

Abstract
Excitation-driven entry of Ca2+ through L-type voltage-gated Ca2+ channels controls gene expression in neurons and a variety of fundamental activities in other kinds of excitable cells. The probability of opening of CaV1.2 L-type channels is subject to pronounced enhancement by cAMP-dependent protein kinase (PKA), which is scaffolded to CaV1.2 channels by A-kinase anchoring proteins (AKAPs). CaV1.2 channels also undergo negative autoregulation via Ca2+-dependent inactivation (CDI), which strongly limits Ca2+ entry. An abundance of evidence indicates that CDI relies upon binding of Ca2+/calmodulin (CaM) to an isoleucine–glutamine motif in the carboxy tail of CaV1.2 L-type channels, a molecular mechanism seemingly unrelated to phosphorylation-mediated channel enhancement. But our work reveals, in cultured hippocampal neurons and a heterologous expression system, that the Ca2+/CaM-activated phosphatase calcineurin (CaN) is scaffolded to CaV1.2 channels by the neuronal anchoring protein AKAP79/150, and that overexpression of an AKAP79/150 mutant incapable of binding CaN (ΔPIX; CaN-binding PXIXIT motif deleted) impedes CDI. Interventions that suppress CaN activity—mutation in its catalytic site, antagonism with cyclosporine A or FK506, or intracellular perfusion with a peptide mimicking the sequence of the phosphatase's autoinhibitory domain—interfere with normal CDI. In cultured hippocampal neurons from a ΔPIX knock-in mouse, CDI is absent. Results of experiments with the adenylyl cyclase stimulator forskolin and with the PKA inhibitor PKI suggest that Ca2+/CaM-activated CaN promotes CDI by reversing channel enhancement effectuated by kinases such as PKA. Hence, our investigation of AKAP79/150-anchored CaN reconciles the CaM-based model of CDI with an earlier, seemingly contradictory model based on dephosphorylation signaling.
Introduction
Voltage-gated Ca2+ channel activity is tuned to cellular need by a variety of processes, with Ca2+-dependent inactivation (CDI) providing one form of Ca2+-driven feedback control. Elimination of CDI from L-type CaV1.2 channels pathologically prolongs action potentials in cardiomyocytes (Alseikhan et al., 2002), illustrating the importance of feedback regulation of Ca2+ channels. CDI also reduces non-L-type CaV2.1 channel activity and thereby contributes to short-term synaptic depression during repetitive stimulation (Mochida et al., 2008).
The Ca2+ sensor for CDI is Ca2+/calmodulin (CaM) (Peterson et al., 1999; Zühlke et al., 1999), which at resting [Ca2+]in is preassociated with the CaV1.2 C terminus (Pitt et al., 2001; Erickson et al., 2001, 2003). Ca2+ entering the cytoplasm through CaV1.2 binds to CaM, initiating Ca2+/CaM interaction with an isoleucine–glutamine (IQ) motif located adjacent to the CaM preassociation site (Zühlke et al., 1999, 2000; Erickson et al., 2003; Kim et al., 2004). Because disruption of the IQ motif impairs CDI, Ca2+/CaM interaction with the IQ motif has been proposed to induce directly a change in channel conformational that corresponds to CDI.
But two sets of observations suggest an alternative to this nonenzymatic model of CDI. First, CaV1.2-anchored Ca2+/CaM often acts via downstream enzymes. Examples include Ca2+-dependent facilitation (CDF) of CaV1.2 by CaM-dependent protein kinase II (CaMKII) (Noble and Shimoni, 1981; Zühlke et al., 2000; Hudmon et al., 2005; Erxleben et al., 2006; Grueter et al., 2006) and coupling of neuronal excitation to activation of transcription factors by kinases and phosphatases (Dolmetsch et al., 2001; Oliveria et al., 2007). Second, Chad and Eckert (1986) found for molluscan neurons that protein kinase A (PKA) activity slowed CaV channel inactivation, and that calcineurin (CaN) opposed this effect, suggesting an enzymatic mechanism for CDI (for review, see Armstrong, 1989). Modulation by PKA of CaV1.2 CDI has since been described in a variety of excitable cell types (Bean et al., 1984; Kalman et al., 1988; Hadley and Lederer, 1991; Meuth et al., 2002). Yet a role for CaN in CDI has remained controversial, in part owing to inconsistent responses to inhibition of CaN (Branchaw et al., 1997; Victor et al., 1997; Lukyanetz et al., 1998; Burley and Sihra, 2000; Zeilhofer et al., 2000; Meuth et al., 2002) and to a lack of appreciation of the role of scaffolding proteins in targeting CaN to CaV1.2.
In hippocampal neurons, Ca2+ and cAMP reciprocally regulate L channel activity by activation of CaN and PKA that are coanchored to CaV1.2 channels by the scaffolding protein AKAP79/150 (human/rodent) (Oliveria et al., 2007). AKAP79/150-anchored CaN suppresses enhancement of neuronal L-type Ca2+ channel activity by coanchored, activated PKA (Oliveria et al., 2007). CaN effectiveness can in part be attributed to its localization within the CaV1.2 macromolecular complex, where CaN is advantageously placed to interact with its activator, Ca2+/CaM. Here we describe experimental results indicating that CaM, the Ca2+ sensor for CDI, triggers CDI in hippocampal neurons by activating AKAP879/150-anchored CaN.
Materials and Methods
Cell culture and transfection of tsA201 cells.
tsA201 cells were transfected with equimolar ratios of cDNAs encoding CaV1.2 channel subunits (α11.2a, β2b, α2δ1a) and other proteins, including AKAP79, using the Effectene transfection reagent (Qiagen). Cells were plated onto glass coverslips and studied 1–3 d after transfection.
Preparation and transfection of primary, short-term cultured hippocampal neurons.
Primary hippocampal neurons were prepared from neonatal Sprague Dawley rats [postnatal day 0 (P0)–P3] as described previously (Gomez et al., 2002; Smith et al., 2006). For Amaxa Nucleofector transfection, P0–P3 neurons were resuspended at 2 × 106 to 3 × 106 cells per transfection, electroporated with 6–8 μg of total DNA (including GFP or CaNH151A-YFP cDNA and/or the 150RNAi construct), plated at 0.25 × 106 to 0.5 × 106 cells per 25 mm glass coverslip, and grown up to 6 d in vitro. Current waveform and tail current time course indicated that these short-term cultured neurons were electrotonically more compact than older neurons, permitting analysis of the inactivation kinetics of pharmacologically isolated L currents.
AKAP150ΔPIX knock-in mice.
AKAP150ΔPIX mice were developed in conjunction with the Rocky Mountain Neurological Disorders Transgenic and Gene Targeting Vectors Core and housed at the University of Colorado Anschutz Medical Campus Center for Comparative Medicine. Briefly, the AKAP150ΔPIX mutation, which removes the conserved PIAIIIT motif (amino acids 655–661), was introduced into the single AKAP150 coding exon in the AKAP5 gene by homologous recombination in C57BL/6J×129 F1 embryonic stem cells using a loxP-flanked neomycin cassette located downstream of the exon. Targeted ES clones were identified by G418 selection for the neomycin resistance gene and PCR screening of genomic DNA, expanded, and injected into blastocysts that were implanted into surrogate females. Heterozygous and homozygous mice carrying the mutation were derived from the resulting F0 chimeric founders by standard breeding with C57BL/6J. AKAP150ΔPIX mice housed in a home-cage environment had no obvious phenotypic alterations in body weight, life span, breeding, or behavior. A full description of the AKAP150ΔPIX knock-in targeting vector and additional details of generation and characterization of AKAP150ΔPIX mice was reported by Sanderson et al. (2012). Dissociated mouse neurons were cultured as for rat neurons.
Patch-clamp recording.
Borosilicate patch pipettes were heat-polished to a resistance, measured in the bath, of 3–6 MΩ. Voltage-clamped currents were measured with an Axopatch 1D amplifier (Molecular Devices), filtered at 2 kHz, and sampled at 10 kHz using Pulse software (HEKA) and an ITC-16 analog-to-digital and digital-to-analog interface (HEKA). Series resistance compensation, capacitance cancellation, and leak subtraction (−P/4 protocol) were used.
