

Abstract
Objective
We aimed to examine if smoking is an independent predictor of oral candidiasis (OC) among HIV-1 infected persons.
Methods
The cross-sectional part of this study evaluated 631 adult dentate HIV-1 seropositive persons examined for OC from 1995 – 2000 at the University of North Carolina Hospitals in Chapel Hill, NC. In the second part, from the above sample, 283 individuals who were free of HIV-associated oral diseases at baseline were followed up for two years to assess incident OC events. Data collected from medical record review, interview questionnaires and clinical examinations were analyzed using chi-square tests and t-tests. Logistic regression models were developed for prevalent OC employing the likelihood ratio test, whereas Poisson regression models were developed for assessing cumulative incidence of OC. These models included a variety of independent variables to adjust for confounding.
Results
Thirteen percent of participants had OC only; 4.6% had OC with Oral Hairy Leukoplakia; and 69.7% had neither. Smoking was associated with OC in all models [prevalent OC - current smokers: logistic regression – Odd Ratio (95% CI) = 2.5 (1.3, 4.8); Incident OC - current smokers: Poisson regression (main effects model) - Incidence Rate Ratio (95% CI) = 1.9 (1.1, 3.8)]. Other Poisson regression models suggested evidence for effect modification between CD4 cell count and incident OC by smoking.
Conclusion
Smoking is an independent risk factor for development of OC in HIV-1 infected persons and the risk of OC is modified by CD4 cell count which measures strength of the immune system.
Keywords: smoking, HIV, Candida, interaction, regression models
INTRODUCTION
Oral candidiasis (OC) is the most frequent opportunistic fungal infection among human immunodeficiency virus (HIV)-infected patients with incident rate/density of 9.3 per 1000 person months and a strong correlation with immune suppression as measured by reduced CD4 cell counts (<200 cells/mm3) (1). Occurrence of OC in HIV-infected persons has been reported globally and it continues to have a major public health impact despite studies demonstrating a declining incidence and prevalence since initiation of HAART, related largely to improving immune status as measured by increasing CD4 counts (2–5). The nature of HIV/AIDS has changed from an acute disease to a chronic disease after introduction of HAART in developed countries. However, in countries where HIV/AIDS is widespread and HAART medications are too expensive or patients are failing HAART, oral disease management and risk remains an important issue. Resurgence of OC following failure of HAART has been reported (6) with prevalence that is similar to the pre-HAART era (7). Despite reports of reduced incidence of OC following HAART, it remains a common opportunistic infection in HIV-infected patients (8–12).
The effect of smoking on OC in populations with HIV/AIDS remains controversial. Whereas some studies have suggested association of smoking with OC (13–17), others failed to find such an association (18), or found it only among patients with higher levels of immune competence (CD4 counts ≥200 cells/mm3) (19). Most of these studies assess the associations between OC and smoking in samples with prevalent OC. Only two studies have reported incidence of OC (both in HIV-1 infected cohorts) – one in a cohort of HIV-positive women (3) and the other in a mixed-gender sample (1). Both of these studies reported greater risk of OC among current smokers after adjusting for several factors. Asymptomatic oral Candida carriage has also been associated with smoking (20).
In conducting this study, our aim was to assess smoking as an independent risk factor for OC among adults with HIV/AIDS and to examine for effect measure modification (EMM) by smoking between OC and any other important risk factor, specifically the immune marker, CD4 cell count. A CD4 cell count determines how well the immune system is working, and it is well-established that a low CD4 count usually indicates a weakened immune system and a higher chance of getting opportunistic infections.
METHODS
We evaluated a cohort of 631 HIV-positive persons at baseline in a longitudinal study with yearly visits to establish independent risk factors/indicators for HIV- associated oral diseases between 1995–2000 at the University of North Carolina (UNC) Hospitals in Chapel Hill, North Carolina in this study approved by the UNC School of Medicine and UNC Hospitals Committee on the Protection of the Rights of Human Subjects and NIH Institutional Review Board. Comprehensive collection of medical and oral disease history and oral clinical examinations was conducted. Assessment of oral manifestations of HIV was based on the published standard presumptive clinical criteria (21) and conducted by a single Oral Medicine trained examiner (LP). A medical record review was then conducted for each participant to ascertain laboratory and medication variables and AIDS case status.
Univariate distributions of variables were assessed, as were bivariate relationships between the covariates. Outcome variable was occurrence of OC. CD4 cell count was our main explanatory variable (continuous variable as well as dichotomized as: < 200 vs. ≥200 cells/mm3; and trichotomized as <200 vs. 200–500 cells mm3 vs. >500 cells mm3 depending on the type of analysis) whereas other explanatory variables included age, sex, education level, smoking history, drug use, sexual orientation, antiretroviral medication use and antifungal medication use. Bivariate associations were assessed using t-tests, ANOVA; correlation coefficients and chi-square tests as needed.
