

Abstract
While in vitro cell based systems have been an invaluable tool in biology, they often suffer from a lack of physiological relevance. The discrepancy between the in vitro and in vivo systems has been a bottleneck in drug development process and biological sciences. The recent progress in microtechnology has enabled manipulation of cellular environment at a physiologically relevant length scale, which has led to the development of novel in vitro organ systems, often termed ‘organ-on-a-chip’ systems. By mimicking the cellular environment of in vivo tissues, various organ-on-a-chip systems have been reported to reproduce target organ functions better than conventional in vitro model systems. Ultimately, these organ-on-a-chip systems will converge into multi-organ ‘body-on-a-chip’ systems composed of functional tissues that reproduce the dynamics of the whole-body response. Such microscale in vitro systems will open up new possibilities in medical science and in the pharmaceutical industry.
1. Introduction
In vitro models of human tissues are invaluable tools for research and drug discovery. Experimentation with in vitro models that mimic the in vivo metabolism and respond to stimuli authentically, i.e. that behave similar to those in vivo, provide the most meaningful results. While some tissue models are well established and used successfully for selected aspects of drug screening (for example the Caco-2 model of the GI-tract epithelium1), other tissues such as the blood brain barrier are more difficult to re-create. It has been shown that the microenvironments in which cells grow play an essential role in providing important mechanical and chemical cues that are needed to promote authentic cellular behavior2.
In recent years, the use of microtechnology has become an indispensable strategy to manipulate cell growth environments. The size scale at which microtechnology operates is highly relevant to living tissues. For example, the most commonly fabricated devices are microfluidic channels of sizes between 10 – 200 μm. In comparison, mammalian cells are 8-30 μm in size, and the diameters of microvascular capillaries range from 10 – 500 μm. Adaptation of the microtechnology in semiconductor industry to the field of tissue engineering resulted in various novel technologies, such as microfluidic cell patterning and manipulation3, hydrogel microfabrication4, and serum-free media formulation for multiple cell types5-7. These techniques have allowed researchers to place a variety of cell types in physiologically realistic proximity to each other and thereby create multi-cell type tissue constructs as well as tissue-tissue boundaries8. The stiffness of the substrate and shear stresses exerted on the cells can be controlled to match the physiological levels. Furthermore, fluid-to-cell ratios within microfluidic devices are much closer to physiological levels than in conventional cell culture dishes. Drug concentrations and fluid residence times can be more accurately mimicked using microfabricated cell growth reactors.
This review will discuss a variety of in vitro microscale tissue models, and describe how these single tissue models can be integrated into multi-tissue devices, often termed ‘body-on-a-chip’ or ‘human-on-a-chip’ devices9, 10. One of the most important advantages of microtechnology in term of body-on-a-chip devices is that the tissue chambers can be connected by a set of microfluidic channels mimicking blood vessels. Testing new drugs for toxic side effects or activated compounds as a result of liver metabolism is one of the most important considerations in drug development11. Body-on-a-chip cell culture platforms can simulate tissue-tissue interactions in a more physiologically realistic manner, improving the efficiency of drug development process.
2. Microfabricated organ models
Microfabrication technology has been applied to mimic various organ systems. Novel strategies have therefore been developed to reproduce certain aspects of the necessary tissue environment in vitro. In vivo tissue traits recreated in vitro so far include tissue geometries12, cell compositions13, biomolecular gradient14, and mechanical movement15. In this section, we describe how microfabrication strategies have been applied to the development of accurate representation of in vivo organs and how they led to more authentic in vitro organ functionalities.
2.1 Microvasculature
Microfluidics, dealing with extremely small quantities of liquid in microscale channels, is an ideal technology for recreating the microenvironment of the vasculature. Several factors are involved in the microenvironment of the blood vessels, including fluidic shear stress, peristaltic movement, chemical gradient, and cell to cell communication. The low Reynolds number typically achieved in a microfluidic system enables precise control of these factors, allowing researchers to study the combinatorial effect of the factors.
The fluid dynamics and transport phenomena inside microfluidic systems can be analyzed theoretically. In simple cases an analytical solution may be obtained but it is more typical to use a computational method. The flow inside a microfluidic channel can be analyzed by solving Navier-Stokes equation, assuming incompressible fluid:
| (1) |
Where u is the velocity field (m/s), ρ is the density (kg/m3), η is the viscosity (Pa·s), p is the pressure (Pa). In the simple case of a long, cylindrical tube with the radius R, the steady-state flow rate can be described by following equation.
| (2) |
Where Δp is the pressure drop across the channel with the length of L and η is viscosity (Pa·s). The equation (2) relates the volumetric flow rate with the pressure drop and the geometry of the channel (the radius and the length). An analogy to an electrical circuit can be made by relating the flow rate (Q) to the current (I), and the pressure drop (Δp) to the voltage drop (ΔV), then an equation similar to the Ohm's law can be written.
| (3) |
In fluid dynamics, the resistance R is dictated mainly by the geometry of the channel. In the case of a rectangular channel with a high aspect ratio (w>>h), the resistance can be described by the following equation:
| (4) |
Analogy of a microfluidic system with an electrical circuit allows a network of microfluidic channels to be analyzed in a similar manner16, 17. For example, the fluidic resistance of a serially connected microfluidic channels is the same as the sum of fluidic resistance of all channels. In a similar manner, the reciprocal of the fluidic resistance of a parallel-connected microfluidic channels is the same as the sum of the reciprocal of the resistance of all channels.
Microfluidic systems allow researchers to precisely control the parameters that define the microenvironment of in vivo tissues, enabling parametric study of relationship between environment and the cellular behavior. Young et al. used a microfluidic device to study the adhesion properties of endothelial cells in the presence of various extracellular matrix (ECM) proteins and fluidic shear stresses18. The fluidic shear stress in a parallel plate is determined by the geometry of the channel and the fluid velocity in a following manner.
| (5) |
where μ is the viscosity, Q is the flow rate, w is the width and h is the height of the channel. A unique advantage of using microfluidics was demonstrated in a study that used a microscale channel with a tapered profile, generating a wide range of shear stresses19. It is noteworthy that in many studies, a combinatorial effect of various environmental factors on cell behavior was studied, for example ECM proteins and shear stress18, signaling molecules and shear stress20, 21
While polydimethylsiloxane (PDMS) is the most popular material for microfluidic systems and carries many advantages, the material properties of the PDMS surface are not biologically relevant22. On the other hand, ECM proteins or hydrogels provide scaffolds that resemble the natural in vivo tissue environment better than PDMS23. Also hydrogels are more porous than PDMS, allowing molecular diffusion inside the hydrogel scaffold24. Therefore, many efforts have been directed toward fabricating hydrogel into a microfluidic scaffold25, 26. Other hydrogels have been used to create microfluidic devices, such as polyethylene glycol diacrylate (PEG-DA)27, and fibrin28. While these approaches focused on creating a rather simple representation of the blood vasculature, a different approach focused on recreating a more complex vasculature network. A sacrificial molding technique was used to create an interconnected network inside a scaffold made of collagen28 or PDMS29.
2.2 Lung and gas transfer
In the lung, the interface between air in the alveoli and blood in the surrounding capillaries is characterized by a bilayer of alveolar epithelial cells and microvascular endothelial cells, as well as surfactant and mucus produced by specialized cells in the epithelium. This interface acts primarily to deliver oxygen to and remove carbon dioxide from the blood, but also acts as a physical barrier to inhaled insults30. The microenvironment for the epithelial-endothelial bilayer includes an air-liquid interface with appropriate partial pressures and dissolved gas concentrations and mechanical stretching resulting from the action of breathing. Several microdevices have been designed to recreate portions of the lung physiology. The physiological epithelial monolayer using an air-liquid interface has been developed31. This air-liquid interface was shown to influence the integrity of the epithelial layer and to increase the production of surfactant, similar to the native epithelium in the lung. A microfluidic device was used to model the airway architecture to simulate abnormal obstruction of small airways and to study the effect of liquid plug propagation and rupture on the alveolar epithelial cells lining the alveoli32. The addition of a surfactant significantly reduced damage to the epithelium, providing evidence that such a device may be useful for evaluating methods for reducing airway damage due to occlusion and clearing of small airways. The addition of mechanical stretch to a device with similar fluid-based airway obstruction produced a device that combined physiological solid mechanical stresses with fluid stresses on alveolar epithelial cells33.