For tsA201 cells, the whole-cell pipette contained the following (in mm): 135 CsCl, 10 EGTA or 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (BAPTA), 10 HEPES, and 4 MgATP, pH adjusted to 7.5 with tetraethylammonium-hydroxide (TEA-OH). For recordings in Ca2+ or Ba2+, the bath solution contained the following (in mm): 125 NaCl, 15 CaCl2 or 15 BaCl2, 10 HEPES, and 20 sucrose, pH adjusted to 7.3 with TEA-OH. For recordings of Na+ currents carried by Ca2+ channels, the whole-cell pipette contained the solution described above, but supplemented with 10 mm BAPTA, and the bath solution contained the following (in mm): 140 NaCl, 5 CsCl, 5 EGTA, 10 HEPES, and 20 sucrose, pH adjusted to 7.3 with TEA-OH. Positive transfectants were identified by GFP, AKAP79-YFP, CaM-YFP, CaN-CFP, or CaN-YFP fluorescence. When unlabeled AKAP79 was transfected, only cells exhibiting membrane-localized CaN were selected for recording. In each tsA201 cell where inactivation rate was studied, the holding potential was −80 mV, and a sequence of 500-ms step depolarizations, from −60 to +70 mV in 10 mV increments, was applied for both Ca2+ and Ba2+ solutions. A fast perfusion system (Warner Instruments) and slower bath perfusion were used in combination to ensure rapid and complete solution exchange. Cells that exhibited an artifactual current–voltage relationship were excluded from further analysis. CdCl2 (200 μm) was applied at the end of each experiment to isolate Ca2+ current and Ba2+ current from leak or other contamination. Na+ current recordings were performed similarly, but not in the same cells as the Ca2+ and Ba2+ current recordings. To provide a reliable basis for comparison of Ca2+ and Ba2+ inactivation rates, currents were generally allowed to stabilize for ∼5 min after establishment of whole-cell recording. In the experiments examining the time course of development for forskolin action inactivation (see Fig. 5), the stabilization period was necessarily omitted.
Figure 5.

Time course of forskolin action supports a role for AKAP79-anchored calcineurin in CDI of CaV1.2 channels expressed in tsA201 cells. A, Whole-cell recording with 5 μm forskolin in the pipette, with breakthrough into whole-cell mode at time 0 min. For cells transfected with 79ΔPIX, the rate of inactivation of Ca2+ current slowed as forskolin entered the cell. Cells transfected with wild-type AKAP79 (79) showed no slowing of CDI during intracellular perfusion with forskolin. #p < 0.05; ##p < 0.01 vs 0.5 min (paired t test); *p < 0.05 vs AKAP79. Ca2+ current densities were as follows (in pA/pF): 79 + FSK, −29.9 ± 5.6; 79ΔPIX + FSK, −25.0 ± 5.0. B, Bath application of FK506 (10 μm) plus cyclosporin A (10 μm) significantly slowed inactivation of forskolin-enhanced CaV1.2 Ca2+currents in cells overexpressing AKAP79. #p < 0.05 (paired t test). Top right, Ca2+ current records before and during drug application are scaled to the same peak amplitude to facilitate comparison of inactivation rate.
For cultured hippocampal pyramidal neurons, the whole-cell pipette contained the following (in mm): 120 CsMeSO4, 30 TEA-Cl, 10 EGTA, 5 MgCl2, 5 Na2ATP, and 10 HEPES, pH 7.2. The extracellular solution contained the following (in mm): 125 NaCl, 10 BaCl2 or CaCl2, 5.85 KCl, 22.5 TEA-Cl, 1.2 MgCl2, 10 HEPES(Na), and 11 d-glucose, pH 7.4, as well as tetrodotoxin (1 μm). To isolate L-type currents, N- and P/Q-type Ca2+ channel currents were blocked by preincubating neurons in extracellular recording solution supplemented with ω-conotoxins (ω-CTx) GVIA (1 μm) and MVIIC (5 μm) for 30 min before recording (Tavalin et al., 2004; Oliveria et al., 2007). Neurons were used for <1 h after preincubation to minimize contamination from unblocked N- and P/Q-type currents [τoff, ∼200 min for ω-CTx-MVIIC unbinding from P/Q-type channels (Sather et al., 1993); ω-CTx-GVIA binding to N-type channels was even more prolonged (McCleskey et al., 1987)]. A holding potential of −60 mV was chosen to inactivate R-type Ca2+ channel current (>80%) (Sochivko et al., 2003). Transfected neurons were identified by GFP or CaNH151A-YFP fluorescence. Only neurons with an access resistance <10 MΩ were studied.
To analyze inactivation time course during 500 ms depolarizations to 0 mV, the decaying phase of each current record was best fit with a double-exponential function; in a very few cases, single exponential fits provided, by eye, equally good fits (PulseFit software; HEKA). For the double-exponential fits, the faster time constant (τfast) provided an index of Ca2+-dependent inactivation, and the slower component represented voltage-dependent inactivation (Adams and Tanabe, 1997). Currents were obtained in Ba2+ and Ca2+ in the same cell, with Ba2+ currents recorded before Ca2+ currents.
Where indicated, forskolin (5 μm), protein kinase inhibitor fragment (PKI; residues 6–22; 5 μm), cyclosporin A (CsA; 5 μm), the CaN autoinhibitory peptide (AIP; residues 457–482; 100 μm), or CaN anchoring antagonist peptide VIVIT (Oliveria et al., 2007; Li et al., 2012) (biotin-GPHPVIVITGPHEE, 10, 25 or 100 μm) was included in the pipette solution. To examine reversibility of CaN inhibition, cyclosporin and FK506 (10 μm each) were added to the bath in some experiments, as indicated in the text. Forskolin, PKI, cyclosporin A, and FK506 were obtained from Sigma-Aldrich; the CaN autoinhibitory peptide was obtained from Biomol; and the VIVIT peptide was obtained from Biomatik.
Ratiometric fluorometry.
Measurements of YFP535/CFP470 were made using a ratiometric fluorimeter (Solamere Technology Group) where the donor fluorophore, CFP, was excited (excitation bandpass, 436 ± 5 nm), and emissions from both CFP (CFP470) and YFP (YFP535) were collected using parallel photomultiplier tubes fronted by a dichroic mirror and bandpass filters (CFP bandpass, 470 ± 15 nm; YFP bandpass, 535 ± 15 nm). In all experiments, simultaneous emission ratio (YFP535/CFP470) and current (ICa) measurements were obtained. CFP was excited (shutter open) for 50 ms periods centered in time on the membrane depolarizations (20 ms steps; 0.067 or 1 Hz), and the emission ratio was measured as the mean YFP535/CFP470 value during each 50 ms depolarization. Concurrent ICa and YFP535/CFP470 signals were acquired and recorded using Pulse software (HEKA). The step depolarization frequency of 0.067 Hz was chosen to establish baseline emission ratio and current values because this was the highest frequency that was indistinguishable from the baseline determined for cells held at −80 mV for an extended period between depolarizing steps.
Fluorescence resonance energy transfer microscopy.
Fluorescence images were acquired 2–3 d after transfection from live tsA201 cells using a Nikon TE-300 inverted microscope equipped with a 175 W xenon illumination source, 100× oil-immersion objective lens, 16 MHz CCD camera (SensiCam QE; Cooke), and dual filter wheels (Sutter Instruments) controlled by SlideBook 4.0 software (Intelligent Imaging Innovation). For sensitized fluorescence resonance energy transfer (FRET) measurements [FRETC; three filter (3F)], an 86002 (JP4) dichroic mirror (Chroma) and three different filter sets (donor/CFP, acceptor/YFP, and raw FRET) were used to capture serially a set of three images from a fixed image plane. The three images were captured using the same exposure time (100 ms in some experiments, 250 ms in others). The three filter sets used were donor [CFP; 436 center excitation wavelength and 10 nm bandwidth (436/10 nm); emission, 470/30 nm], acceptor (YFP; excitation, 500/20 nm; emission, 535/30 nm), and FRET (raw FRET; excitation, 436/10 nm; emission, 535/30 nm). Light that has passed through the FRET filter set is contaminated by donor bleed-through (average fraction, 0.50) and acceptor cross-excitation (average fraction, 0.02); fractional contamination by bleed-through and cross-excitation were determined in separate experiments using cells that expressed CFP- or YFP-tagged constructs alone. Corrected, sensitized FRET (FRETC) images were obtained by subtracting the contamination components, pixel by pixel, from raw FRET images using the following equation (adapted from Gordon et al., 1998): FRETC = rawFRET − (0.50 * CFP) − (0.02 * YFP).
To obtain estimates of effective FRET efficiency (Eeff) from images, Slidebook 4.0 was used to draw masks that isolated in-focus CaN-CFP fluorescence. From masked raw FRET, CFP, and YFP images, the FRET ratio (FR) was extracted as FR = [rawFRET − (0.5 * CFP)]/(0.02 * YFP).
Eeff was then calculated as Eeff, 3F = (FR − 1) * (εYFP440/εCFP440), where εYFP440 and εCFP440 represent the average molar extinction coefficients for YFP and CFP over the bandpass of the CFP excitation filter, respectively. The measurement Eeff takes into account cell-to-cell variation in expression of YFP and CFP, so that this FRET index is effectively independent of donor and acceptor concentration (Erickson et al., 2003; Oliveria et al., 2007).
