

Abstract
Aims/hypothesis
Obesity is associated with aldosterone excess, hypertension and the metabolic syndrome, but the relative contribution of aldosterone to obesity-related complications is debated. We previously demonstrated that aldosterone impairs insulin secretion, and that genetic aldosterone deficiency increases glucose-stimulated insulin secretion in vivo. We hypothesised that elimination of endogenous aldosterone would prevent obesity-induced insulin resistance and hyperglycaemia.
Methods
Wild-type and aldosterone synthase-deficient (As−/−) mice were fed a high-fat (HF) or normal chow diet for 12 weeks. We assessed insulin sensitivity and insulin secretion using clamp methodology and circulating plasma adipokines, and examined adipose tissue via histology.
Results
HF diet induced weight gain similarly in the two groups, but As−/− mice were protected from blood glucose elevation. HF diet impaired insulin sensitivity similarly in As−/− and wild-type mice, assessed by hyperinsulinaemic–euglycaemic clamps. Fasting and glucose-stimulated insulin were higher in HF-fed As−/− mice than in wild-type controls. Although there was no difference in insulin sensitivity during HF feeding in As−/− mice compared with wild-type controls, fat mass, adipocyte size and adiponectin increased, while adipose macrophage infiltration decreased. HF feeding significantly increased hepatic steatosis and triacylglycerol content in wild-type mice, which was attenuated in aldosterone-deficient mice.
Conclusions/interpretation
These studies demonstrate that obesity induces insulin resistance independently of aldosterone and adipose tissue inflammation, and suggest a novel role for aldosterone in promoting obesity-induced beta cell dysfunction, hepatic steatosis and adipose tissue inflammation.
Keywords: Adiponectin, Aldosterone, Aldosterone synthase, Diabetes, Fatty liver, Insulin, Insulin resistance, Insulin secretion, Renin-angiotensin-aldosterone system
Introduction
Obesity is associated with aldosterone excess [1, 2], hypertension [1, 3] and the metabolic syndrome [4], but the relative contribution of aldosterone to the development of obesity-related complications is not known. The renin–angiotensin–aldosterone system (RAAS) is classically activated under conditions of low sodium intake, but also appears to be activated inappropriately in patients with obesity and resistant hypertension [5, 6]. Pharmacological RAAS blockade improves glucose homeostasis in some clinical studies, although the mechanism of this beneficial effect is not well-delineated [7, 8].
Tissue insulin resistance and failure of pancreatic beta cell compensation are the chief metabolic defects in type 2 diabetes progression [9, 10]. The effect of exogenous aldosterone on insulin sensitivity has been demonstrated extensively in vivo and in vitro. In this context, excess aldosterone impairs peripheral insulin sensitivity via the mineralocorticoid receptor (MR) in rodents by activating oxidative stress and inflammatory pathways, impairing insulin receptor signalling, and inhibiting glucose transporter trafficking [5]. MR antagonism with spironolactone or eplerenone prevents obesity- and aldosterone-induced impairment in adipose tissue and skeletal muscle glucose uptake in animal models [11, 12]. However, the glucocorticoids, corticosterone and cortisol, can also bind to and activate the MR, raising the possibility that steroids other than aldosterone contribute to the adverse complications of obesity.
Previous clinical studies have shown that, in addition to insulin resistance, a beta cell defect is a critical component of type 2 diabetes progression [9, 10, 13, 14]. Recent genome-wide association studies also suggest an important role for insulin secretory pathways as genetic risk factors for type 2 diabetes [15]. The effect of aldosterone on insulin secretion has not been investigated extensively, but there is evidence that modest hyperaldosteronism is associated with an impaired beta cell response in humans [16]. Aldosterone’s effect on insulin secretion may occur indirectly by depleting potassium [17]. We recently investigated the effect of genetic aldosterone deficiency on glucose metabolism in aldosterone synthase-deficient (As−/−) mice [18]. Genetic aldosterone deficiency resulted in increased insulin secretion in vivo without altered insulin sensitivity, even when potassium and renin activity were normalised with a high-sodium diet. Studies in isolated murine islets and the MIN6 clonal beta cell line have shown that aldosterone directly impairs insulin secretion in an MR-independent manner.
Because obesity is associated with inappropriately elevated aldosterone concentrations [1, 2, 5], endogenous aldosterone may increase the risk of progression to type 2 diabetes by impairing insulin resistance, insulin secretion, or both. In the present study, we used As−/− mice, which lack aldosterone and have corticosterone concentrations comparable to wild-type mice [18], to test the hypothesis that endogenous aldosterone impairs glucose-stimulated insulin secretion and promotes insulin resistance and adipose inflammation in a diet-induced obesity model.
Methods
Animals
All experiments were approved by the Vanderbilt Institutional Animal Care and Use Committee, and NIH principle of laboratory animal care were followed. Male As−/− and wild-type littermates on a C57BL/6J background (Jackson Laboratories, Bar Harbor, ME, USA; backcrossed ≥12 generations) from heterozygous As+/− breeding pairs were obtained as previously described [18]. At the age of 6 weeks, mice were randomised to either a normal chow diet or a high-fat (HF) diet with 60% fat by calorie content (BioServ, Frenchtown, NJ) for 12 weeks with free access to water ad libitum and housed in a temperature-controlled facility with a 12 h light/dark cycle.
Body composition measurements
Lean body mass, free fluid and fat mass were measured in conscious mice using nuclear magnetic resonance (NMR) spectroscopy (MiniSpec LF50; Bruker Optics, The Woodlands, TX, USA) before catheter implantation. Gonadal fat was dissected and weighed after study completion as an additional quantitative measure of body fat. Systolic blood pressure was measured using automated tail-cuff impedance plethysmography (BP-2000 Blood Pressure Analysis System; Visitech Systems, Apex, NC, USA) in unanaesthetised, trained mice, prewarmed for 5 min at 37°C.