Recently, a PDMS system that incorporated both an air-facing epithelial cell monolayer and a liquid-facing endothelial cell monolayer of the alveolar-capillary air-liquid interface was produced15. The two layer device was designed to allow controlled mechanical stretching of the endothelial-epithelial bilayer, mimicking the mechanical cues present in the lung during breathing. When mechanical stretching was applied to the bilayer system under flow, organ-level responses to bacteria, adhesion of neutrophils, and pathogen phagocytosis were recreated. The extremely high gas permeability of PDMS commonly used for these types of devices limits the on-device control and measurement of the gas concentrations in the media and air phases on the device, both of which are important for recreating the microenvironment of the alveoli. Iterative computational fluid dynamics and experimental flow visualization were used to design microfluidic paths and internal structures to control the liquid side flow in a silicon-based device (Figure 1(A))34.
Figure 1.

Microfabricated organ systems mimicking various organ tissues. (A) Lung on a chip device modeling an alveolus and layout of fluid side of lung-based body-on-a-chip device fabricated in silicon. Reprinted from Long et al.34 with permission from Springer. (B) BBB on a chip, consisting of two perpendicular channels separated by a membrane. Reprinted with permission from Booth et al.60. (C) The contractility of heart tissue is measure using the muscular thin film (MTF). Reprinted with permission from Grosberg et al.79. (D) A microfluidic bioreactor for 3D liver tissue engineering. Reprinted with permission from Domansky et al40. (E) Microscale hydrogel scaffold mimicking the intestinal villi geometry. Reprinted with permission from Sung et al.41. (F) Cantilever for detecting myotube contraction. Above: SEM micrograph of silicon cantilever array at 60x magnification. (scale bar = 5004m), Below: Confocal micrograph detailing top down view of a single cultured myotube on a cantilever. (scale bar = 204m). Reproduced with permission from Wilson et al.86. (G) Microvascular network in 3D tissue scaffold made of collagen matrix. Reprinted with permission from Zheng et al.26.
2.3 Liver metabolism and in vitro metabolism
The first-pass metabolism refers to the metabolism of a drug during oral absorption. Before a drug reaches the systemic circulation, it goes through the intestine and the liver, where it is metabolized by intestinal enzymes, microbial enzymes, and hepatic enzymes. The liver is strategically located behind the gastrointestinal (GI) tract to detoxify xenobiotics, and receives blood from the GI tract through the portal vein. Since it has as profound effect on the final effect of a drug, reproducing the liver metabolism in vitro has been of great interest. The liver metabolism can be divided into two types of reactions; phase I and phase II metabolism. The phase I metabolism includes hydrolysis, oxidation, and reduction reactions while the phase II metabolism mainly consists of conjugation reactions35. Cytochrome P450 enzymes play dominant roles in the phase I reaction, while various enzymes such as uridine diphosphoglucuronosyl transferase (UGT), glutathione S-transferase (GST), and sulfotransferase (ST) are involved in the phase II reactions.
Various in vitro systems for reproducing the liver metabolism have been developed. While liver slices36 or primary hepatocytes37 are considered to demonstrate metabolic profiles similar to the liver over a short period of time, the scarcity of the model system and the difficulty with using the system hinders them from being widely used models, especially in high-throughput settings. Cell lines such as HepG238 or microsomes39 - cellular subfraction separated from the liver tissue - are easier to use but may not accurately reflect the actual liver metabolism. It has been known that the functional unit of the liver, the acinus, expresses different set of proteins depending on the locations within the unit, and it has been speculated that it might be related to the gradient of oxygen concentration present in the liver tissue40. Reproducing the oxygen gradient resulted in heterogeneous distribution of P450 enzyme activity, consistent with the distribution of the actual liver14. Khetani et al. demonstrated that reproducing the co-culture pattern of primary hepatocytes and stromal cells improved various liver-specific functions13. Culturing rat hepatocytes in 3-dimensional configuration and exposing the culture to fluidic flow also improved liver functions41, 42. Mimicking the endothelial-hepatocyte interface of the liver sinusoid induced rat hepatocytes to organize into bile canaliculi along hepatic-cord like structures43.
It has been well demonstrated that microfabricated organ systems can improve liver functions of cultured hepatocytes, and improved liver function is likely to result in a more accurate prediction of the metabolic profile of drugs. However, adaptation into a high-throughput format is important,44 and newly introduced systems need to be validated against in vivo hepatic clearance data before it can be implemented into a drug development process.
2.4 Gastrointestinal (GI) tract and absorption
Since the oral route is generally a preferred method of drug administration, and the absorption of drugs in the GI tract plays a dominant role in determining the bioavailability of the pharmacokinetic-pharmacodynamic profile of the drugs, predicting the oral drug absorption kinetics early in the drug development process is important. The in vivo environment of the GI tract is extremely complex, consisting of various factors. The GI tract in human male adult is several meters long, with circular tissue geometry. The lumen is separated by several layers of tissues containing mucosa, muscle, and blood vessels. The inside lining of the epithelial layer of the small intestine is covered with villi, which increase the absorptive surface area1. One distinctive feature of the GI tract is that it is under complex mechanical movement, including segmental contraction, peristaltic wave, and microscopic villi motility45. Another feature of the GI tract is that it is occupied by a large number of microbes, co-existing with intestinal epithelial cells46.
The two major in vitro methods for predicting drug absorption are the Caco-2 model47 and the parallel artificial membrane permeability assay (PAMPA)48. They mainly test the permeability of drugs based on passive diffusion across membranes, and neglect the other mechanisms of drug transport and complex interaction with microbiota. For example, the Caco-2 cell monolayer model was able to predict the absorption coefficient of rapidly and completely absorbed drugs, while the prediction for slowly and incompletely absorbed drugs were inaccurate49.
Several attempts to reproduce the microenvironment of the GI tract have been reported. Sung et al. developed a novel hydrogel microfabrication method to create collagen scaffold mimicking the shape of intestinal villi, and cultured Caco-2 cells into a 3-dimensional villi shape12. Using this 3D villi scaffold, permeability coefficients were measured and were shown to be closer to in vivo values than the conventional 2D model50. The synergistic effect of crypt topography and extracellular matrix (ECM) proteins on the Caco-2 cells was investigated by fabricating a collagen scaffold mimicking the crypt structure51. Kim et al. developed a microfluidic device for co-culturing eukaryotic HeLa cells and bacteria (E.coli) by compartmentalizing the device with a pneumatic valve to study the interaction of microbes and the epithelial cells52. Using the elastic nature of PDMS, a device operated with vacuum was developed to simulate the peristaltic movement of the GI tract53.
2.5 Blood-brain-barrier (BBB) and central nervous system (CNS)
The blood brain barrier is a complex biological structure involved in the protection and maintenance of the central nervous system (CNS) against exogenous compounds present in the blood. This multi-layer structure acts as a restrictive membrane separating the blood from the cerebrospinal liquid but needs to be selectively permeable to essentials compounds such as selected sugar, amino-acids, electrolytes, and water. The BBB is made primarily of three different cell types embedded in extracellular matrix which together form the neurovascular units54. The microvascular vessels with endothelial cells are lined by pericytes and astrocytes (glial cells). The large number of tight junctions present in the brain endothelium results in a membrane with high values of trans-endothelial electrical resistance (TEER). Moreover, most of the exchanges between the two compartments are under the control of specialized membrane transporters such as P-gycoprotein. These characteristics make the access to the brain difficult for drugs and the BBB is a major target for drug development in the pharmaceutical industry.