FRET was additionally estimated using an acceptor photobleach (PB) method. PB measurements were obtained from the same cells used to make the 3F measurements. PB FRET was measured as the difference between CFP (donor) fluorescence intensity before and after photobleach of YFP (acceptor). Photobleach was achieved by several minutes of continuous illumination at 535 nm, and was virtually complete. Eeff was calculated from acceptor photobleach images as Eeff, PB = 1 − (FDA/FD), where FDA and FD indicate donor intensity before and after photobleaching, respectively [Eeff calculations are adapted from the study by Erickson et al. (2003)].
Statistical analysis.
Statistical analyses were performed using Student's t test, ANOVA, or Bonferroni's test. All error bars indicate SEM.
Results
AKAP79 and the CaV1.2 IQ motif sustain CaM–CaN signaling
We showed previously that CaV1.2 currents enhanced by the adenylyl cyclase agonist forskolin can be de-enhanced through CaN activation during episodes of increased stimulation frequency (Oliveria et al., 2007). To test whether CaM docked at the IQ motif is the activator of anchored CaN, we recorded forskolin-enhanced CaV1.2 currents from tsA201 cells while monitoring CaM–CaN interaction in response to an increase in step-depolarization frequency from 0.067 to 1 Hz. CaM stimulation of CaN was monitored by the CaM-YFP535/CaN-CFP470 fluorescence emission ratio in response to CFP436 excitation, which provides a FRET-based readout of changes in CaM–CaN interaction. Increasing depolarization frequency in voltage-clamped, transfected tsA201 cells caused a rapid increase in CaM-YFP535/CaN-CFP470, which appeared to proceed as fast as, or slightly faster than, de-enhancement of current (Fig. 1A). When intracellular EGTA was replaced with BAPTA, neither the change in YFP535/CFP470 ratio nor de-enhancement was observed upon increase in depolarization frequency (Fig. 1B). Differential sensitivity to BAPTA versus EGTA indicates that the change in YFP535/CFP470 occurs in response to increased [Ca2+] in the channel's nanoenvironment. These findings, along with previous ones showing that de-enhancement was fully prevented by intracellular dialysis with a peptide that mimics the CaN autoinhibitory domain and blocks CaN (Oliveria et al., 2007), together suggest that in our system Ca2+/CaM activates CaN, which in turn acts to de-enhance activity of CaV1.2 channels.
Figure 1.

In transfected tsA201 cells, AKAP79 and the IQ motif in CaV1.2 sustain CaM–CaN signaling during heightened channel activity. A–D, After establishing a steady baseline for both forskolin-enhanced CaV1.2 Ca2+ current amplitude (ICa) and YFP535/CFP470 emission ratio in response to step depolarization once every 15 s (0.067 Hz), depolarization frequency was increased to 1 Hz. Cells expressing channel subunits, AKAP79, CaN-CFP, and CaM-YFP (A), internally perfused with 10 mm intracellular BAPTA in place of 10 mm EGTA (B), expressing AKAP79PIX (79ΔPIX) in place of AKAP79 (C), and expressing CaV1.2IQ/EQ in place of wild-type CaV1.2 (D) are shown. Left, Ca2+ current records evoked before (0 s) and after 600 s of 1 Hz depolarizations. Right, Time course of response to increased depolarization frequency for both Ca2+ current (black) and YFP535/CFP470 (gold). Ca2+ current was normalized to the value obtained at 0 s; when necessary, time courses were corrected for the rate of channel rundown at 0.067 Hz. ΔYFP535/CFP470 indicates the change in the emission ratio relative to the average value at 0.067 Hz. The white envelope against the shaded background represents SEM. E, Left, Micrographs showing subcellular localization of CaN-CFP, CaM-YFP, and corrected, sensitized FRET gated to donor intensity [FRETC (CFP); pseudocolor]. Right, Pseudocolor images representing FRET donor intensity before (CFPpre) and after (CFPpost) acceptor photobleaching. F, Effective FRET efficiency calculated from both 3F and also YFP-acceptor PB measurements in some conditions. CaV1.2, β2b, α2δ, and AKAP79 were coexpressed with fluorescent CaN-CFP and CaM-YFP, except when CaV1.2 was omitted (−CaV1.2). In the BAPTA-AM experiments, cells were bathed in 100 μm BAPTA-AM for 60 min. n > 10 for all conditions. *p < 0.001 (black) vs 3F control; *p < 0.001 (gray) vs PB control (by both ANOVA and Bonferroni's test). G, Left, Micrographs showing subcellular localization of CaN-CFP, CaM-YFP, and corrected, sensitized FRET gated to donor intensity [FRETC (CFP); pseudocolor] in cells coexpressing unlabeled CaV1.2, β2b, α2δ, and AKAP79. CaV1.2IQ/EQ and 79ΔPIX were expressed in place of wild-type CaV1.2 and AKAP79 as indicated. Right, Three-filter-set effective FRET efficiency. n > 10 for all conditions. **p < 0.001 vs control (by both ANOVA and Bonferroni's test).
Unlike the strong CaN activation signal we observed in cells expressing CaV1.2 channels with wild-type AKAP79, CaN activation was defective when the AKAP79–CaN interaction was disrupted via deletion of the CaN-binding AKAP79 PxIxIT-like motif (79ΔPIX). In cells expressing 79ΔPIX, the extent of apparent CaM–CaN interaction declined subsequent to the increase in depolarization frequency (Fig. 1C), revealing a detectable amount of CaM–CaN association even when the channel was only activated at low frequency. Currents recorded from channels lacking scaffolded CaN retained a residual degree of de-enhancement, as though they were weakly regulated by the phosphatase even in the absence of AKAP79 anchoring.
When the CaV1.2 IQ motif-CaM interaction was disrupted by the well-characterized IQ-to-EQ mutation (IQ/EQ) (Zühlke et al., 1999; Erickson et al., 2003; Kim et al., 2004), the strength of the CaM–CaN interaction signal also declined as depolarization frequency was increased (Fig. 1D). As for cells expressing 79ΔPIX, this decline reveals detectable CaM–CaN association near CaV1.2IQ/EQ channels during low-frequency channel activation. Intriguingly, for channels containing the IQ/EQ mutation, increased stimulation frequency relieved channel inhibition that was present during the preceding period of low-frequency channel activity, as seen by a transient increase in current amplitude. The effects of the IQ/EQ and 79ΔPIX mutations indicate that IQ–CaM and AKAP79–CaN interactions are both necessary to sustain effective CaN-mediated opposition to PKA enhancement of channel activity.
CaM and CaN preassemble near CaV1.2 at resting Ca2+
Because CaN was able to partially suppress CaV1.2 activity during high-frequency depolarization in 79ΔPIX cells (Fig. 1C), we tested whether CaM and CaN are able to associate near the channel independent of AKAP79. To do this, we imaged FRET between CaM-YFP and CaN-CFP using two independent methods of measuring effective FRET efficiency: 3F and PB. When CaM-YFP and CaN-CFP were coexpressed with channel subunits and AKAP79, we observed FRET between CaM and CaN in live cells that was strongest near the plasma membrane (Fig. 1E,F). In contrast, FRET was absent when the pore-forming CaV1.2 channel subunit was not expressed or when cells were loaded with BAPTA-AM. Since the CaV1.2 C terminus contains both the IQ motif and a modified leucine zipper motif that helps support CaV1.2–AKAP79 interaction, one possible explanation for these observations is that the channel C terminus promotes CaM–CaN FRET between IQ-docked CaM and AKAP79-scaffolded CaN. However, CaN–CaM FRET persisted when FRET measurements were performed in cells expressing either CaV1.2IQ/EQ or 79ΔPIX (Fig. 1G), indicating that in tsA201 cells, neither the CaN-binding site on AKAP79 nor the IQ motif is necessary for CaM–CaN preassemble within the CaV1.2 complex at resting [Ca2+]in.
Calcineurin phosphatase activity triggers CDI of CaV1.2 channels
Given these results and the established role of CaM binding to the IQ motif in CaV1.2 CDI, we revisited the possibility that CaN might contribute to CDI. During 500-ms-step depolarization, AKAP79-associated CaV1.2 channels exhibited strong ion-dependent inactivation (measured as 1/τfast) with Ca2+ as the current carrier, but much less inactivation when current was carried by Ba2+ (Fig. 2A). CDI was most evident when Ca2+ influx was greatest (Brehm and Eckert, 1978; Lee et al., 1985): depolarization that evoked smaller Ca2+ current elicited little CDI, while depolarization that evoked maximal Ca2+ current (peak of the current–voltage relationship) elicited maximum CDI.
Figure 2.