Multivariable analyses
We assessed the association between outcome and independent variables using bivariate logistic regression, proportional odds models and Poisson regression analyses. Binary unconditional logistic regression is used for two-level dependent variables (OC present/not present). The logistic regression analysis was conducted on the sample with complete information for all variables for the prevalence study (n=623). Poisson regression analyses were used to assess incident rate ratios (IRR) for incident OC using only those participants that did not have any HIV-associated oral disease at baseline (n=283). Regression diagnostics were performed, and the utility of the models was evaluated by outputting predicted scores and residuals.
Analytical use of smoking variable: To conserve power in the incidence OC study, we assessed smoking as a two-level variable (current smokers vs. current non-smokers). For the prevalent OC study, we first analyzed smoking as a three-level variable (current, former, never) to develop the binary logistic regression model and assess the role of smoking as an independent predictor of OC. We then repeated these logistic regression analyses using smoking as a two-level variable (current smokers vs. current non-smokers) to ascertain if the relationship between smoking and OC was similar in both sets of analyses. By using the same definition of smoking as an exposure variable in both analyses (Poisson regression and logistic regression), we could then make appropriate comparisons between the models to make inferences about the role of smoking as an independent predictor of OC.
The overall goal of each of the multivariable analyses was to control for potential confounders, and to find the best fitting, most parsimonious and biologically reasonable model to describe the relationship between OC and a set of independent explanatory variables in which CD4 cell count was always included. All final main-effect models were adjusted for: current CD4 cell count and antiretroviral medication. We arrived at the final main-effects model to obtain the most useful model from a hierarchical series of models after starting from a full model that was adjusted for: age, sex, race/ethnicity, education level, sexual orientation, recreational drug use, baseline CD4 cell count, and antifungal medication. To select between the hierarchically well-formulated models, we used the likelihood ratio test carrying out all statistical analyses in SAS® (v9.1 Cary, NC, USA). Upon developing the final main-effects models, we tested for statistical interaction between smoking and CD4 cell count. The principal of a hierarchically well-formulated model implies that for any given variable in the model, all lower-order components of the variables must also be contained in the model. We inferred statistical significance using two sided p-values at 0.05 level (a-priori: 0.1 for interaction terms) (22).
Where present, interactions were assessed through the interaction contrast (IC) defined as the relative excess risk for interaction. Employing low CD4 cell count (<200 cells/mm3) and current smoking as the two exposure variables, we calculated IC = R11 − R01 − R10 + R00; where R01 and R10 are the risk estimates for exposure to any one of the exposures, R11 is the risk estimate for doubly exposed, and R00 is the unitary value for the referent (doubly unexposed) group (22).
RESULTS
Figure 1 shows the predicted incidence rates for OC among current smokers and nonsmokers by CD4 cell count (cells/mm3) as a continuous variable demonstrating declining rates with increasing CD4 cell count among both smokers and non-smokers though the difference between smokers and non-smokers does not disappear completely. Figure 2 shows the incidence rates for OC by CD4 cell count groups and smoking status. Table 1 shows the results from main-effects models for logistic regression and Poisson regression analysis after adjusting for a series of covariates mentioned in the methods section. Assessment of smoking as a three level variable suggests current smokers had a 250% increase in risk for prevalent OC compared to those who had never smoked [OR (95% CI): 2.5 (1.3, 4.8)]. Although the risk among former smokers was slightly elevated, it was not statistically significantly different from those who had never smoked. Furthermore, similar levels of elevated risk among current smokers was maintained when they were compared to never smokers and former smokers combined [OR: 2.3 (1.4, 3.7)]. Similarly, current smokers had almost a 190% greater risk for developing new OC compared to current non-smokers [IRR: 1.9 (1.1, 3.8)] in the main-effects model. These results suggest that smoking is an independent risk factor for OC.
Figure 1.
