboj.7600742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Ndoye O, Ba MC, Niang EH, Rebillard O, Tofighi M, Veinmann P, Moretti JL. Dakar Med. 2005;50(3):157–159. [PubMed] [Google Scholar]
- [48].Koh IU, Lim JH, Joe MK, Kim WH, Jung MH, Yoon JB, Song J. FEBS J. 2010;277(10):2304–2317. doi: 10.1111/j.1742-4658.2010.07646.x. [DOI] [PubMed] [Google Scholar]
- [49].Cho KN, Sukhthankar M, Lee SH, Yoon JH, Baek SJ. Eur. J. Cancer. 2007;43(16):2404–2412. doi: 10.1016/j.ejca.2007.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Huang RP, Fan Y, de Belle I, Niemeyer C, Gottardis MM, Mercola D, Adamson ED. Int. J. Cancer. 1997;72(1):102–109. doi: 10.1002/(sici)1097-0215(19970703)72:1<102::aid-ijc15>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- [51].Liu C, Yao J, Mercola D, Adamson E. J. Biol. Chem. 2000;275(27):20315–20323. doi: 10.1074/jbc.M909046199. [DOI] [PubMed] [Google Scholar]
- [52].Calogero A, Lombari V, De Gregorio G, Porcellini A, Ucci S, Arcella A, Caruso R, Gagliardi FM, Gulino A, Lanzetta G, Frati L, Mercola D, Ragona G. Cancer Cell Int. 2004;4(1):1. doi: 10.1186/1475-2867-4-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Goguel AF, Fouquet F, Duverger A, Arvelo F, Jacrot M, Poupon MF, Bernheim A. Cancer Genet. Cytogenet. 1995;80(1):47–54. doi: 10.1016/0165-4608(94)00154-4. [DOI] [PubMed] [Google Scholar]