Currently, the replication of the BBB is performed in transmembrane-well plates. Brain microvascular endothelial cells are cultivated on the top side of the membrane while astrocytes with or without pericytes are cultivated on the bottom side55. Porcine, bovine, rat or murine primary cells have been used as cell sources for the BBB model, and recently human stem cells have been proposed to generate blood brain endothelial cells, astrocytes and neurons56. Commercially available membranes suffer from high flow resistance due to low to modest porosity and irregular pore distribution which does not allow a close interaction between cell types. Microfabricated membranes can address these issues by reducing the global thickness and create high porosity with regular distribution of pores. Shayan et al. have developed a nanofabricated membrane with controlled pore size and low thickness (3 μm) allowing cell culture57. They demonstrated significant reduction of the flow resistance across the synthetized membrane and maintenance of metabolic activity and viability for at least three days.
Mechanical stimuli such as shear stress induced by the blood flow appear to be involved in the differentiation status of the endothelium58. A hollow fiber bioreactor has been developed to overcome the absence of fluid flow and shear in the standard transwell model59, where shear stress has been shown to induce overexpression of genes and proteins of cytoskeleton, tight-junctions and transporters. However, the interaction between the different cell types was limited by the thickness of the fiber (150 um), and the time required to reach steady state transendothelial resistance was longer than the transwell membrane. A microfluidic based system has recently been developed to combine flow stimulation, integrated electrodes for resistance measurement and transparency for observation60. A porous polycarbonate membrane was sandwiched between two PDMS layers containing channels and culture chambers separating two compartments to allow dynamic culture.
Investigation of neuronal biology and CNS functionality has been limited by the lack of pertinent tools to reproduce physiologically accurate models of the neuronal microenvironment. Development of microfabricated platforms dedicated to neurobiology has opened new perspectives allowing precise spatio-temporal control of cellular environment61. For example, the utilization of micropatterned surface has allowed the guidance and polarization of axonal/dendrite outgrowth in dissociated neuronal culture62, 63. The study of the axonal biology inside a microfluidic chamber was presented by Taylor et al61. The authors developed a compartmentalized device allowing the physical separation of the neuronal body (somal side) with the axonal extension (axonal side). The platform demonstrated a significant advantage compared to the traditional methods by permitting the isolation and growth of the CNS axon, polarized through microchannels without somal and dendritic contamination. Another approach offered by this compartmentalized chambers is the possibility to co-cultivate neuron with glial cells (for example oligodendrocytes).
Multi Electrode Array or microelectrode arrays (MEA) are widely used for multi-neuron electrical recordings and have been used to model disease such as Alzheimer's disease64. They have been recently improved by the addition of microfluidic culture chambers and micro patterning techniques65. One of the limitations of the MEA system has been the difficulty of isolating the electrical signal of a single cell. By combining an MEA with replica molded PDMS channels and wells, Tinturé et al.66 proposed a one-to-one electrode-neuron recording system. A promising example of this technology was applied to neurons cultivated in a dual-compartment device placed on an MEA recording system67. The two neuronal populations were interconnected by microchannel networks allowing the neurite extension, and recording of the electrical activity combined with a statistical connection map showed functional connection between the two populations. The use of MEA devices for in vitro neuronal culture systems has been recently reviewed68. This technology offers a significant insight in neurobiology by potentially permitting the interaction of different neuronal population with a simultaneous recording system.
2.6 Cardiac systems
The use of perfused mammalian hearts for the study of cardiac physiology, contractile function and pathology was pioneered by Oscar Langendorff in the late 19th century69. The system is still currently used in functional in vitro studies where the effects of pathological and chemical challenges on the contractile ability of the tissue can be characterized70. However, its size, physiological structure and the need for intricate supporting equipment make it an impractical system for integrated chip based models. A number of systems have been developed using single cardiomyocytes to evaluate the forces generated by these cells. These systems were based on the observation of deformation of an elastic substrate71, pillars72 or an attached bead73; or a piezoelectric force transducer74. However, results from a study using a small number of cardiomyocytes in an array indicated that single cells may not be suitable for testing contraction function of the heart75.
While single cell assays are valuable for evaluating the function of a single cell, larger microtissues of many cardiomyocytes better mimic the action of cardiomyocytes in the heart. The forces generated by sheets or films of cardiomyocytes have been measured by growing the cardiomyocytes on PDMS cantilevers76, PDMS films mounted on posts77, and PDMS films attached at one end78, 79. The film configuration of cardiomyocytes has also been incorporated onto a diaphragm such that when the cardiomyocytes contrated, the diaphragm deformed and produced a change in pressure in the chamber80. Microtissues in a 3D configuration have been tested in a system of coupled vertical cantilever posts, in which a microtissue between two cantilevers bent the beams toward each other81. Microelectrode arrays have been used extensively for the measurement of the electrical activity of cardiomyocyte cultures and their use has been reviewed recently82. An extension of the system to enable patterning of the cardiomyocyte was used to evaluate drug effects on conduction velocity and the action potential refractory period in cardiomyocyte cultures83.
2.7 Muscle
Techniques for establishing in vitro cultures of primary muscle cells from both human and rodent sources have been available for over 30 years84. Myoblasts in culture retain the hypertrophic ability they possess in vivo and are therefore able to fuse, forming primary myotubes capable of functional contractile activity85. To date, the movement towards more biomimetic and sophisticated in vitro muscle models has centered on methods for improving the cellular architecture of the various culture systems, as well as developing the means to effectively measure, characterize and maximize the functional output of the seeded cells86.
Three-dimensional muscle culture systems center on the use of either synthetic87 or biopolymer88 exogenous matrices as scaffolds in which to seed the desired cell population. In culture models utilizing 3D biopolymer matrices, cells reorganize along the lines of principal strain, provided by cellular contraction against fixed posts, and fuse to form parallel arrays of myotubes capable of performing directed work89. Attachment of the culture model's fixed posts to a force transducer89 or microscopic measurement of the matrix movement in the system90 then allows for calculation of the specific force generated by the cells in culture. While the level of sophistication in these systems is impressive, the presence of the biopolymer matrix makes integration of other complimentary cell systems difficult.
Two dimensional culture models are far more amenable to integration into more complex body-on-a-chip type systems. Orientation of muscle cells in such models is relatively simple, relying on patterning of culture surfaces to provide the desired cellular architecture91, 92. More problematic in these cultures is generating methods to effectively measure and quantify the functional output of the cells. The use of cantilever chips in 2D culture environments represents an elegant method for measuring the contractile activity of cultured myotubes(Figure 1(F))93. When attempting to develop complex, multi-organ in vitro systems, such models are attractive since further patterning of these chips should allow for organized and controllable integration of other supporting cell types including Schwann cells, motoneurons and sensory neurons.
2.8 Neuromuscular junctions
The ability for dissociated primary muscle cells and motor neurons to generate functional neuromuscular transmission in vitro was first reported using chick cells by Fischbach in 197094. Culture systems utilizing rodent cells were later developed and widely adopted due to the considerable advantages associated with the use of mammalian cells for studying NMJ physiology and function95.
The ability for primary rodent neurons and myotubes to form neuromuscular contacts in vitro has been demonstrated5. Electrophysiological recordings from such cultures suggest the existence of functional neuromuscular transmission, and spontaneous contraction of the cultured myotubes is observed and shown to be blocked by treatment with the non-depolarising neuromuscular blocker D-tubocurarine96. Culture systems using human and rodent derived embryonic stem cells (ESCs) with primary muscle and C2C12 muscle cell line sources have been developed6. These cell culture methods have the potential to be used as functional in vitro neuromuscular junction systems as means to test the response of novel therapeutic compounds in both healthy and diseased synapses. However, despite these advances in cell culture methods to produce and demonstrate neuromuscular junction formation, these systems have yet to be incorporated into a system suitable for use in a body-on-a-chip device.
3. Microfluidic model of whole animals
Ultimately, microfabricated organ systems can be integrated to simulate the whole-body response to drugs or pathological challenge. Although individual organ systems are not the perfect mimic of the in vivo tissues, such integrated model systems can be a valuable tool for studying multi-organ interactions. Interaction between two organ systems has been demonstrated by several groups97-99 and interaction between three or four organs have also been reported12, 100, 101, but a true dynamic interaction between more than four organs has yet to be demonstrated. In this section we describe the concept of pharmacokinetics and the experimental approach to simulate multi-organ interactions in vitro.