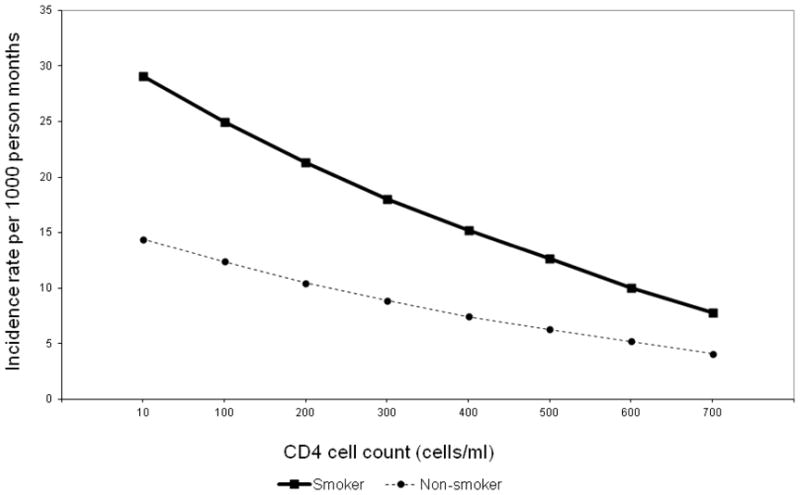
Predicted incidence rates for OC among current smokers and nonsmokers by CD4 cell count (cells/mm3) at event: final models corresponding to each disease outcome, adjusted for antiretroviral therapy, using CD4 cell count as a continuous variable.
Figure 2.
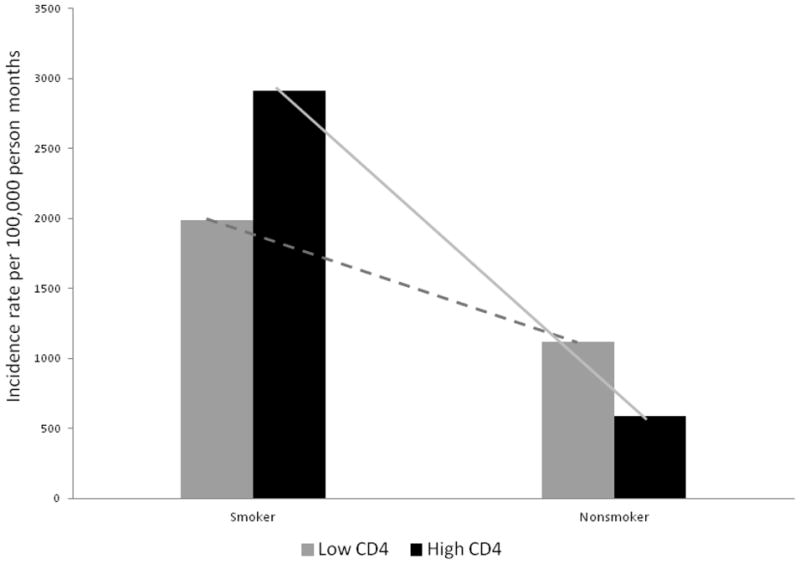
Observed incidence rates for OC among current smokers and nonsmokers by CD4 cell count (categorized) at event: Smoker-Low CD4 cell count:1990.05; Smoker-High CD4 cell count: 2912.62; Non-smoker-Low CD4 cell count: 1116.07; and Non-smoker-Low CD4 cell count: 590.41 per 100,000 person-months.
Table 1.
Summary of risk factors/indicator associations with HIV associated oral diseases as seen in final multivariable main-effects models (adjusted for age, sex, race/ethnicity, education level, sexual orientation, recreational drug use, and antifungal medication (also baseline CD4 cell count in Poisson regression). Risk estimates are presented as adjusted ORs (prevalent disease) and IRR (incident disease).
| Prevalent OC | Incident OC | |||
|---|---|---|---|---|
| Factor level | Binary unconditional Logistic regression (n=623) | Factor level | Binary unconditional Logistic regression (n=623) | Poisson regression (n=283) |
| Current smoker | 2.5 (1.3, 4.8) | Current smoker | 2.3 (1.4, 3.7) | 1.9 (1.1, 3.8) |
| Former smoker | 1.2 (0.5, 2.7) | Never/Former smoker | 1 | 1 |
| Never smoked | 1 | |||
Upon further testing, we found evidence for statistical interaction between CD4 cell count and smoking status in the final Poisson regression model mentioned above [variable (type-3 p-value pr>Chi-sq): CD4 cell count (0.0002), antiretroviral medication (0.0066); current smoker (0.0244); CD4 * current smoker (0.0339)]. Therefore, the emphasis of assessing smoking as a risk factor should address not the main-effects IRR presented above, but on the within group IRRs calculated from the interaction model. Table 2 shows calculated IRRs from the Poisson regression interaction models showing the modification of the association between CD4 and OC by smoking, suggesting greater risk of OC among current smokers. IC was calculated as: IC (R11 − R01 − R10 − R00) = 4.76 − 2.51 − 1.89 + 1.00 = 1.36 implying positive departure from additivity or transadditivity (i.e. super additivity) of EMM in the multiplicative scale (22).
Table 2.