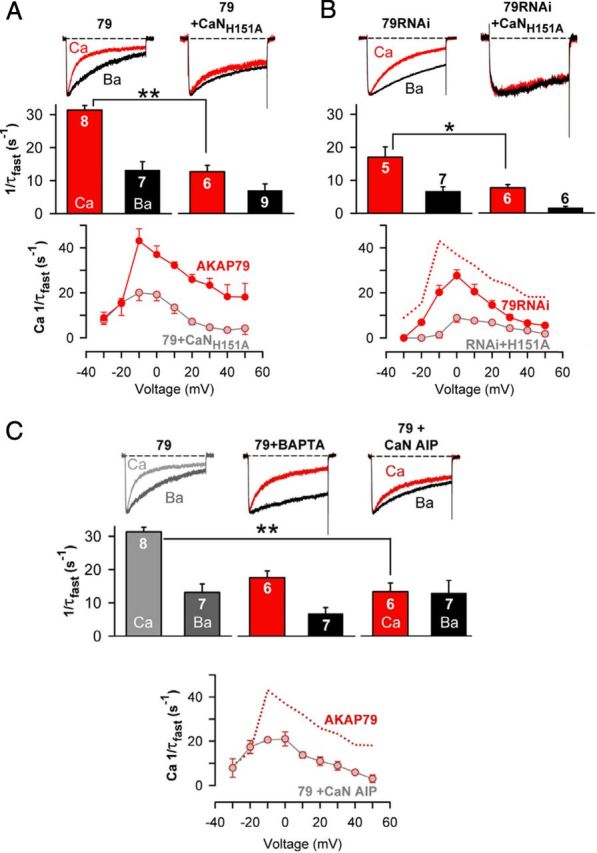
Localized calcineurin phosphatase activity triggers CDI of CaV1.2 channels heterologously expressed in tsA201 cells. A, Top, Ca2+ (red) and Ba2+ (black) currents recorded from the same cell by a 500 ms depolarization from −80 to +10 mV. Ca2+ and Ba2+ currents are shown scaled to the same peak inward current amplitude to facilitate comparison of inactivation rate. In addition to the three channel subunits, AKAP79 was overexpressed either alone (79) or with CaNH151A. Middle, Inactivation rates for Ca2+ current (red bars) and Ba2+ current (black bars). Number of individual cells recorded (n) is marked on the bars. Bottom, Ca2+ current inactivation rates over the range of test potentials that elicited inward current in cells overexpressing AKAP79 alone or AKAP79 + CaNH151A. **p < 0.001 (Student's t test). B, As in A, except that endogenous AKAP79 was suppressed (79RNAi), either alone or in combination with CaNH151A overexpression. *p < 0.05 (Student's t test). C, CaV1.2 Ca2+ (red) and Ba2+ (black) current records and inactivation rates with 10 mm intracellular BAPTA in place of 10 mm EGTA, or with the CaN AIP (100 μm) perfused into the cell via the patch pipette. Data for AKAP79-overexpressing cells, shown in gray, are reproduced from A for comparison. **p < 0.001 (ANOVA and Bonferroni's test). Rates of inactivation of Ca2+ current over the range of test potentials that elicited inward current in AKAP79-expressing cells that were exposed to the CaN AIP. Dotted red line reproduces from A the voltage dependence of Ca2+ current inactivation measured from control AKAP79-expressing cells without the CaN AIP. Ca2+ current densities were as follows (in pA/pF): 79, −37.1 ± 6.3; 79 + CaNH151A, −33.8 ± 7.4; 79RNAi, −35.0 ± 13; 79RNAi + CaNH151A, −27.4 ± 6.4; 79 + BAPTA, −31.1 ± 7.1; 79 + CaN AIP, −32.8 ± 8.6. No forskolin was used in the experiments.
CDI was significantly slowed when a catalytically inactive mutant of CaN (CaNH151A) (Mondragon et al., 1997) was transfected along with AKAP79 and the CaV1.2 subunits. Suppression of AKAP79 expression by a well-characterized RNAi construct (Hoshi et al., 2005) also slowed CDI, but this was less effective than CaNH151A (Fig. 2B). Combining CaNH151A overexpression with RNAi suppression of AKAP79, thus disrupting both CaN activity and anchoring, prevented CDI (Fig. 2B). These results suggest that when it could be scaffolded to channels by AKAP79, endogenous CaN remained partially effective in regulating channel activity despite competing against overexpressed CaNH151A. CDI was also reduced by an alternative means of inhibiting CaN: in cells transfected with AKAP79 and CaV1.2 subunits, intracellular perfusion with the CaN autoinhibitory peptide significantly depressed CDI (Fig. 2C). CDI was suppressed, albeit incompletely, by the Ca2+ chelator BAPTA (Fig. 2C), indicating that Ca2+ entry activates CDI and that the Ca2+ sensor that initiates CDI must be located close to the point of Ca2+ entry. Comparison of the full suppression by BAPTA of CaN-mediated channel de-enhancement during high-frequency stimulation (Fig. 1D) with the partial slowing of CDI during 500 ms depolarizations (Fig. 2C, middle) suggests that these two means of measuring CDI reveal different aspects of a single underlying mechanism. In either case, our results indicate that AKAP-anchored CaN, like IQ-bound Ca2+/CaM, signals within the channel nanodomain.
Ba2+ is a weak agonist for CDI
Inactivation of Ba2+ current carried by CaV1.2 is typically taken to represent voltage-dependent inactivation. But compared to inactivation of Ba2+ currents, Na+ currents through CaV1.2 channels exhibited much slower inactivation rates across the entire range of depolarizing pulses that evoked inward current (Fig. 3). Hence, it seems likely that Ba2+ acts as a weak agonist for CDI. This idea is supported by the observation that CaNH151A slowed inactivation of Ba2+ current (Fig. 2A,B) but not of Na+ current (Fig. 3A).
Figure 3.
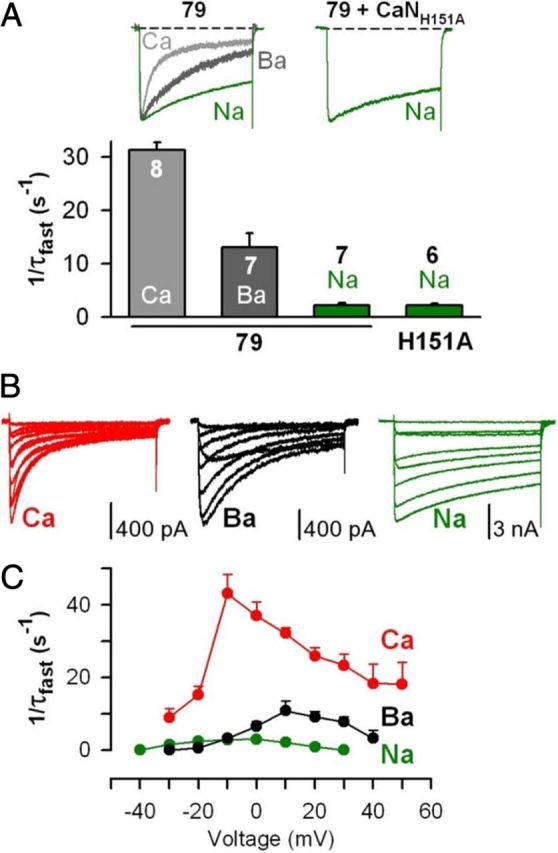
Ba2+ acts as a weak agonist for CDI of CaV1.2 channels in tsA201 cells. A, Top, Recordings of Na+ current (green) evoked by 500 ms depolarization from −80 to −10 mV in cells in which AKAP79 was overexpressed either alone (79) or with CaNH151A. Control CaV1.2 Ca2+ (light gray) and Ba2+ (dark gray) current records reproduced from Figure 2A. Ca2+, Ba2+, and Na+ currents are shown scaled to the same peak inward current amplitude. Bottom, Inactivation rates measured for Ca2+ (light gray bar), Ba2+ (dark gray bar), and Na+ (green bars) currents carried by CaV1.2 channels in cells overexpressing AKAP79 (79) or AKAP79 and CaNH151A. Ca2+ and Ba2+ data reproduced from Figure 2A. B, Families of Ca2+, Ba2+, and Na+ currents through CaV1.2 channels, recorded over a range of test potentials that elicited inward current in cells overexpressing AKAP79. C, For Ca2+, Ba2+, and Na+ currents carried by CaV1.2 channels, inactivation rates obtained over a range of test potentials applied to cells overexpressing AKAP79. No forskolin was used in the experiments.