3.1 Concept of pharmacokinetics
After administration, drugs go through a complex process involving absorption, distribution, metabolism and elimination (ADME). This complex process results in a time-dependent change of drug concentration in the target tissue, which affects the final pharmacological effect of drugs. The pharmacokinetics (PK) refers to the time-dependent profile of a substance in a living system102. While PK plays an important role in determining the pharmacological effect of a drug11, there is no in vitro system that can accurately predict the PK of drugs in human. Currently available in vitro systems can recapitulate only a part of the entire process. For example, in vitro systems for predicting gut absorption kinetics and in vitro systems for predicting the liver metabolism are frequently being used in pharmaceutical research, but in vitro systems that can predict the dynamics of gut absorption followed by the liver metabolism have only begun to be reported in the last few years.
A mathematical modeling technique, called PK modeling, enables prediction of the drug concentration from a given dose. PK modeling is a technique widely used in pharmaceutical industry for dose optimization, and various forms of PK models exist depending on the complexity of the model. One form of a PK modeling approach, called physiologically-based pharmacokinetic (PBPK) modeling, segregates the body into organ compartments, which are connected via hypothetical blood flows. The mass balance for drug concentration in each compartment results in a set of ordinary differential equation that can be solved numerically103. Compared to other simpler forms of PK models, PBPK models provide a mechanistic basis for the model. The practical limitation of using a PBPK model for prediction of drug concentration comes from the difficulty of obtaining experimental data and finding model parameters, although various mathematical techniques to circumvent this problem exist104. Physical replication of PBPK models are an ideal in vitro platform to measure parameters, test hypotheses, and develop novel dosing strategies (Figure 2)10. As microfluidics allow precise control of flow and connection of multiple compartments, compartmentalized microfluidic systems can serve as an in vitro platform of a mathematical PBPK model105. The concept of using microfluidic systems as a physical representation of a PBPK model has been demonstrated106.
Figure 2.
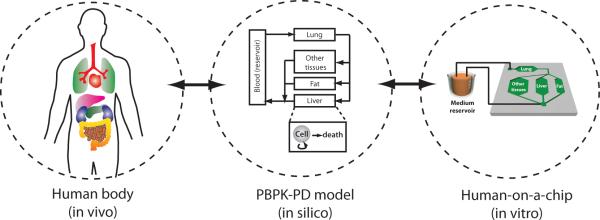
Concept of physiologically-based pharmacokinetic (PBPK) model as a mathematical representation of the human body, and human-on-a-chip as a physical replication of a PBPK model.
3.2 Microfluidic systems for reproducing organ interactions
The simplest method to reproduce interactions between multiple organs is to incubate multiple cell types in a common overlying media and allow the cells to communicate via soluble signals. Using this concept, primary human hepatocytes and mouse fibroblasts were cultured together in a device termed integrated discrete multiple cell co-culture (IdMOC)107. Using this device, hepatic metabolism and subsequent cytotoxicity of various compounds could be observed. However, this device lacked the time-dependent dynamics of organ interaction, as all components were submerged in a common medium, providing a homogeneous environment.
The Ahluwali group in Pisa University, Italy published a series of papers on developing a bioreactor system for reproducing multi-organ interactions108. Using this bioreactor system, an interaction between the hepatocytes (liver) and adipose tissue (fat) was observed99. By analyzing various parameters representing the liver metabolic activity, it was demonstrated that the connection of the liver and the fat compartments led to an enhancement of the liver metabolic activity. More recently the researchers extended their system to mimic a three-way interaction between the hepatocytes (liver), adipose tissue (fat), and endothelial cells (blood vessel) on glucose metabolism100 (Figure 3(A)). By controlling the glucose level in the perfused medium, normal and hyper- glycaemia were mimicked, and the response of each tissue model to insulin was observed by measuring the metabolites related to lipid and glucose metabolism.
Figure 3.

(A) Schematics of 3-way connected culture, containing the hepatocytes, endothelial cells, adipose tissue. Reprinted with permission from Iori et al.100. (B) Schematic of two sequentially perfused chambers (GI tract-liver). Reprinted with permission from van Midwoud et al.97. (C) A multi-channel 3D microfluidic cell culture system (3D-μFCCS), containing four connected chambers on a chip. Reprinted with permission from Zhang et al.118. (D) A microfluidic device for reproducing multi-organ interaction, containing three chambers connected with fluidic channels. Reprinted with permission from Sung et al.106.
Recenlty, several research groups have developed microfluidic systems to reproduce multi-organ interactions and the dynamics of a drug's action in the body. Van Midwoud et al. previously developed a novel perfusion system to incubate precision cut liver slices and observed improvement in drug metabolizing activity109. They extended their work to incubate a liver slice and an intestinal slice, and connected the two compartments with fluidic channels to mimic first-pass metabolism97 (Figure 3(B)). The interplay between the two organs was demonstrated by exposing the device to bile acid, which induced expression of fibroblast growth factor 15 in the intestinal slice and subsequent down regulation of cytochrome P450 7A1 in the liver slice.
Mao et al. developed a microfluidic system that reproduced liver metabolism and subsequent liver toxicity by connecting compartments containing PEG (polyethylene glycol)-encapsulated liver microsome and liver cell (HepG2) culture, and a solid-phase extraction system to purify the reaction product110. 5’-diphosphate-glucuronosyltransferase (UGT) metabolism of acetaminophen and subsequent liver toxicity was characterized using this system. A device with a similar concept was reported earlier by another research group98 (Figure 3(C)).
In the study by Imura et al., microscale models of the intestine (Caco-2 cells), the liver (HepG2 cells), and breast cancer (MCF-7 cells) were integrated to create what they termed ‘micro total bioassay system’111. The activity of anticancer agents and estrogen-like substances were assayed using this system, and the researchers further improved the system by adding another compartment for gastrointestinal degradation prior to the intestinal absorption112. This compartment employed a synthetic digestive juice to reproduce the digestive process in the stomach by mixing gastric juice, drug sample and alkaline solution.
3.3 Microfluidics for reproducing the whole-body response
The concept of using a microfluidic device as a physical representation of a PBPK model was proposed by Shuler in the early 2000s. Being a physical representation of a PBPK model, the microfluidic system can serve as a physical model for reproducing whole-body response and multi-organ interactions. In 2004, Sin et al. reported the fabrication of a three-chamber ‘microscale cell culture analog’ integrated with an oxygen sensor113. Three chambers were fabricated on a 1 inch square silicon chip, representing the lung, liver and other tissues, and were connected by fluidic channels representing the blood flow. Although this study mainly focused on the fabrication and the operation of the device, it was the first reported example of using a microfluidic system to study multi-organ interactions. The advantage of using microfluidics was that it was possible to control the flow rate so that the fluid residence time in each chamber was set to be the residence time of corresponding organs in the human body. This kind of precise control was not achievable in previous studies that did not use microfluidics114. In a study using the same device, the mechanism of naphthalene toxicity was studied and revealed that the liver metabolism played an important role in the observed lung toxicity of naphthalene115. This was the first demonstration that microfluidic systems can reproduce the multi-organ interaction.
This concept was further developed to fabricate a four-chamber device to study the efficacy of drug mixtures on multidrug resistance cancers101. In this study, the four chambers represented the liver, bone marrow, uterine cancer, and MDR variant of uterine cancer. The liver chamber was included to simulate the liver metabolism, and the bone marrow chamber was included to simulate the toxic side effects of chemotherapeutic agents. The pharmacokinetic profile in the microfluidic device was predicted by developing a PBPK model based on the device configuration. A microfluidic module to simulate oral absorption was added to the device and acetaminophen was used to demonstrate the liver toxicity of acetaminophen after oral absorption and liver metabolism116. The silicon-based device was also used to incorporate 3D cell culture models to better simulate the in vivo tissue environment117. The device was further improved to better simulate the tissue environment by incorporating a cylindrical hydrogel scaffold and connecting the scaffold with fluidic channels106 (Figure 3(D)). This configuration was intended to mimic the tissue mass surrounded by blood vessels. Building a PBPK model based on the device and fitting the model parameters to experimental data revealed some biological insight into the mechanism of cytotoxicity.