OC-Effect measure modification: CD4+ cell count and smoking: crude levels, bivariable and multivariable Poisson regression models (IRR: incident rate ratio), testing from the model developed as shown in Table 1.
| Grouping | IRR: Bivariable | IRR: Final model* | Incremental IRR: Final model ** | % Change(Fi – Bi)/Fi |
|---|---|---|---|---|
| Low CD4 - Smoker | 5.16 | 4.76 | 1.9 | -8.4 |
| Low CD4 - non-Smoker | 1.73 | 2.51 | 1.33 | 31.1 |
| Higher CD4 – Smoker | 1.56 | 1.89 | 1.89 | 17.5 |
| Higher CD4 – non-Smoker | 1 | 1 | 1 | --- |
Incremental IRR calculated as a ratio of stratum IRR with the IRR of the previous stratum in the table (i.e. 1.89/1 = 1.89; 2.51/1.89=1.33; 4.76/2.51=1.9).
IC = 4.76 − 2.51 − 1.89 + 1.00 = 1.36 implying positive departure from additivity or transadditivity (superadditivity) of effect measure modification in the multiplicative scale. Low CD4 is <200 cells/mm3; High CD4 is ≥200 cells/mm3
DISCUSSION
In this longitudinal HIV-infected patient cohort study, we have demonstrated that smoking is an independent risk factor for OC. However, the relationship between OC, immune suppression (CD4 count) and smoking is not straight forward, but rather the risk for OC varies across different CD4 count levels dependent upon current smoking status. Our confidence in inferring causal association between smoking and OC, and suggesting potential biological interaction between CD4 cell count and smoking status stems from the finding of significant EMM in the incident disease study from assessing IRs in the Poisson regression models, and biological plausibility in various studies (15).
Smoking has short and long-term effects on many important aspects of the inflammatory and immune responses in the oral cavity (23). Tobacco smoke affects both cell-mediated and humoral immunity (24, 25). Smoking impedes neutrophil transmigration across the oral and periodontal microvasculature, suppresses neutrophil cell spreading, chemokinesis, chemotaxis, and phagocytosis, and results in protease release from neutrophils that may be an important mechanism in tissue destruction. In a normal oral mucosa fibroblast in vitro model, fibroblasts exposed once to whole cigarette smoke, produced significant morphological and functional deregulation and resulted in a significant inhibition of cell adhesion, a decrease in the number of β1-integrin-positive cells, increased lactate dehydrogenase activity in the target cells, and reduced growth which may explain the higher predisposition of tobacco users to oral infections (26).
Tobacco smoking enhances oral candida colonization due to induction of increased epithelial keratinisation (27), reductions in salivary immunoglobulin A levels (28), and depression of polymorphonuclear leukocyte function (29). The association of smoking with increased risk of clinical infection, or candidiasis, relates to an increased fungal burden, reduced numbers of Langerhans cells and likely reduction of immunoglobulins (15). Both bacterial and fungal populations are altered with tobacco exposure. Exposure to direct and passive tobacco smoking has significant impact on a variety of gingival and oropharyngeal flora (30). The flora of smokers contains fewer aerobic and anaerobic organisms with interfering activity against bacterial pathogens and harbors more potential pathogens as compared with the flora of non-smokers. Tobacco smoke compromises the antibacterial function of leukocytes, including neutrophils, monocytes, T-cells, and B-cells, providing a mechanistic explanation for increased infection risk (23).
The in vitro effect of cigarette smoke condensate on ten clinical isolates of C. albicans obtained from nonsmoking volunteers, as well as a culture collection strain, showed a temporal increase in the secretion rates of enzymes, particularly when yeast cells were exposed to cigarette smoke condensate for 48–72 h (31). Similarly, among denture wearers, adhesion to acrylic and cell surface hydrophobicity increased with exposure period to cigarette smoke condensate, leading authors to conclude that cigarette smoke may promote significant enhancement in the secretion of candidal histolytic enzymes and adherence to denture surfaces, thereby promoting oral yeast carriage and possible infection. The more heavily an individual smokes, the more likely there will be Candida in the oral cavity (32). Clinically, heavy smokers (smoked more than a pack a day) were shown to have a significant 6 fold increased odds of candidiasis in patients with multiple oral leukoplakias compared to lighter smokers (33), implying some level of dose-response effect may be present.
Among patients with HIV infection, the severity and chronicity of OC has been largely attributed to the HIV-associated immune deficiency (as expressed by reduced CD4 counts), but may also relate to the virulence factors of the candidal pathogen. Adherence of the fungus to oral mucosal tissues is the first step in colonization and disease initiation, with extracellular hydrolases (proteinases and phospholipases) being major facilitators of tissue invasion. After colonization and adhesion of Candida to the epithelial surface, potent proteolytic enzymes or toxins and an inflammatory response to Candida antigens destroy tissue that results in the subsequent mucosal lesion (34).