AKAP79-targeted PKA modulates CDI
AKAP79/150-anchored PKA and CaN act as opponents in regulating L channel opening (Oliveria et al., 2007), predicting that active PKA might counter CaN-dependent CDI. Forskolin stimulation of PKA did not affect CaV1.2 inactivation (Fig. 4), perhaps because AKAP79 coanchoring of CaN allows the catalytically faster phosphatase to overcome PKA activity in the nanoenvironment of each channel (Oliveria et al., 2007). At a basal level of PKA activity, CDI was slightly slowed by expression of the CaN-anchoring-defective 79ΔPIX mutant, but stimulating PKA activity with forskolin in cells expressing 79ΔPIX slowed Ca2+ current inactivation nearly to the rate for Ba2+ (Fig. 4). Therefore it seems that in the presence of anchored and active PKA, CaN requires AKAP79/150 to overcome current enhancement by PKA and effectuate CDI. If CDI does indeed represent reversal of PKA phosphorylation of the channel by CaN, then CDI should also be sensitive to disruption of PKA enhancement. In accord with this idea, CDI was significantly slower when PKA activity was inhibited with PKI. However, inhibition of PKA did not eliminate CDI, as though PKA was not solely responsible for phosphorylation of the channel complex.
Figure 4.

AKAP79-anchored PKA opposes CDI of CaV1.2 channels in tsA201 cells. Top, Superimposed Ca2+ (red) and Ba2+ (black) currents carried through CaV1.2 channels. Bottom, Inactivation rates for Ca2+ and Ba2+ currents. Currents were recorded during forskolin (FSK; 5 μm) activation of PKA, with overexpressed mutant AKAP79 lacking the CaN binding site (79ΔPIX) with no forskolin present, with a combination of 79ΔPIX and forskolin, or during PKI (5 μm) inhibition of PKA activity (no forskolin). *p < 0.05 (Student's t test); **p < 0.001 vs AKAP79 + wild-type CaV1.2 (by both ANOVA and Bonferroni's test). Control data for AKAP79-overexpressing cells, shown in gray, are reproduced from Figure 2A for comparison. Ca2+ current densities were as follows (in pA/pF): FSK, −25.7 ± 7.2; 79ΔPIX, −28.5 ± 4.7; 79ΔPIX + FSK, −30.1 ± 3.9; PKI, −25.5 ± 6.5.
AKAP79-anchored calcineurin mediates CDI of CaV1.2 channels by opposing PKA
As an alternative means of studying PKA opposition to CaN, we studied the dynamics of development of this process in individual cells. Following patch rupture and initiation of whole-cell recording, internal perfusion with forskolin progressively slowed CDI in tsA201 cells transfected with 79ΔPIX, but not in cells transfected with wild-type AKAP79 (Fig. 5A). During intracellular perfusion of wild-type AKAP79 cells with forskolin, bath application of the CaN antagonists CsA and FK506, which inhibit CaN through distinct cyclophilin and FKBP pathways, was able to reduce CDI (Fig. 5B). The acute effect on CDI of this combination of CsA and FK506 was reversed upon washout. Together, the results support the idea that PKA, stimulated by elevation of cAMP with forskolin, can oppose CaN-mediated inactivation of Ca2+ current.
CaN contributes to CDI of L-type Ca2+ channels in cultured hippocampal neurons
As powerful as reconstitution in heterologous expression systems is for investigating mechanisms of channel regulation, models ultimately need to be tested in the biological system of interest, in this case, hippocampal neurons. We found previously that AKAP-anchored CaN powerfully regulates L-type Ca2+ currents in hippocampal neurons (Oliveria et al., 2007), so we next examined whether CaN might do so via CDI.
Pharmacologically isolated L-type Ca2+ current recorded from short-term cultured rat hippocampal neurons show robust CDI characterized by rapid inactivation in Ca2+ compared to Ba2+. Consistent with a role for AKAP79/150 in CDI, inactivation was much slower when expression of the rodent ortholog of human AKAP79, AKAP150, was suppressed with a previously characterized RNAi construct (Hoshi et al., 2005; Oliveria et al., 2007) (Fig. 6A). Suppression of CDI by 150RNAi was fully rescued by coexpression of AKAP79 in rat neurons, but no rescue of CDI was observed in neurons cotransfected with 79ΔPIX (Fig. 6B). In addition, the strong CDI observed in wild-type mouse neurons was completely absent in neurons cultured from 150ΔPIX knock-in mice (Sanderson et al., 2012) (Fig. 6C).
Figure 6.

AKAP79/150-anchored CaN is required for CDI of L-type Ca2+ channels in dissociated, short-term cultured hippocampal neurons. A, For rat neurons, pharmacologically isolated L channel Ca2+ (red) and Ba2+ (black) currents recorded from the same neuron, evoked by 500 ms depolarization from −60 to 0 mV. Peak Ca2+ current is normalized to peak Ba2+ current. Control neurons were transfected with GFP and the empty pSilencer vector. 150RNAi neurons were transfected with a short hairpin construct to suppress endogenous AKAP150 expression. Images of short-term cultured hippocampal neurons are a differential interference contrast (DIC) image of an untransfected hippocampal neuron (left) and a fluorescence image of hippocampal neuron expressing 150RNAi along with cytoplasmic GFP to mark transfected neurons (150RNAi; right). B, Cultured rat neurons were transfected with 150RNAi and either AKAP79 [79 wild-type (wt); rescue] or AKAP79ΔPIX (79ΔPIX; failed rescue). Images of cultured neurons are a fluorescence image of a hippocampal neuron expressing 150RNAi and AKAP79 (left) and a fluorescence image of a rat cultured hippocampal neuron expressing 150RNAi and either AKAP79ΔPIX or AKAP150ΔPIX. C, For mouse short-term cultured hippocampal neurons, pharmacologically isolated L channel Ca2+ (red) and Ba2+ (black) currents were recorded from the same neuron. DIC images of neurons from a wild-type mouse and a transgenic AKAP150ΔPIX mouse are shown. ***p < 0.0001 vs control (Student's t test). For inactivation rates, n values are indicated on the bars. Ca2+ current densities were as follows (in pA/pF): control, −32.2 ± 2.3; 150RNAi, −32.8 ± 2.0; 79 wt + 150RNAi, −29.2 ± 2.5; 79ΔPIX + 150RNAi, −28.7 ± 1.9; wt Mouse, −30.0 ± 2.1; 150ΔPIX Mouse, −31.9 ± 2.5.
To confirm a requirement for AKAP-anchored CaN in neuronal CDI, we used an acute means to antagonize CaN anchoring. Internal perfusion with a peptide that mimics the AKAP150 anchoring sequence for CaN, VIVIT (Oliveria et al., 2007; Li et al., 2012), competitively and dose-dependently suppressed CDI of Ca2+ current, leaving Ba2+ current unaltered (Fig. 7A). At the highest concentration tested (100 μm in the whole-cell pipette), VIVIT reduced the rate of inactivation in Ca2+ to that measured for Ba2+ current. To establish a requirement for CaN phosphatase activity in CDI of neuronal L channels, we used two different approaches: transfection of cultured neurons with CaNH151A and internal perfusion with CsA. Both experimental manipulations strongly reduced the rate of inactivation of Ca2+ current carried by neuronal L channels (Fig. 7B).
Figure 7.

Acute inhibition of CaN anchoring and activity suppresses CDI in hippocampal neurons. A, VIVIT peptide, a competitive inhibitor of CaN anchoring on AKAP150, dose-dependently suppresses CDI in dissociated, short-term cultured hippocampal neurons. VIVIT was perfused into neurons via the whole-cell patch pipette. Control neurons were recorded with 0.1% DMSO in the patch pipette, which was the concentration of DMSO in the VIVIT experiments. B, Calcineurin phosphatase activity is required for Ca2+-dependent inactivation of L-type channels in dissociated, short-term cultured hippocampal neurons. Top, Comparison of scaled currents evoked by 500 ms depolarization in Ca2+ versus Ba2+ for control, neurons transfected with CaNH151A, and neurons internally perfused with cyclosporin A (5 μm). Bottom, Corresponding inactivation rates (1/τfast). Control neurons were perfused with a whole-cell patch pipette solution containing 0.1% DMSO, which is the same concentration present in the cyclosporin A experiments. Results for CaNH151A were compared to the control neurons in Figure 6 (no DMSO), while results with CsA were compared to the DMSO control. *p = 0.01; **p < 0.005; ***p < 0.0001 vs control neurons (Student's t test). Ca2+ current densities were as follows (in pA/pF): control, −31.3 ± 3.5; 10 μm VIVIT, −31.9 ± 5.0; 25 μm VIVIT, −31.5 ± 4.9; 100 μm VIVIT, −29.2 ± 4.0; CaNH151A, −30.4 ± 2.2; and CsA, −32.6 ± 1.5.