Microfluidic systems to simulate the whole-body response have often been termed ‘human-on-a-chip’, or ‘animal-on-a-chip’. Zhang et al. reported a multi-channel, 3D microfluidic cell culture system (3D-μFCCS), which contained 3D aggregates of different cells to mimic multiple organs in the body118. Four human cell lines were chosen to represent the liver, lung, kidney and adipose tissue. Although the researchers did not report the observation of multi-organ interaction using this device, a notable advantage of this system was the use of gelatin microspheres to selectively deliver growth factors to a specific organ (the lung in this case). This type of technique can be useful for creating organ-specific environments, while maintaining fluidic connection between the compartments. A bio-printing method was developed to seed a cell-laden hydrogel into a microfluidic device to mimic liver metabolism119. A similar technique was used to create a dual-tissue microfluidic chip, containing the liver cells (HepG2) and mammary epithelial cells (M10)120. This device was used to test the effect of the radiation and radiation shielding effect of the pro-drug amifostine. Although these systems aim to reproduce interaction between multiple organs, they are mostly limited to reproducing the interaction between two organs, and the demonstration of more complex interactions has not yet been reported yet. However, these preliminary systems and relevant technologies can serve as a basis to achieve more complex systems in the future.
4. Remaining challenges
Microfluidic body-on-a-chip systems have been shown to simulate parts of the human metabolism. The devices are suitable for screening new drug candidates, testing the toxicity of environmental contaminants, and conducting studies in the area of nutritional sciences. However, some challenges remain to be solved before such systems become more widely used. First, the usefulness of the devices will be enhanced if they include more organs than have been shown so far, as well as make the organ systems more functionally relevant. Currently four cell-containing chambers is the maximum number of chambers that has been included in a single chip. Incorporating more cell types into a single microchip will require more robust manipulation of fluid and cells, as well as the ability to monitor multiple cell types simultaneously. Adaptation into a high throughput format is also essential. Application of passive-mode flows such as gravity-induced flow may be a solution to achieving high throughput format106. Second, many of the developed systems still rely on the use of immortalized cell lines. Although these cell lines can give an estimate of tissue behavior, especially if they are human cell lines, they do not always exhibit all metabolic reactions found in vivo. Using primary cells, tissue samples or cells derived from stem cells will enhance the authenticity of the systems. The use of tissue samples will allow clinicians to test combinations of drugs on individual samples, thereby enabling individualized medicine for patients.
Another critical requirement for the success of multi-organ systems is the development of a blood surrogate that supports the growth of all tissues within the system. Since the cell culture media is re-circulated within the devices, all cells are exposed to the same medium components. The immobilization of growth factors within particular chambers and their controlled release could be one way to solve this problem118. Similarly, the removal of waste products from the medium stream will need to be accomplished. A periodic replacement of part of the volume of the recirculated medium could be a solution to the problem of waste buildup and the resulting cytotoxicity as it emulates natural processes (for example kidney) for removal of toxic metabolic byproducts. Hickman published the first serum-free, defined culture system for neuronal systems,121 which has been applied to cardiac83, hippocampal neurons122, MNs123, sensory neurons124, muscle125, and NMJ formation6, 7.
Body-on-a-chip devices will realize their full potential when bioanalytics are incorporated into the microfluidic chip. Many on-chip analytical approaches have been developed, including electrochemical electrodes, optical sensors, label-free detection of molecules, field-effect sensors, and cantilevers that sense changes in mass. These sensors are geared towards sensing small sample volumes, and sometimes include sample preparation and separation. For example, Kim and Shuler have developed in situ optical detection system that can be integrated with a microfluidic device to analyze dynamics of cell viability126, 127. This in situ detection and analysis system will yield more detailed information about the dynamics of multi-organ interactions. Ideally, and in order to be applied to a high-throughput format, these on-chip detection units should also be cost-effective.
5. Conclusion
An advantage that body-on-a-chip systems provide to drug toxicity testing is that metabolic reactions, even those that have not been discovered yet are incorporated within the devices via the cultured tissues. Comparing the data from experiments with multi-organ devices with those derived from mathematical PBPK models will allow for the verification and improvement of our understanding of specific metabolic pathways. A discrepancy will point to missing links that can be investigated further. Further, comparing data from body-on-a-chip devices that were operated with animal tissues with results obtained from animal models will validate the body-on-a-chip concept and give confidence that results from the body-on-a-chip devices with human cells would be applicable to humans prior to testing on humans. For this purpose, a combination of mathematical and experimental approaches is essential for the effective study of the dynamics of multi-organ interaction. We believe that such devices will assume an increasingly important role in pharmaceutical and medical sciences.
5. Acknowledgments
JHS gratefully acknowledges support from National Research Foundation of Korea (NRF, Grant no. 2012-0003408), KFRI (Korea Food Research Institute, grant no: E0121705), 2012 Hongik University Research Fund. James Hickman's work is funded under NIH grant numbers R01NS050452 and R01EB009429
References
- 1.Artursson P, Palm K, Luthman K. Advanced drug delivery reviews. 2001;46:27–43. doi: 10.1016/s0169-409x(00)00128-9. [DOI] [PubMed] [Google Scholar]
- 2.Abbott A. Nature. 2003;424:870–872. doi: 10.1038/424870a. [DOI] [PubMed] [Google Scholar]