The relationship of Candida virulence to both smoking and HIV infection are not fully understood. While a recent study comparing oral swab candidal isolates by HIV infection status demonstrated isolates from HIV-positive patients were predominantly genotype A and had significantly increased expression of proteinase, phospholipase and hemolytic activities, as well as a greater ability to adhere, compared with HIV-negative individuals, no significant differences in virulence factor expression in isolates colonizing or infecting HIV-positive individuals were seen (35). Authors suggested HIV infection might lead to preferential selection of C. albicans strains with altered virulence determinants that make them more pathogenic; however, a relationship has not yet been established between the genotypes and the virulence factors, or with clinical infection (35). In a study of patients without HIV-infection, enhanced virulence was found in C. albicans from tongue and buccal mucosa of chronic smokers, but oral candida loads did not differ between smokers and non-smokers in that study population (36).
EMM may be assessed through statistical interaction in multivariable models. Examining independent risk factors and interplay between them for occurrence of new cases of OC offers an opportunity of addressing temporality to allow confident interpretation of causality. Therefore, we examined EMM between CD4 cell counts and current smoking towards incident OC. As seen in Table 2, for the two-variable interaction – outcome association, the bivariate IRR corresponds to what would be “crude” IRR for one variable-outcome association. Full model IRR shows the relationship when all covariates exist whereas the final model IRR assesses the relationship in the most parsimonious model. In each of the three situations, there is an incremental risk of incident OC when exposed to smoking and/or low CD4 count. Therefore, among those with higher CD4 count, current smokers exhibit greater risk of OC compared to nonsmokers. For example, in the final model, compared to nonsmokers, smokers were 1.89 times as likely to get OC in the higher CD4 groups.
The general trend of increasing risk for OC with increasing exposure to smoking and low CD4 cell count was exhibited in bivariate analyses, as well as the final model (Table 2). The risk for OC was greatest among current smokers with low CD4 cell count compared to current non-smokers with higher CD4 cell count, a group that can be viewed as not being exposed to smoking or low CD4 cell count (i.e. doubly unexposed – R00). Compared to this doubly unexposed group, exposure to either smoking or low CD4 cell count or both increased the risk for OC. This finding demonstrates that although the magnitude of risk of OC differs depending upon exposure types, risk of OC is higher among current smokers thereby strengthening the argument that smoking is a strong independent risk factor for OC development.
We noted a potential dose-response association as joint exposures to lower CD4 count and current smoking was considered i.e. the risk for doubly exposed low CD4 cells/smoking (final model IRR 4.76) was substantially greater than singly exposed (IRR 2.51 & 1.89) which, in turn, was also substantially greater than the doubly unexposed (IRR 1.0 ~ referent group). This trend was similar for bivariate as well as the fully adjusted models. In assessing this change of risk across exposure categories, it is important to understand how much increased risk each exposure offers. The column “incremental risk” in Table 2 addresses this issue by ordering a sequence of increasing exposure and dividing the risk of outcome in greater exposure categories (upper rows) by lesser exposure categories (lower rows). This assessment suggested that compared to doubly unexposed, the exposure to smoking almost doubled the risk of OC (incremental IRR – IIRR: 1.89). Compared to this group, in absence of current smoking, exposure to lower CD4 cell count imparts another 33% increased risk (IIRR: 1.33). However, compared to current nonsmokers with low CD4 cell count, the risk of OC is nearly doubled by exposure to smoking (IIRR: 1.9). These results demonstrate that for each CD4 cell count category, exposure to smoking nearly doubles the risk of OC.
Confounding can be estimated by assessing the departure of estimated adjusted risk from their crude values after adjusting for confounders. The next question we addressed in these analyses was the possibility of differential magnitude of confounding in different joint-exposure strata. Although not generally discussed in the literature, it is assumed that each strata of joint-exposures involved in EMM are confounded homogenously. Based on this assumption, we would expect that the bivariate IRRs for each joint-exposure strata would be equidistant from the IRRs of the adjusted model. The last column in Table 2 addresses this issue. We assessed the percent change in risk estimates from their bivariate values (column % change) by assessing the difference between the two estimates as a proportion of the estimate of the final model.
The percent change column in Table 2 demonstrates that confounding by the common factors in the model may impact different strata of joint-exposures differently in terms of both direction and magnitude of confounding. For example, the percent change is positive for higher CD4-smoker and low CD4-nonsmoker strata, but negative for the doubly exposed group. Possible explanation of heterogeneity of confounding across factors involved in EMM may include different thresholds or sets of biological mechanisms at play under different exposure status and co-action of other factors or a role for unmeasured confounding factors.