Discussion
In neurons and a heterologous expression system that incorporates the important neuronal scaffolding molecule AKAP79/150, we found that Ca2+/CaM induces CDI of L channels through activation of CaN. A role for CaN in CDI is supported by our observations of significantly slowed inactivation by (1) overexpression of catalytically inactive CaN (CaNH151A), (2) internal dialysis with the CaN autoinhibitory peptide, and (3) application of the CaN antagonist CsA. The fact that CaNH151A had no effect on inactivation of CaV1.2 Na+ current indicates that this CaN mutant acts selectively to depress CDI. Expression of L channel CDI in neurons requires anchoring of CaN by AKAP79/150, whereas in the tsA201 expression system, CaN anchoring by AKAP79/150 is not absolutely required but is needed to support rapid, fully developed CDI. Evidence implicating CaN-anchoring by AKAP79/150 in CDI includes (1) elimination of CDI by 150RNAi knockdown and also by replacement with CaN-binding-defective AKAP79ΔPIX, (2) elimination of CDI in a 150ΔPIX knock-in mouse, and (3) competitive inhibition of CaN anchoring by VIVIT. Overall, interventions that impaired CaN action had more complete effects in neurons than in tsA201 cells, possibly reflecting more natural spatial organization and signaling stoichiometry, or yet-to-be identified regulatory factors.
Dephosphorylation signaling in CDI: permissive and direct models
Although Ca2+/CaM activation of CaN phosphatase activity appears to be a required step for CDI of L channels in hippocampal pyramidal neurons, our results do not exclude the possibility that conformational change of CaM docked to CaV1.2 can directly inactivate channel activity in some cellular contexts. An extensive body of work has highlighted the centrality of Ca2+/CaM in CDI (for review, see Calin-Jageman and Lee, 2008), but none of this work has taken into consideration the L channel scaffolding protein AKAP79/150, the absence of which minimizes CaN action on L channels. The logic underlying the two different experimental measures of CDI used here—cumulative inactivation during high-frequency depolarization reported by progressive reduction in Ca2+ current amplitude versus rate of inactivation during 500 ms depolarization—suggests a pair of alternative models for the action of dephosphorylation in CDI. In a permissive model, dephosphorylation by CaN must precede downstream steps that carry out CDI, such as conformational rearrangement of Ca2+/CaM on the C-terminal regulatory domains (PreIQ, IQ) of CaV1.2; dephosphorylation by CaN is a prerequisite for downstream steps to effectuate CDI. Alternatively, in a direct model of CaN′s role in CDI, dephosphorylation is envisioned as the terminal regulatory step in CDI, and Ca2+/CaM acts exclusively through CaN. In either case, the advantageous placement of CaN via anchoring on AKAP79/150 promotes CaN-dependent CDI. For highly abundant neuronal signaling molecules such as CaN (∼1% of protein in brain neurons), AKAP-based scaffolding may not only serve to localize enzymes to their targets, but via steric protection may also help shield substrates from extraneous phosphorylation signaling. It remains to be determined whether these models or related ones apply in excitable cells other than hippocampal neurons. For either model of CaN action, a dephosphorylation-based mechanism of CDI adds to the richness of signaling mechanisms that regulate L-type Ca2+ channels in neurons.
Ser/Thr kinases that oppose CaN-dependent CDI
In hippocampal pyramidal neurons, β2 adrenergic receptors are known to regulate L-type Ca2+ current through PKA (Gray and Johnston, 1987; Davare et al., 2001; Hoogland and Saggau, 2004), and in thalamocortical relay neurons, β2 adrenergic receptor activation slows CDI (Rankovic et al., 2011). But β2 adrenergic receptor activity is not the only potential means of activating PKA: a major adenylyl cyclase isotype in hippocampal pyramidal neurons is the Ca2+-stimulable type 8, which is known to bind to AKAP79/150 (Willoughby et al., 2010). Thus, Ca2+ influx via L channels could locally stimulate adenylyl cyclase type 8 production of cAMP and hence PKA activity. Our results in cells expressing a CaN-anchoring-defective AKAPΔPIX construct, demonstrate that activation of PKA through forskolin stimulation of adenylyl cyclase reduced CDI, suggesting that phosphorylation by PKA opposes dephosphorylation by CaN. Inhibition of PKA by PKI also impacted CDI, but this treatment, too, slowed CDI. That both stimulation and inhibition of PKA slow CDI might be reconciled to some degree by the following: while PKA kinase activity reverses CaN phosphatase action and thus suppresses CDI, PKI inhibition of kinase activity may result in few channels remaining in a phosphorylated state and so few remain available to undergo CaN-dependent CDI. However, when compared to CaN inhibition, incomplete suppression of CDI by PKI suggests contributions from other signaling processes.
CDF of L channels is also a phosphosignaling-based process and one that overlaps CDI, as CDF can be detected as a reduction in CDI (Wu et al., 2001; Hudmon et al., 2005; Erxleben et al., 2006; Grueter et al., 2006). CDF is mediated by channel-associated Ca2+/CaMKII activity, which is not only congruent with a phosphatase-based mechanism for CDI, but also indicates that kinases other than PKA may be involved in CDI. Indeed, because our experiments showed that a high concentration of PKI was unable to prevent CDI entirely, as predicted if PKA were solely responsible for recovery from CaN-dependent inactivation, it seems likely that additional kinases may contribute to phosphorylation of the CaN-sensitive CDI substrate. CaMKII is one likely candidate, as it associates with CaV1.2 channels and enhances their activity. If CaMKII cooperates with PKA to oppose CaN action, it is intriguing to consider how Ca2+/CaM molecules residing in the channel's nanodomain are able to initiate signaling to CaMKII or CaN and thereby initiate either CaMKII-dependent facilitation or CaN-dependent inactivation (Saucerman and Bers, 2008).
Previous evidence for CaN-dependent CDI
Considering that an enzymatic mechanism for CDI of CaV channels was proposed over two decades ago (Chad and Eckert, 1986; for review, see Armstrong, 1989), why has a role for CaN in CDI fallen into disfavor? One likely reason is that channel-associated CaN activity is difficult to suppress. CsA and FK506 are the most commonly used CaN inhibitors, but CsA and FK506 were much less effective in preventing CDI when compared to the CaN AIP (Figs. 2C, 5B). This may be related to the mechanism of action of CsA and FK506, which must first bind to immunophilins and then inhibit CaN activity as drug-immunophilin complexes. Because such complexes are much larger (>18 kDa) than the CaN AIP (a 25-mer polypeptide of 2.9 kDa), it is possible that CaN may be protected from inhibition by drug-immunophilin complexes when the phosphatase is tightly associated with the channel. In some cases, the effectiveness of CaN-inhibiting drugs like CsA has also been found to be limited by immunophilin expression level (Kung and Halloran, 2000; Mitchell et al., 2002). In our experiments in tsA201 cells, CaNH151A expression did not fully suppress CDI unless AKAP79/150 targeting of CaN was also disrupted, indicating that CaN-dependent channel inactivation is more resistant to perturbation when CaN is scaffolded to the channel. Although AKAP79/150 is expressed in every cell type where CaV1.2 is found and in most heterologous expression systems used to study CaV1.2 channels, including HEK293 and tsA201cells, the level of AKAP79/150 expression varies widely between cell types and might therefore explain partial or conflicting effects of CaN inhibitors on CDI.
Another confounding factor arises from the reliance upon Ba2+ to isolate CDI. But Ba2+ weakly supports CDI (Fig. 3), making it more challenging to work out the molecular mechanism of this process. Many divalent cations are able to activate CaM, their effectiveness as substitutes for Ca2+ largely depending on how closely they approximate Ca2+ in ionic radius (Chao et al., 1984). The difference in size between Ba2+ and Ca2+ generally makes Ba2+ a poor substitute for Ca2+, but several factors may facilitate Ba2+ binding to CaM within the channel complex: (1) [Ba2+], like [Ca2+], reaches high levels in the nanodomain near the channel mouth, (2) Ba2+ is poorly chelated by EGTA or BAPTA, and (3) Ba2+ is slowly sequestered by cellular transporters. Ba2+ has been found to activate other Ca2+-sensitive processes including neurotransmitter release (McMahon and Nicholls, 1993), Ca2+ release from internal stores (Satoh et al., 1987), contraction of skeletal (Stephenson and Thieleczek, 1986) and smooth muscle (Satoh et al., 1987), gene expression (Curran and Morgan, 1986), and cAMP production by Ca2+-sensitive adenylyl cyclases (Gu and Cooper, 2000).