- 3.Paguirigan AL, Beebe DJ. BioEssays : news and reviews in molecular, cellular and developmental biology. 2008;30:811–821. doi: 10.1002/bies.20804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Khademhosseini A, Langer R. Biomaterials. 2007;28:5087–5092. doi: 10.1016/j.biomaterials.2007.07.021. [DOI] [PubMed] [Google Scholar]
- 5.Das M, Rumsey JW, Bhargava N, Stancescu M, Hickman JJ. Biomaterials. 2010;31:4880–4888. doi: 10.1016/j.biomaterials.2010.02.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guo X, Gonzalez M, Stancescu M, Vandenburgh HH, Hickman JJ. Biomaterials. 2011;32:9602–9611. doi: 10.1016/j.biomaterials.2011.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guo X, Das M, Rumsey J, Gonzalez M, Stancescu M, Hickman J. Tissue engineering. Part C, Methods. 2010;16:1347–1355. doi: 10.1089/ten.tec.2010.0040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.El-Ali J, Sorger PK, Jensen KF. Nature. 2006;442:403–411. doi: 10.1038/nature05063. [DOI] [PubMed] [Google Scholar]
- 9.Esch MB, Sung JH, Shuler ML. Journal of biotechnology. 2010;148:64–69. doi: 10.1016/j.jbiotec.2010.02.020. [DOI] [PubMed] [Google Scholar]
- 10.Sung JH, Esch MB, Shuler ML. Expert opinion on drug metabolism & toxicology. 2010;6:1063–1081. doi: 10.1517/17425255.2010.496251. [DOI] [PubMed] [Google Scholar]
- 11.Kola I, Landis J. Nature reviews. Drug discovery. 2004;3:711–715. doi: 10.1038/nrd1470. [DOI] [PubMed] [Google Scholar]
- 12.Sung JH, Yu J, Luo D, Shuler ML, March JC. Lab on a chip. 2011;11:389–392. doi: 10.1039/c0lc00273a. [DOI] [PubMed] [Google Scholar]
- 13.Khetani SR, Bhatia SN. Nature biotechnology. 2008;26:120–126. doi: 10.1038/nbt1361. [DOI] [PubMed] [Google Scholar]
- 14.Allen JW, Khetani SR, Bhatia SN. Toxicological sciences : an official journal of the Society of Toxicology. 2005;84:110–119. doi: 10.1093/toxsci/kfi052. [DOI] [PubMed] [Google Scholar]
- 15.Huh D, Matthews BD, Mammoto A, Montoya-Zavala M, Hsin HY, Ingber DE. Science. 2010;328:1662–1668. doi: 10.1126/science.1188302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ajdari A. Comptes Rendus Physique. 2003;5:539–546. [Google Scholar]
- 17.Oh KW, Lee K, Ahn B, Furlani EP. Lab on a chip. 2012;12:515–545. doi: 10.1039/c2lc20799k. [DOI] [PubMed] [Google Scholar]
- 18.Young EW, Wheeler AR, Simmons CA. Lab on a chip. 2007;7:1759–1766. doi: 10.1039/b712486d. [DOI] [PubMed] [Google Scholar]
- 19.Gutierrez E, Groisman A. Analytical chemistry. 2007;79:2249–2258. doi: 10.1021/ac061703n. [DOI] [PubMed] [Google Scholar]
- 20.van der Meer AD, Vermeul K, Poot AA, Feijen J, Vermes I. American journal of physiology. Heart and circulatory physiology. 2010;298:H719–725. doi: 10.1152/ajpheart.00933.2009. [DOI] [PubMed] [Google Scholar]
- 21.Shin Y, Jeon JS, Han S, Jung GS, Shin S, Lee SH, Sudo R, Kamm RD, Chung S. Lab on a chip. 2011;11:2175–2181. doi: 10.1039/c1lc20039a. [DOI] [PubMed] [Google Scholar]
- 22.Berthier E, Young EW, Beebe D. Lab on a chip. 2012;12:1224–1237. doi: 10.1039/c2lc20982a. [DOI] [PubMed] [Google Scholar]
- 23.Cushing MC, Anseth KS. Science. 2007;316:1133–1134. doi: 10.1126/science.1140171. [DOI] [PubMed] [Google Scholar]
- 24.Frisk T, Rydholm S, Andersson H, Stemme G, Brismar H. Electrophoresis. 2005;26:4751–4758. doi: 10.1002/elps.200500478. [DOI] [PubMed] [Google Scholar]
- 25.Choi NW, Cabodi M, Held B, Gleghorn JP, Bonassar LJ, Stroock AD. Nature materials. 2007;6:908–915. doi: 10.1038/nmat2022. [DOI] [PubMed] [Google Scholar]
- 26.Zheng Y, Chen J, Craven M, Choi NW, Totorica S, Diaz-Santana A, Kermani P, Hempstead B, Fischbach-Teschl C, Lopez JA, Stroock AD. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:9342–9347. doi: 10.1073/pnas.1201240109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cuchiara MP, Allen AC, Chen TM, Miller JS, West JL. Biomaterials. 2010;31:5491–5497. doi: 10.1016/j.biomaterials.2010.03.031. [DOI] [PubMed] [Google Scholar]
- 28.Golden AP, Tien J. Lab on a chip. 2007;7:720–725. doi: 10.1039/b618409j. [DOI] [PubMed] [Google Scholar]
- 29.Bellan LM, Singh SP, Henderson PW, Porri TJ, Craighead HG, Spector JA. Soft Matter. 2009;5:1354–1357. [Google Scholar]
- 30.Tam A, Wadsworth S, Dorscheid D, Man SF, Sin DD. Therapeutic advances in respiratory disease. 2011;5:255–273. doi: 10.1177/1753465810396539. [DOI] [PubMed] [Google Scholar]
- 31.Nalayanda DD, Puleo C, Fulton WB, Sharpe LM, Wang TH, Abdullah F. Biomedical microdevices. 2009 doi: 10.1007/s10544-009-9325-5. [DOI] [PubMed] [Google Scholar]
- 32.Huh D, Fujioka H, Tung YC, Futai N, Paine R, 3rd, Grotberg JB, Takayama S. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:18886–18891. doi: 10.1073/pnas.0610868104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Douville NJ, Tung YC, Li R, Wang JD, El-Sayed ME, Takayama S. Analytical chemistry. 2010;82:2505–2511. doi: 10.1021/ac9029345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Long C, Finch C, Esch M, Anderson W, Shuler M, Hickman J. Annals of biomedical engineering. 2012;40:1255–1267. doi: 10.1007/s10439-012-0513-8. [DOI] [PubMed] [Google Scholar]
- 35.Brandon EF, Raap CD, Meijerman I, Beijnen JH, Schellens JH. Toxicology and applied pharmacology. 2003;189:233–246. doi: 10.1016/s0041-008x(03)00128-5. [DOI] [PubMed] [Google Scholar]
- 36.Ekins S. Drug metabolism reviews. 1996;28:591–623. doi: 10.3109/03602539608994019. [DOI] [PubMed] [Google Scholar]
- 37.Cross DM, Bayliss MK. Drug metabolism reviews. 2000;32:219–240. doi: 10.1081/dmr-100100574. [DOI] [PubMed] [Google Scholar]
- 38.Westerink WM, Schoonen WG. Toxicology in vitro : an international journal published in association with BIBRA. 2007;21:1581–1591. doi: 10.1016/j.tiv.2007.05.014. [DOI] [PubMed] [Google Scholar]
- 39.Lu C, Li P, Gallegos R, Uttamsingh V, Xia CQ, Miwa GT, Balani SK, Gan LS. Drug metabolism and disposition: the biological fate of chemicals. 2006;34:1600–1605. doi: 10.1124/dmd.106.010793. [DOI] [PubMed] [Google Scholar]
- 40.Camp JP, Capitano AT. Biotechnology progress. 2007;23:1485–1491. doi: 10.1021/bp070308v. [DOI] [PubMed] [Google Scholar]
- 41.Domansky K, Inman W, Serdy J, Dash A, Lim MH, Griffith LG. Lab on a chip. 2010;10:51–58. doi: 10.1039/b913221j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sivaraman A, Leach JK, Townsend S, Iida T, Hogan BJ, Stolz DB, Fry R, Samson LD, Tannenbaum SR, Griffith LG. Current drug metabolism. 2005;6:569–591. doi: 10.2174/138920005774832632. [DOI] [PubMed] [Google Scholar]
- 43.Nakao Y, Kimura H, Sakai Y, Fujii T. Biomicrofluidics. 2011;5:22212. doi: 10.1063/1.3580753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee MY, Dordick JS. Current opinion in biotechnology. 2006;17:619–627. doi: 10.1016/j.copbio.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 45.Gayer CP, Basson MD. Cellular signalling. 2009;21:1237–1244. doi: 10.1016/j.cellsig.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Clarke MB, Sperandio V. American journal of physiology. Gastrointestinal and liver physiology. 2005;288:G1105–1109. doi: 10.1152/ajpgi.00572.2004. [DOI] [PubMed] [Google Scholar]