EMM between smoking and CD4 cell count in determining OC demonstrated in our analysis suggests a type 2 response type (22), i.e. this association can be described as causal association for OC with single plus joint coactions by low CD4 cell count and smoking because compared to the doubly unexposed, the risk of OC was greater in those with low CD4 cell groups as well as among those who smoked. Therefore, each factor may cause OC when the other is absent, i.e. they are not necessary conditions for OC (OC can occur in other situations).
Under the sufficient cause model, low CD4 cell count can be classified as a necessary cause for OC in this study because OC occurred among nonsmokers. This relationship between CD4 cell count and smoking in determining OC can be classified as sufficient cause type B (22) where one factor is necessary (low CD4 cell count) and the other is not (smoking). However, risk of OC was greater even among higher CD4 cell count persons who smoked. This apparent counter-intuitive outcome can be explained by the fact that to conserve power, we defined the threshold of low CD4 cell count at 200 cells/mm3 where the OC risk is highest. Immune compromise is still significant at CD4 cell counts higher than this threshold which could explain the risk seen for higher CD4 cell count persons, many of whom would be at greater risk due to CD4 cell count being not high enough to substantially reduce the risk of OC (Figure 1). This fact is also expected to drive the incidence of OC among nonsmokers. However, in the presence of low CD4 cell count as well as smoking exposure, the risk for OC almost doubles as seen by the IRR figures in Table 2. These values suggest coparticipation between low CD4 cell count and smoking to cause OC. This interaction is also defined as causal coactions or synergism.
The effect of smoking on OC in populations with HIV/AIDS has been controversial. Our analysis has been able to provide evidence that in HIV/AIDS, smoking is an independent risk factor for OC in general and that this risk is greatest among those with low CD4 cell count. Furthermore, we have been able to quantify these increased risks - on an average, the risk for developing new OC is double among smokers compared to never-smokers/non-smokers (Tables 1 & 2).
These results have direct clinical application. In general, health hazards due to smoking may undermine benefits of HIV treatment on morbidity and mortality. Over 40 % of persons with HIV are current smokers. (37) Considering the results of this study, it is clear that incorporating smoking cessation advice and smoking-related education should be an important component of clinical management of HIV/AIDS patients with OC as well as an important component of OC prevention efforts.
CONCLUSION
We have been able to demonstrate that smoking is an independent risk factor for OC in HIV-1 infected persons and this risk is modified by the CD4 cell count. The risk of OC among those with low CD4 cell count who smoke is almost four and half times greater than among those having high CD4 cell count and who do not smoke. This suggests that the lower the CD4 cell count level, the more smoking enhances the individuals risk for OC.
Acknowledgments
Support
This investigation was supported by USPHS Grant 5T32DE07191, P30-HD27360 and R29DE11369 from the National Institute of Dental and Craniofacial Research, National Institutes of Health, Bethesda, MD 20892 USA.
The views expressed in this article are that of the authors and do not necessarily represent the views of the National Institutes of Health or the United States Government. This investigation was supported by USPHS Grant 5T32DE07191, P30-HD27360 and R29DE11369 from the National Institute of Dental and Craniofacial Research, National Institutes of Health, Bethesda, MD 20892 USA.
References
- 1.Chattopadhyay A, Caplan DJ, Slade GD, Shugars DC, Tien HC, Patton LL. Incidence of oral candidiasis and oral hairy leukoplakia in HIV-infected adults in North Carolina. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2005;99(1):39–47. doi: 10.1016/j.tripleo.2004.06.081. [DOI] [PubMed] [Google Scholar]