Conclusions
In cultured hippocampal pyramidal neurons, CaN and PKA appear to work by opposing mechanisms of CDI/de-enhancement versus enhancement. Because multiple phosphorylation sites may be involved, and other kinases such as CaMKII or phosphatases such as PP2A (Xu et al., 2010), CaN versus PKA expressed as CDI versus enhancement may prove to be an oversimplification. The AKAP79/150 PxIxIT-like motif and CaV1.2 IQ motif play supporting roles in positioning CaN and CaM near the source of Ca2+, but preassembly of CaM with CaN may also help speed enzymatic CDI. The affinity of AKAP79/150 for CaN is relatively low (KD ≈ 0.5 μm), perhaps anchoring CaN just tightly enough to target it in cells while also permitting sufficiently rapid release from the AKAP to promote regulation of nearby substrates (Li et al., 2012).
Footnotes
This work was supported by NIH Grants F30-NS051963, T32-AA007464, R01-MH080291, and R01-HL088548.
The authors declare no competing financial interest.
References
- Adams B, Tanabe T. Structural regions of the cardiac Ca channel α1C subunit involved in Ca-dependent inactivation. J Gen Physiol. 1997;110:379–389. doi: 10.1085/jgp.110.4.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alseikhan BA, DeMaria CD, Colecraft HM, Yue DT. Engineered calmodulins reveal the unexpected eminence of Ca2+ channel inactivation in controlling heart excitation. Proc Natl Acad Sci U S A. 2002;99:17185–17190. doi: 10.1073/pnas.262372999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong DL. Calcium channel regulation by calcineurin, a Ca2+-activated phosphatase in mammalian brain. Trends Neurosci. 1989;12:117–122. doi: 10.1016/0166-2236(89)90168-9. [DOI] [PubMed] [Google Scholar]
- Bean BP, Nowycky MC, Tsien RW. β-Adrenergic modulation of calcium channels in frog ventricular heart cells. Nature. 1984;307:371–375. doi: 10.1038/307371a0. [DOI] [PubMed] [Google Scholar]
- Branchaw JL, Banks MI, Jackson MB. Ca2+- and voltage-dependent inactivation of Ca2+ channels in nerve terminals of the neurohypophysis. J Neurosci. 1997;17:5772–5781. doi: 10.1523/JNEUROSCI.17-15-05772.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brehm P, Eckert R. Calcium entry leads to inactivation of calcium channel in Paramecium. Science. 1978;202:1203–1206. doi: 10.1126/science.103199. [DOI] [PubMed] [Google Scholar]
- Burley JR, Sihra TS. A modulatory role for protein phosphatase 2B (calcineurin) in the regulation of Ca2+ entry. Eur J Neurosci. 2000;12:2881–2891. doi: 10.1046/j.1460-9568.2000.00178.x. [DOI] [PubMed] [Google Scholar]
- Calin-Jageman I, Lee A. CaV1 L-type Ca2+ channel signaling complexes in neurons. J Neurochem. 2008;105:573–583. doi: 10.1111/j.1471-4159.2008.05286.x. [DOI] [PubMed] [Google Scholar]
- Chad JE, Eckert R. An enzymatic mechanism for calcium current inactivation in dialysed Helix neurones. J Physiol. 1986;378:31–51. doi: 10.1113/jphysiol.1986.sp016206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao SH, Suzuki Y, Zysk JR, Cheung WY. Activation of calmodulin by various metal cations as a function of ionic radius. Mol Pharmacol. 1984;26:75–82. [PubMed] [Google Scholar]
- Curran T, Morgan JI. Barium modulates c-fos expression and post-translational modification. Proc Natl Acad Sci U S A. 1986;83:8521–8524. doi: 10.1073/pnas.83.22.8521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davare MA, Avdonin V, Hall DD, Peden EM, Burette A, Weinberg RJ, Horne MC, Hoshi T, Hell JW. A β2 adrenergic receptor signaling complex assembled with the Ca2+ channel CaV1.2. Science. 2001;293:98–101. doi: 10.1126/science.293.5527.98. [DOI] [PubMed] [Google Scholar]
- Dolmetsch RE, Pajvani U, Fife K, Spotts JM, Greenberg ME. Signaling to the nucleus by an L-type calcium channel-calmodulin complex through the MAP kinase pathway. Science. 2001;294:333–339. doi: 10.1126/science.1063395. [DOI] [PubMed] [Google Scholar]
- Erickson MG, Alseikhan BA, Peterson BZ, Yue DT. Preassociation of calmodulin with voltage-gated Ca2+ channels revealed by FRET in single living cells. Neuron. 2001;31:973–985. doi: 10.1016/s0896-6273(01)00438-x. [DOI] [PubMed] [Google Scholar]
- Erickson MG, Liang H, Mori MX, Yue DT. FRET two-hybrid mapping reveals function and location of L-type Ca2+ channel CaM preassociation. Neuron. 2003;39:97–107. doi: 10.1016/s0896-6273(03)00395-7. [DOI] [PubMed] [Google Scholar]
- Erxleben C, Liao Y, Gentile S, Chin D, Gomez-Alegria C, Mori Y, Birnbaumer L, Armstrong DL. Cyclosporin and Timothy syndrome increase mode 2 gating of CaV1.2 calcium channels through aberrant phosphorylation of S6 helices. Proc Natl Acad Sci U S A. 2006;103:3932–3937. doi: 10.1073/pnas.0511322103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez LL, Alam S, Smith KE, Horne E, Dell'Acqua ML. Regulation of A-kinase anchoring protein 79/150-cAMP-dependent protein kinase postsynaptic targeting by NMDA receptor activation of calcineurin and remodeling of dendritic actin. J Neurosci. 2002;22:7027–7044. doi: 10.1523/JNEUROSCI.22-16-07027.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon GW, Berry G, Liang XH, Levine B, Herman B. Quantitative fluorescence resonance energy transfer measurements using fluorescence microscopy. Biophys J. 1998;74:2702–2713. doi: 10.1016/S0006-3495(98)77976-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray R, Johnston D. Noradrenaline and β-adrenoceptor agonists increase activity of voltage-dependent calcium channels in hippocampal neurons. Nature. 1987;327:620–622. doi: 10.1038/327620a0. [DOI] [PubMed] [Google Scholar]
- Grueter CE, Abiria SA, Dzhura I, Wu Y, Ham AJ, Mohler PJ, Anderson ME, Colbran RJ. L-type Ca2+ channel facilitation mediated by phosphorylation of the beta subunit by CaMKII. Mol Cell. 2006;23:641–650. doi: 10.1016/j.molcel.2006.07.006. [DOI] [PubMed] [Google Scholar]
- Gu C, Cooper DM. Ca2+, Sr2+, and Ba2+ identify distinct regulatory sites on adenylyl cyclase (AC) types VI and VIII and consolidate the apposition of capacitative cation entry channels and Ca2+-sensitive ACs. J Biol Chem. 2000;275:6980–6986. doi: 10.1074/jbc.275.10.6980. [DOI] [PubMed] [Google Scholar]
- Hadley RW, Lederer WJ. Ca2+ and voltage inactivate Ca2+ channels in guinea-pig ventricular myocytes through independent mechanisms. J Physiol. 1991;444:257–268. doi: 10.1113/jphysiol.1991.sp018876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoogland TM, Saggau P. Facilitation of L-type Ca2+ channels in dendritic spines by activation of β2 adrenergic receptors. J Neurosci. 2004;24:8416–8427. doi: 10.1523/JNEUROSCI.1677-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshi N, Langeberg LK, Scott JD. Distinct enzyme combinations in AKAP signalling complexes permit functional diversity. Nat Cell Biol. 2005;7:1066–1073. doi: 10.1038/ncb1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudmon A, Schulman H, Kim J, Maltez JM, Tsien RW, Pitt GS. CaMKII tethers to L-type Ca2+ channels, establishing a local and dedicated integrator of Ca2+ signals for facilitation. J Cell Biol. 2005;171:537–547. doi: 10.1083/jcb.200505155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalman D, O'Lague PH, Erxleben C, Armstrong DL. Calcium-dependent inactivation of the dihydropyridine-sensitive calcium channels in GH3 cells. J Gen Physiol. 1988;92:531–548. doi: 10.1085/jgp.92.4.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Ghosh S, Nunziato DA, Pitt GS. Identification of the components controlling inactivation of voltage-gated Ca2+ channels. Neuron. 2004;41:745–754. doi: 10.1016/s0896-6273(04)00081-9. [DOI] [PubMed] [Google Scholar]
- Kung L, Halloran PF. Immunophilins may limit calcineurin inhibition by cyclosporine and tacrolimus at high drug concentrations. Transplantation. 2000;70:327–335. doi: 10.1097/00007890-200007270-00017. [DOI] [PubMed] [Google Scholar]