- 47.Hubatsch I, Ragnarsson EG, Artursson P. Nature protocols. 2007;2:2111–2119. doi: 10.1038/nprot.2007.303. [DOI] [PubMed] [Google Scholar]
- 48.Reis JM, Sinko B, Serra CH. Mini reviews in medicinal chemistry. 2010;10:1071–1076. doi: 10.2174/1389557511009011071. [DOI] [PubMed] [Google Scholar]
- 49.Stevenson CL, Augustijns PF, Hendren RW. International journal of pharmaceutics. 1999;177:103–115. doi: 10.1016/s0378-5173(98)00331-7. [DOI] [PubMed] [Google Scholar]
- 50.Yu J, Peng S, Luo D, March JC. Biotechnology and bioengineering. 2012 doi: 10.1002/bit.24518. [DOI] [PubMed] [Google Scholar]
- 51.Wang L, Murthy SK, Barabino GA, Carrier RL. Biomaterials. 2010;31:7586–7598. doi: 10.1016/j.biomaterials.2010.06.036. [DOI] [PubMed] [Google Scholar]
- 52.Kim J, Hegde M, Jayaraman A. Lab on a chip. 2010;10:43–50. doi: 10.1039/b911367c. [DOI] [PubMed] [Google Scholar]
- 53.Kim HJ, Huh D, Hamilton G, Ingber DE. Lab on a chip. 2012;12:2165–2174. doi: 10.1039/c2lc40074j. [DOI] [PubMed] [Google Scholar]
- 54.Hawkins BT, Davis TP. Pharmacological reviews. 2005;57:173–185. doi: 10.1124/pr.57.2.4. [DOI] [PubMed] [Google Scholar]
- 55.Hatherell K, Couraud PO, Romero IA, Weksler B, Pilkington GJ. Journal of neuroscience methods. 2011;199:223–229. doi: 10.1016/j.jneumeth.2011.05.012. [DOI] [PubMed] [Google Scholar]
- 56.Lippmann ES, Azarin SM, Kay JE, Nessler RA, Wilson HK, Al-Ahmad A, Palecek SP, Shusta EV. Nature biotechnology. 2012 doi: 10.1038/nbt.2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shayan G, Felix N, Cho Y, Chatzichristidi M, Shuler ML, Ober CK, Lee KH. Tissue engineering. Part C, Methods. 2012 doi: 10.1089/ten.TEC.2011.0598. [DOI] [PubMed] [Google Scholar]
- 58.Cucullo L, Hossain M, Puvenna V, Marchi N, Janigro D. BMC neuroscience. 2011;12:40. doi: 10.1186/1471-2202-12-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Janigro D, Leaman SM, Stanness KA. Pharmaceutical science & technology today. 1999;2:7–12. doi: 10.1016/s1461-5347(98)00110-2. [DOI] [PubMed] [Google Scholar]
- 60.Booth R, Kim H. Lab on a chip. 2012;12:1784–1792. doi: 10.1039/c2lc40094d. [DOI] [PubMed] [Google Scholar]
- 61.Taylor AM, Jeon NL. Current opinion in neurobiology. 2010;20:640–647. doi: 10.1016/j.conb.2010.07.011. [DOI] [PubMed] [Google Scholar]
- 62.Stenger DA, Hickman JJ, Bateman KE, Ravenscroft MS, Ma W, Pancrazio JJ, Shaffer K, Schaffner AE, Cribbs DH, Cotman CW. Journal of neuroscience methods. 1998;82:167–173. doi: 10.1016/s0165-0270(98)00047-8. [DOI] [PubMed] [Google Scholar]
- 63.Molnar PK, Bhargava J, Das N, Hickman M, J. J. In: Methods in Molecular Biology. Molnar PH, J. J., editors. Humana Press; New York: 2007. [Google Scholar]
- 64.Varghese K, Molnar P, Das M, Bhargava N, Lambert S, Kindy MS, Hickman JJ. PloS one. 2010;5:e8643. doi: 10.1371/journal.pone.0008643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Morin F, Nishimura N, Griscom L, Lepioufle B, Fujita H, Takamura Y, Tamiya E. Biosensors & bioelectronics. 2006;21:1093–1100. doi: 10.1016/j.bios.2005.04.020. [DOI] [PubMed] [Google Scholar]
- 66.Claverol-Tinturé E, Rosell X, Cabestany J. Neurocomputing. 2007;70:2716–2722. [Google Scholar]
- 67.Kanagasabapathi TT, Ciliberti D, Martinoia S, Wadman WJ, Decre MM. Frontiers in neuroengineering. 2011;4:13. doi: 10.3389/fneng.2011.00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Johnstone AF, Gross GW, Weiss DG, Schroeder OH, Gramowski A, Shafer TJ. Neurotoxicology. 2010;31:331–350. doi: 10.1016/j.neuro.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 69.Langendorff O. Pflügers Archiv European Journal of Physiology. 1895;61:291–332. [Google Scholar]
- 70.Bell RM, Mocanu MM, Yellon DM. Journal of molecular and cellular cardiology. 2011;50:940–950. doi: 10.1016/j.yjmcc.2011.02.018. [DOI] [PubMed] [Google Scholar]
- 71.Jacot JG, McCulloch AD, Omens JH. Biophysical journal. 2008;95:3479–3487. doi: 10.1529/biophysj.107.124545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tanaka Y, Morishima K, Shimizu T, Kikuchi A, Yamato M, Okano T, Kitamori T. Lab on a chip. 2006;6:230–235. doi: 10.1039/b512099c. [DOI] [PubMed] [Google Scholar]
- 73.Yin S, Zhang X, Zhan C, Wu J, Xu J, Cheung J. Biophysical journal. 2005;88:1489–1495. doi: 10.1529/biophysj.104.048157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lin G, Palmer RE, Pister KS, Roos KP. IEEE transactions on bio-medical engineering. 2001;48:996–1006. doi: 10.1109/10.942589. [DOI] [PubMed] [Google Scholar]
- 75.Kaneko T, Kojima K, Yasuda K. The Analyst. 2007;132:892–898. doi: 10.1039/b704961g. [DOI] [PubMed] [Google Scholar]
- 76.Kim J, Park J, Na K, Yang S, Baek J, Yoon E, Choi S, Lee S, Chun K, Park S. Journal of biomechanics. 2008;41:2396–2401. doi: 10.1016/j.jbiomech.2008.05.036. [DOI] [PubMed] [Google Scholar]
- 77.Alford PW, Feinberg AW, Sheehy SP, Parker KK. Biomaterials. 2010;31:3613–3621. doi: 10.1016/j.biomaterials.2010.01.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Grosberg A, Nesmith AP, Goss JA, Brigham MD, McCain ML, Parker KK. Journal of pharmacological and toxicological methods. 2012;65:126–135. doi: 10.1016/j.vascn.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Grosberg A, Alford PW, McCain ML, Parker KK. Lab on a chip. 2011;11:4165–4173. doi: 10.1039/c1lc20557a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Linder P, Trzewik J, Ruffer M, Artmann GM, Digel I, Kurz R, Rothermel A, Robitzki A, Temiz Artmann A. Medical & biological engineering & computing. 2010;48:59–65. doi: 10.1007/s11517-009-0552-y. [DOI] [PubMed] [Google Scholar]
- 81.Boudou T, Legant WR, Mu A, Borochin MA, Thavandiran N, Radisic M, Zandstra PW, Epstein JA, Margulies KB, Chen CS. Tissue engineering. Part A. 2012;18:910–919. doi: 10.1089/ten.tea.2011.0341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jones IL, Livi P, Lewandowska MK, Fiscella M, Roscic B, Hierlemann A. Analytical and bioanalytical chemistry. 2011;399:2313–2329. doi: 10.1007/s00216-010-3968-1. [DOI] [PubMed] [Google Scholar]
- 83.Natarajan A, Stancescu M, Dhir V, Armstrong C, Sommerhage F, Hickman JJ, Molnar P. Biomaterials. 2011;32:4267–4274. doi: 10.1016/j.biomaterials.2010.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bischoff R. The Anatomical record. 1974;180:645–661. doi: 10.1002/ar.1091800410. [DOI] [PubMed] [Google Scholar]