- 2.Patton LL, McKaig R, Strauss R, Rogers D, Eron JJ., Jr Changing prevalence of oral manifestations of human immuno-deficiency virus in the era of protease inhibitor therapy. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2000;89(3):299–304. doi: 10.1016/s1079-2104(00)70092-8. [DOI] [PubMed] [Google Scholar]
- 3.Greenspan D, Gange SJ, Phelan JA, Navazesh M, Alves ME, MacPhail LA, Mulligan R, Greenspan JS. Incidence of oral lesions in HIV-1-infected women: reduction with HAART. J Dent Res. 2004;83(2):145–50. doi: 10.1177/154405910408300212. [DOI] [PubMed] [Google Scholar]
- 4.Yang YL, Lo HJ, Hung CC, Li Y. Effect of prolonged HAART on oral colonization with Candida and candidiasis. BMC Infect Dis. 2006;6:8. doi: 10.1186/1471-2334-6-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nittayananta W, Chanowanna N, Winn T. Mode of HIV transmission associated with risk of oral lesions in HIV-infected subjects in Thailand. J Oral Pathol Med. 2010;39(2):195–200. doi: 10.1111/j.1600-0714.2009.00839.x. [DOI] [PubMed] [Google Scholar]
- 6.Miziara ID, Weber R. Oral candidosis and oral hairy leukoplakia as predictors of HAART failure in Brazilian HIV-infected patients. Oral Dis. 2006;12(4):402–7. doi: 10.1111/j.1601-0825.2005.01214.x. [DOI] [PubMed] [Google Scholar]
- 7.Patton LL. Sensitivity, specificity, and positive predictive value of oral opportunistic infections in adults with HIV/AIDS as markers of immune suppression and viral burden. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2000;90(2):182–8. doi: 10.1067/moe.2000.108799. [DOI] [PubMed] [Google Scholar]
- 8.Miziara ID, Filho BC, Weber R. Oral lesions in Brazilian HIV-infected children undergoing HAART. Int J Pediatr Otorhinolaryngol. 2006;70(6):1089–96. doi: 10.1016/j.ijporl.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 9.Obuekwe ON, Onunu AN. Gender and oral manifestations of HIV infection among adult Nigerians. Afr J Reprod Health. 2006;10(2):81–9. [PubMed] [Google Scholar]
- 10.Okoje VN, Obiechina AE, Aken’Ova YA. Orofacial lesions in 126 newly diagnosed HIV/AIDS patients seen at the University College Hospital, Ibadan. Afr J Med Med Sci. 2006;35(1):97–101. [PubMed] [Google Scholar]
- 11.Sharma G, Pai KM, Suhas S, Ramapuram JT, Doshi D, Anup N. Oral manifestations in HIV/AIDS infected patients from India. Oral Dis. 2006;12(6):537–42. doi: 10.1111/j.1601-0825.2006.01232.x. [DOI] [PubMed] [Google Scholar]
- 12.Taiwo OO, Okeke EN, Jalo PH, Danfillo IS. Oral manifestation of HIV/AIDS in Plateau state indigenes, Nigeria. West Afr J Med. 2006;25(1):32–7. doi: 10.4314/wajm.v25i1.28242. [DOI] [PubMed] [Google Scholar]
- 13.Van Meter F, Gallo JW, Carcia-Rojas G, Tan MM, Silverman S., Jr A study of oral candidiasis in HIV-positive patients. J Dent Hyg. 1994;68(1):30–4. [PubMed] [Google Scholar]
- 14.Chattopadhyay A, Gray LR, Patton LL, Caplan DJ, Slade GD, Tien HC, Shugars DC. Salivary secretory leukocyte protease inhibitor and oral candidiasis in human immunodeficiency virus type 1-infected persons. Infect Immun. 2004;72(4):1956–63. doi: 10.1128/IAI.72.4.1956-1963.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Soysa NS, Ellepola AN. The impact of cigarette/tobacco smoking on oral candidosis: an overview. Oral Dis. 2005;11(5):268–73. doi: 10.1111/j.1601-0825.2005.01115.x. [DOI] [PubMed] [Google Scholar]
- 16.Palacio H, Hilton JF, Canchola AJ, Greenspan D. Effect of cigarette smoking on HIV-related oral lesions. J Acquir Immune Defic Syndr Hum Retrovirol. 1997;14(4):338–42. doi: 10.1097/00042560-199704010-00005. [DOI] [PubMed] [Google Scholar]
- 17.Sroussi HY, Villines D, Epstein J, Alves MC, Alves ME. Oral lesions in HIV-positive dental patients - one more argument for tobacco smoking cessation. Oral Dis. 2007;13(3):324–8. doi: 10.1111/j.1601-0825.2006.01289.x. [DOI] [PubMed] [Google Scholar]
- 18.Masipa JN, Hauman CH, Raubenheimer EJ. Oral carriage of Candida species in patients visiting the Medunsa Dental Clinic. J Dent Assoc S Afr. 1992;47(9):407–9. [PubMed] [Google Scholar]
- 19.Slavinsky J, 3rd, Myers T, Swoboda RK, Leith JE, Hager S, Fidel PL., Jr Th1/Th2 cytokine profiles in saliva of HIV-positive smokers with oropharyngeal candidiasis. Oral Microbiol Immunol. 2002;17(1):38–43. doi: 10.1046/j.0902-0055.2001.00080.x. [DOI] [PubMed] [Google Scholar]