- Lee KS, Marban E, Tsien RW. Inactivation of calcium channels in mammalian heart cells: joint dependence on membrane potential and intracellular calcium. J Physiol. 1985;364:395–411. doi: 10.1113/jphysiol.1985.sp015752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Pink MD, Murphy JG, Stein A, Dell'Acqua ML, Hogan PG. Balanced interactions of calcineurin with AKAP79 regulate Ca2+-calcineurin-NFAT signaling. Nat Struct Mol Biol. 2012;19:337–345. doi: 10.1038/nsmb.2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukyanetz EA, Piper TP, Sihra TS. Calcineurin involvement in the regulation of high-threshold Ca2+ channels in NG108–15 (rodent neuroblastoma x glioma hybrid) cells. J Physiol. 1998;510:371–385. doi: 10.1111/j.1469-7793.1998.371bk.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCleskey EW, Fox AP, Feldman DH, Cruz LJ, Olivera BM, Tsien RW, Yoshikami D. ω-Conotoxin: direct and persistent blockade of specific types of calcium channels in neurons but not muscle. Proc Natl Acad Sci U S A. 1987;84:4327–4331. doi: 10.1073/pnas.84.12.4327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon HT, Nicholls DG. Barium-evoked glutamate release from guinea-pig cerebrocortical synaptosomes. J Neurochem. 1993;61:110–115. doi: 10.1111/j.1471-4159.1993.tb03543.x. [DOI] [PubMed] [Google Scholar]
- Meuth S, Pape HC, Budde T. Modulation of Ca2+ currents in rat thalamocortical relay neurons by activity and phosphorylation. Eur J Neurosci. 2002;15:1603–1614. doi: 10.1046/j.1460-9568.2002.01999.x. [DOI] [PubMed] [Google Scholar]
- Mitchell PO, Mills ST, Pavlath GK. Calcineurin differentially regulates maintenance and growth of phenotypically distinct muscles. Am J Cell Physiol. 2002;282:C984–C992. doi: 10.1152/ajpcell.00483.2001. [DOI] [PubMed] [Google Scholar]
- Mochida S, Few AP, Scheuer T, Catterall WA. Regulation of presynaptic CaV2.1 channels by Ca2+ sensor proteins mediates short-term synaptic plasticity. Neuron. 2008;57:210–216. doi: 10.1016/j.neuron.2007.11.036. [DOI] [PubMed] [Google Scholar]
- Mondragon A, Griffith EC, Sun L, Xiong F, Armstrong C, Liu JO. Overexpression and purification of human calcineurin alpha from Escherichia coli and assessment of catalytic functions of residues surrounding the binuclear metal center. Biochemistry. 1997;36:4934–4942. doi: 10.1021/bi9631935. [DOI] [PubMed] [Google Scholar]
- Noble S, Shimoni Y. The calcium and frequency dependence of the slow inward current ‘staircase’ in frog atrium. J Physiol. 1981;310:57–75. doi: 10.1113/jphysiol.1981.sp013537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveria SF, Dell'Acqua ML, Sather WA. AKAP79/150 anchoring of calcineurin controls neuronal L-type Ca2+ channel activity and nuclear signaling. Neuron. 2007;55:261–275. doi: 10.1016/j.neuron.2007.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson BZ, DeMaria CD, Adelman JP, Yue DT. Calmodulin is the Ca2+ sensor for Ca2+-dependent inactivation of L-type calcium channels. Neuron. 1999;22:549–558. doi: 10.1016/s0896-6273(00)80709-6. [DOI] [PubMed] [Google Scholar]
- Pitt GS, Zühlke RD, Hudmon A, Schulman H, Reuter H, Tsien RW. Molecular basis of calmodulin tethering and Ca2+-dependent inactivation of L-type Ca2+ channels. J Biol Chem. 2001;276:30794–30802. doi: 10.1074/jbc.M104959200. [DOI] [PubMed] [Google Scholar]
- Rankovic V, Landgraf P, Kanyshkova T, Ehling P, Meuth SG, Kreutz MR, Budde T, Munsch T. Modulation of calcium-dependent inactivation of L-type Ca2+ channels via β-adrenergic signaling in thalamocortical relay neurons. PLoS One. 2011;6:e27474. doi: 10.1371/journal.pone.0027474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson JL, Gorski JA, Gibson ES, Lam P, Freund RK, Chick WS, Dell'Acqua ML. AKAP150-anchored calcineurin regulates synaptic plasticity by limiting synaptic incorporation of Ca2+-permeable AMPA receptors. J Neurosci. 2012 doi: 10.1523/JNEUROSCI.3326-12.2012. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sather WA, Tanabe T, Zhang JF, Mori Y, Adams ME, Tsien RW. Distinctive biophysical and pharmacological properties of class A (BI) calcium channel α1 subunits. Neuron. 1993;11:291–303. doi: 10.1016/0896-6273(93)90185-t. [DOI] [PubMed] [Google Scholar]
- Satoh S, Kubota Y, Itoh T, Kuriyama H. Mechanisms of the Ba2+-induced contraction in smooth muscle cells of the rabbit mesenteric artery. J Gen Physiol. 1987;89:215–237. doi: 10.1085/jgp.89.2.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saucerman JJ, Bers DM. Calmodulin mediates differential sensitivity of CaMKII and calcineurin to local Ca2+ in cardiac myocytes. Biophys J. 2008;95:4597–4612. doi: 10.1529/biophysj.108.128728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KE, Gibson ES, Dell'Acqua ML. cAMP-dependent protein kinase postsynaptic localization regulated by NMDA receptor activation through translocation of an A-kinase anchoring protein scaffold protein. J Neurosci. 2006;26:2391–2402. doi: 10.1523/JNEUROSCI.3092-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sochivko D, Chen J, Becker A, Beck H. Blocker-resistant Ca2+ currents in rat CA1 hippocampal pyramidal neurons. Neuroscience. 2003;116:629–638. doi: 10.1016/s0306-4522(02)00777-7. [DOI] [PubMed] [Google Scholar]
- Stephenson DG, Thieleczek R. Activation of the contractile apparatus of skinned fibres of frog by the divalent cations barium, cadmium and nickel. J Physiol. 1986;380:75–92. doi: 10.1113/jphysiol.1986.sp016273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavalin SJ, Shepherd D, Cloues RK, Bowden SE, Marrion NV. Modulation of single channels underlying hippocampal L-type current enhancement by agonists depends on the permeant ion. J Neurophysiol. 2004;92:824–837. doi: 10.1152/jn.00700.2003. [DOI] [PubMed] [Google Scholar]
- Victor RG, Rusnak F, Sikkink R, Marban E, O'Rourke B. Mechanism of Ca2+-dependent inactivation of L-type Ca2+ channels in GH3 cells: direct evidence against dephosphorylation by calcineurin. J Membr Biol. 1997;156:53–61. doi: 10.1007/s002329900187. [DOI] [PubMed] [Google Scholar]
- Willoughby D, Masada N, Wachten S, Pagano M, Halls ML, Everett KL, Ciruela A, Cooper DM. AKAP79/150 interacts with AC8 and regulates Ca2+-dependent cAMP synthesis in pancreatic and neuronal systems. J Biol Chem. 2010;285:20328–20342. doi: 10.1074/jbc.M110.120725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Dzhura I, Colbran RJ, Anderson ME. Calmodulin kinase and a calmodulin-binding ‘IQ’ domain facilitate L-type Ca2+ current in rabbit ventricular myocytes by a common mechanism. J Physiol. 2001;535:679–687. doi: 10.1111/j.1469-7793.2001.t01-1-00679.x. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Xu H, Ginsburg KS, Hall DD, Zimmermann M, Stein IS, Zhang M, Tandan S, Hill JA, Horne MC, Bers D, Hell JW. Targeting of protein phosphatases PP2A and PP2B to the C-terminus of the L-type calcium channel CaV1.2. Biochemistry. 2010;49:10298–10307. doi: 10.1021/bi101018c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeilhofer HU, Blank NM, Neuhuber WL, Swandulla D. Calcium-dependent inactivation of neuronal calcium channel currents is independent of calcineurin. Neuroscience. 2000;95:235–241. doi: 10.1016/s0306-4522(99)00434-0. [DOI] [PubMed] [Google Scholar]
- Zühlke RD, Pitt GS, Deisseroth K, Tsien RW, Reuter H. Calmodulin supports both inactivation and facilitation of L-type calcium channels. Nature. 1999;399:159–162. doi: 10.1038/20200. [DOI] [PubMed] [Google Scholar]
- Zühlke RD, Pitt GS, Tsien RW, Reuter H. Ca2+-sensitive inactivation and facilitation of L-type Ca2+ channels both depend on specific amino acid residues in a consensus calmodulin binding motif in the alpha 1C subunit. J Biol Chem. 2000;275:21121–21129. doi: 10.1074/jbc.M002986200. [DOI] [PubMed] [Google Scholar]