- 85.Vandenburgh H. Tissue engineering. Part B, Reviews. 2010;16:55–64. doi: 10.1089/ten.teb.2009.0445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wilson K, Das M, Wahl KJ, Colton RJ, Hickman J. PloS one. 2010;5:e11042. doi: 10.1371/journal.pone.0011042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Levenberg S, Rouwkema J, Macdonald M, Garfein ES, Kohane DS, Darland DC, Marini R, van Blitterswijk CA, Mulligan RC, D'Amore PA, Langer R. Nature biotechnology. 2005;23:879–884. doi: 10.1038/nbt1109. [DOI] [PubMed] [Google Scholar]
- 88.Hinds S, Bian W, Dennis RG, Bursac N. Biomaterials. 2011;32:3575–3583. doi: 10.1016/j.biomaterials.2011.01.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Khodabukus A, Baar K. Tissue engineering. Part C, Methods. 2012;18:349–357. doi: 10.1089/ten.TEC.2011.0364. [DOI] [PubMed] [Google Scholar]
- 90.Vandenburgh H, Shansky J, Benesch-Lee F, Barbata V, Reid J, Thorrez L, Valentini R, Crawford G. Muscle & nerve. 2008;37:438–447. doi: 10.1002/mus.20931. [DOI] [PubMed] [Google Scholar]
- 91.Huang NF, Patel S, Thakar RG, Wu J, Hsiao BS, Chu B, Lee RJ, Li S. Nano letters. 2006;6:537–542. doi: 10.1021/nl060060o. [DOI] [PubMed] [Google Scholar]
- 92.Molnar P, Wang W, Natarajan A, Rumsey JW, Hickman JJ. Biotechnology progress. 2007;23:265–268. doi: 10.1021/bp060302q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Das M, Wilson K, Molnar P, Hickman JJ. Nature protocols. 2007;2:1795–1801. doi: 10.1038/nprot.2007.229. [DOI] [PubMed] [Google Scholar]
- 94.Fischbach GD. Science. 1970;169:1331–1333. doi: 10.1126/science.169.3952.1331. [DOI] [PubMed] [Google Scholar]
- 95.Daniels MP, Lowe BT, Shah S, Ma J, Samuelsson SJ, Lugo B, Parakh T, Uhm CS. Microscopy research and technique. 2000;49:26–37. doi: 10.1002/(SICI)1097-0029(20000401)49:1<26::AID-JEMT4>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 96.Das M, Rumsey JW, Gregory CA, Bhargava N, Kang JF, Molnar P, Riedel L, Guo X, Hickman JJ. Neuroscience. 2007;146:481–488. doi: 10.1016/j.neuroscience.2007.01.068. [DOI] [PubMed] [Google Scholar]
- 97.van Midwoud PM, Merema MT, Verpoorte E, Groothuis GM. Lab on a chip. 2010;10:2778–2786. doi: 10.1039/c0lc00043d. [DOI] [PubMed] [Google Scholar]
- 98.Ma B, Zhang G, Qin J, Lin B. Lab on a chip. 2009;9:232–238. doi: 10.1039/b809117j. [DOI] [PubMed] [Google Scholar]
- 99.Guzzardi MA, Domenici C, Ahluwalia A. Tissue engineering. Part A. 2011;17:1635–1642. doi: 10.1089/ten.TEA.2010.0541. [DOI] [PubMed] [Google Scholar]
- 100.Iori E, Vinci B, Murphy E, Marescotti MC, Avogaro A, Ahluwalia A. PloS one. 2012;7:e34704. doi: 10.1371/journal.pone.0034704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Tatosian DA, Shuler ML. Biotechnology and bioengineering. 2009;103:187–198. doi: 10.1002/bit.22219. [DOI] [PubMed] [Google Scholar]
- 102.Gerlowski LE, Jain RK. Journal of pharmaceutical sciences. 1983;72:1103–1127. doi: 10.1002/jps.2600721003. [DOI] [PubMed] [Google Scholar]
- 103.Sung JH, Dhiman A, Shuler ML. Journal of pharmaceutical sciences. 2009;98:1885–1904. doi: 10.1002/jps.21536. [DOI] [PubMed] [Google Scholar]
- 104.Aarons L. British journal of clinical pharmacology. 2005;60:581–583. doi: 10.1111/j.1365-2125.2005.02560.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Moraes C, Mehta G, Lesher-Perez SC, Takayama S. Annals of biomedical engineering. 2012;40:1211–1227. doi: 10.1007/s10439-011-0455-6. [DOI] [PubMed] [Google Scholar]
- 106.Sung JH, Kam C, Shuler ML. Lab on a chip. 2010;10:446–455. doi: 10.1039/b917763a. [DOI] [PubMed] [Google Scholar]
- 107.Li AP, Uzgare A, Laforge YS. Chemico-biological interactions. 2012;199:1–8. doi: 10.1016/j.cbi.2012.05.003. [DOI] [PubMed] [Google Scholar]
- 108.Vozzi F, Mazzei D, Vinci B, Vozzi G, Sbrana T, Ricotti L, Forgione N, Ahluwalia A. Biotechnology and bioengineering. 2011;108:2129–2140. doi: 10.1002/bit.23164. [DOI] [PubMed] [Google Scholar]
- 109.van Midwoud PM, Groothuis GM, Merema MT, Verpoorte E. Biotechnology and bioengineering. 2010;105:184–194. doi: 10.1002/bit.22516. [DOI] [PubMed] [Google Scholar]
- 110.Mao S, Gao D, Liu W, Wei H, Lin JM. Lab on a chip. 2012;12:219–226. doi: 10.1039/c1lc20678h. [DOI] [PubMed] [Google Scholar]
- 111.Imura Y, Sato K, Yoshimura E. Analytical chemistry. 2010;82:9983–9988. doi: 10.1021/ac100806x. [DOI] [PubMed] [Google Scholar]
- 112.Imura Y, Yoshimura E, Sato K. Analytical sciences : the international journal of the Japan Society for Analytical Chemistry. 2012;28:197–199. doi: 10.2116/analsci.28.197. [DOI] [PubMed] [Google Scholar]
- 113.Sin A, Chin KC, Jamil MF, Kostov Y, Rao G, Shuler ML. Biotechnology progress. 2004;20:338–345. doi: 10.1021/bp034077d. [DOI] [PubMed] [Google Scholar]
- 114.Ghanem A, Shuler ML. Biotechnology progress. 2000;16:471–479. doi: 10.1021/bp000047o. [DOI] [PubMed] [Google Scholar]
- 115.Viravaidya K, Sin A, Shuler ML. Biotechnology progress. 2004;20:316–323. doi: 10.1021/bp0341996. [DOI] [PubMed] [Google Scholar]
- 116.Mahler GJ, Esch MB, Glahn RP, Shuler ML. Biotechnology and bioengineering. 2009;104:193–205. doi: 10.1002/bit.22366. [DOI] [PubMed] [Google Scholar]
- 117.Sung JH, Shuler ML. Lab on a chip. 2009;9:1385–1394. doi: 10.1039/b901377f. [DOI] [PubMed] [Google Scholar]
- 118.Zhang C, Zhao Z, Abdul Rahim NA, van Noort D, Yu H. Lab on a chip. 2009;9:3185–3192. doi: 10.1039/b915147h. [DOI] [PubMed] [Google Scholar]
- 119.Chang R, Emami K, Wu H, Sun W. Biofabrication. 2010;2:045004. doi: 10.1088/1758-5082/2/4/045004. [DOI] [PubMed] [Google Scholar]
- 120.Snyder JE, Hamid Q, Wang C, Chang R, Emami K, Wu H, Sun W. Biofabrication. 2011;3:034112. doi: 10.1088/1758-5082/3/3/034112. [DOI] [PubMed] [Google Scholar]
- 121.Schaffner AE, Barker JL, Stenger DA, Hickman JJ. Journal of neuroscience methods. 1995;62:111–119. doi: 10.1016/0165-0270(95)00063-1. [DOI] [PubMed] [Google Scholar]
- 122.Varghese K, Das M, Bhargava N, Stancescu M, Molnar P, Kindy MS, Hickman JJ. Journal of neuroscience methods. 2009;177:51–59. doi: 10.1016/j.jneumeth.2008.09.022. [DOI] [PubMed] [Google Scholar]
- 123.Das M, Molnar P, Devaraj H, Poeta M, Hickman JJ. Biotechnology progress. 2003;19:1756–1761. doi: 10.1021/bp034076l. [DOI] [PubMed] [Google Scholar]
- 124.Rumsey JW, Das M, Bhalkikar A, Stancescu M, Hickman JJ. Biomaterials. 2010;31:8218–8227. doi: 10.1016/j.biomaterials.2010.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Das M, Gregory CA, Molnar P, Riedel LM, Wilson K, Hickman JJ. Biomaterials. 2006;27:4374–4380. doi: 10.1016/j.biomaterials.2006.03.046. [DOI] [PubMed] [Google Scholar]
- 126.Choi JR, Sung JH, Shuler ML, Kim D. Optics letters. 2010;35:1374–1376. doi: 10.1364/OL.35.001374. [DOI] [PubMed] [Google Scholar]
- 127.Sung JH, Choi JR, Kim D, Shuler ML. Biotechnology and bioengineering. 2009;104:516–525. doi: 10.1002/bit.22413. [DOI] [PubMed] [Google Scholar]