- 20.Campisi G, Pizzo G, Milici ME, Mancuso S, Margiotta V. Candidal carriage in the oral cavity of human immunodeficiency virus-infected subjects. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2002;93(3):281–6. doi: 10.1067/moe.2002.120804. [DOI] [PubMed] [Google Scholar]
- 21.EEC Clearinghouse on Oral Problems Related to HIV Infection WHO Collaborating Center on Oral Manifestations of the Immunodeficiency. Virus. Classification and diagnostic criteria for oral lesions in HIV infection. J Oral Pathol Med. 1993;22:289–91. [PubMed] [Google Scholar]
- 22.Rothman KJ, Greenland S. Modern epidemiology. 2. Philadelphia: Lippincott Williams & Wilkins; 1998. [Google Scholar]
- 23.Palmer RM, Wilson RF, Hasan AS, Scott DA. Mechanisms of action of environmental factors-tobacco smoking. J Clin Periodontol. 2005;32(Suppl 6):180–195. doi: 10.1111/j.1600-051X.2005.00786.x. [DOI] [PubMed] [Google Scholar]
- 24.Wallace JM, Oishi JS, Barbers RG, Simmons MS, Tashkin DP. Lymphocytic subpopulation profiles in bronchoalveolar lavage fluid and peripheral blood from tobacco and marijuana smokers. Chest. 1994;105:847–852. doi: 10.1378/chest.105.3.847. [DOI] [PubMed] [Google Scholar]
- 25.Quinn SM, Zhang JB, Gunsolley JC, Schenkein JG, Schenkein HA, Tew JG. Influence of smoking and race on immunoglobulin G subclass concentrations in early-onset periodontitis patients. Infect Immun. 1996;64:2500–2505. doi: 10.1128/iai.64.7.2500-2505.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Semlali A, Chakir J, Rouabhia M. Effects of whole cigarette smoke on human gingival fibroblast adhesion, growth, and migration. J Toxicol Environ Health A. 2011;74(13):848–62. doi: 10.1080/15287394.2011.570230. [DOI] [PubMed] [Google Scholar]
- 27.Banoczy J, Gintner Z, Dombi C. Tobacco use and oral leukoplakia. J Dent Educ. 2001;65:322–7. [PubMed] [Google Scholar]
- 28.Bennet KR, Reade PC. Salivary immunoglobulin A levels in tobacco smokers and patients with minor aphthous ulceration. Oral Surg Oral Med Oral Pathol. 1982;53:461–5. doi: 10.1016/0030-4220(82)90457-1. [DOI] [PubMed] [Google Scholar]
- 29.Berman J, Sudbery PE. Candida albicans: a molecular revolution built on lessons from budding yeast. Nat Rev Genet. 2002;3:918–30. doi: 10.1038/nrg948. [DOI] [PubMed] [Google Scholar]
- 30.Brook I. The impact of smoking on oral and nasopharyngeal bacterial flora. J Dent Res. 2011;90(6):704–710. doi: 10.1177/0022034510391794. [DOI] [PubMed] [Google Scholar]
- 31.Baboni FB, Barp D, Izidoro AC, Samaranayake LP, Rosa EA. Enhancement of Candida albicans virulence after exposition to cigarette mainstream smoke. Mycopathologia. 2009;168(5):227–35. doi: 10.1007/s11046-009-9217-5. [DOI] [PubMed] [Google Scholar]
- 32.Shin ES, Chung SC, Kim YK, Lee SW, Kho HS. The relationship between oral Candida carriage and the secretor status of blood group antigens in saliva. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2003;96(1):48–53. doi: 10.1016/s1079-2104(03)00160-4. [DOI] [PubMed] [Google Scholar]
- 33.Chiu CT, Li CF, Li JR, Wang J, Chuang CY, Chiang WF, Huang SC, Chang SW. Candida invasion and influences in smoking patients with multiple oral leucoplakias--a retrospective study. Mycoses. 2011;54(5):e377–83. doi: 10.1111/j.1439-0507.2010.01927.x. [DOI] [PubMed] [Google Scholar]
- 34.Budtz-Jörgensen E. Etiology, pathogenesis, therapy, and prophylaxis of oral yeast infections. Acta Odontol Scand. 1990;48(1):61–9. doi: 10.3109/00016359009012735. [DOI] [PubMed] [Google Scholar]
- 35.Mane A, Gaikwad S, Bembalkar S, Risbud A. Increased expression of virulence attributes in oral Candida albicans isolates from human immunodeficiency virus-positive individuals. J Med Microbiol. 2012;61:285–90. doi: 10.1099/jmm.0.036269-0. [DOI] [PubMed] [Google Scholar]
- 36.de Azevedo Izidoro AC, Semprebom AM, Baboni FB, Rosa RT, Machado MA, Samaranayake LP, Rosa EA. Low virulent oral Candida albicans strains isolated from smokers. Arch Oral Biol. 2012;57(2):148–53. doi: 10.1016/j.archoralbio.2011.08.016. [DOI] [PubMed] [Google Scholar]
- 37.Lifson AR, Lando HA. Smoking and HIV: Prevalence, Health Risks, and Cessation Strategies. Curr HIV/AIDS Rep. 2012 Sep;9(3):223–30. doi: 10.1007/s11904-012-0121-0. [DOI] [PubMed] [Google Scholar]