Hyperglycaemic clamps
Carotid arterial and jugular venous catheters were implanted for euglycaemic or hyperglycaemic clamps ≥3 days before study, and clamps were conducted in the Vanderbilt Mouse Metabolic Center as previously described [18, 19]. Before clamps, mice were fasted for 5 h starting at ~08:00 h. During the clamps, the glucose infusion rate (GIR) was varied to achieve whole-blood glucose concentration of 250 mg/dl (13.9 mmol/l) through the jugular venous catheter. Blood samples were collected at 5–10 min intervals for 120 min. Heparinised and saline-washed erythrocytes were infused to replace blood loss. Mice were killed at the end of the study under phenobarbital anaesthesia, and tissue samples were collected in liquid nitrogen and stored at −80°C. Blood was collected in heparinised tubes and immediately processed and stored as plasma at −80°C.
Hyperinsulinaemic–euglycaemic clamps with somatostatin
Because insulin levels varied widely among HF feeding groups, we infused somatostatin to suppress endogenous insulin and administered exogenous insulin to achieve hyperinsulinaemia [19]. Somatostatin (8 μgkg−1min−1 or 4.9 μmolkg−1min−1; Bachem, Bubendorf, Switzerland), insulin (5 mUkg−1min−1; Novo-Nordisk, Princeton, NJ, USA), glucagon (2.6 nmolkg−1min−1; Eli Lilly, Indianapolis, IN, USA) and washed erythrocytes (5.5 μl/min) were infused from t=0 to 165 min. In pilot studies in rodents, these somatostatin, insulin and glucagon infusion rates were determined to achieve plasma insulin levels equivalent to those achieved with the standard insulin infusion rate of 4 mUkg−1min−1 in the absence of somatostatin. Blood glucose was monitored every 10 min, and the GIR was varied to maintain blood glucose at ~6.9 mmol/l (125 mg/dl). Blood was taken at −15, 0 min and 80–120 min to determine insulin and C-peptide. To determine tissue-specific glucose uptake, a bolus of 444 kBq of 2-deoxy[14C]glucose (2-DG; Perkin Elmer, Waltham, MA, USA) was administered intravenously at 120 min. Blood was sampled at 122–145 min, and tissues were collected at study completion for 2-DG measurements. After the last sample, mice were killed under anaesthesia, and tissues were collected and stored at −80°C.
Determination of adiponectin oligomers
Adiponectin oligomers were detected by western blot methods using a modification of previously reported methods [20]. Briefly, 1 μl plasma without heat denaturation was diluted with a non-reducing Laemmli sample buffer, separated by 4–20% SDS-PAGE, transferred to polyvinylidene fluoride membranes, and blocked in Odyssey blocking buffer (LI-COR, Lincoln, NE, USA) at room temperature for 1 h. The membrane was then incubated with an anti-rabbit antibody against the globular domain of mouse adiponectin (1:1,000 dilution; Abcam, Cambridge, MA, USA) at 4°C overnight. The membrane was incubated with an IRDye800-conjugated goat anti-rabbit secondary antibody at room temperature for 1 h, and fluorescence signals were detected using the LI-COR Odyssey Infrared Imaging System.
Measurement of liver and muscle triacylglycerol content
Similar amounts of liver and muscle (vastus lateralis) tissue were accurately weighed and digested in 100 μl fresh KOH (3 mol/l, in 60% ethanol) at 70°C for 1 h and then at room temperature overnight. Digested solutions were brought up to 500 μl with 2 mol/l TRIS/HCl (pH 7.5). Diluted samples were incubated with a GPO triacylglycerol reagent (Pointe Scientific, Canton, MI, USA), and sample absorbance was read at a wavelength of 500 nm. Liver lipid was extracted using Folch methodology, and triacylglycerol and diacylglycerol were separated by thin-layer chromatography. Results were normalised to sample weight.
Histology and immunohistochemistry
Frozen liver sections (5 μm) were fixed with 10% ice-cold formalin for 10 min, stained with prewarmed Oil Red O solution for 10 min at 60°C, and differentiated with 85% propylene glycol solution for 3 min. Sections were stained with haematoxylin for 30 min and mounted with glycerine jelly medium. Adipose tissues were fixed in 4% paraformaldehyde for 48 h and then transferred to 70% ethanol for 24 h. Paraffin-embedded 5 μm sections were then deparaffinised in xylene and rehydrated in descending-graded ethanol. A monoclonal rat antibody against F4/80 (1:2,000 dilution; Abcam) was used to stain macrophages with diaminobenzidine detection according to the manufacturer’s protocol (Vectastain kit; Vector labs, Burlingame, CA, USA). F4/80-positive cells were counted as described previously [21]. Briefly, ten different high-power fields (×20) from each slide were analysed, and the total number of nuclei and the number of nuclei of F4/ 80-positive cells were counted and expressed as a percentage of positive cells. Crown-like structures in the gonadal fat were counted in ten randomly selected high-power fields (×20) per animal. Paraffin-embedded pancreas sections (6 μm) were stained with Verhoeff van Gieson stain. Islets (≥20/mouse) were imaged at high power (×20), and cross-sectional area was quantified using Image J (NIH, Bethesda, MD, USA).
Laboratory assays
In all clamp protocols, glucose refers to arterial whole blood glucose (Accu-Chek; Roche Diagnostics, Basel, Switzerland). Plasma samples collected during the clamp studies were used for insulin and C-peptide assays. Plasma cytokines were measured using a Luminex Mouse Metabolic Magnetic Bead Panel Kit (catalogue no MMHMAG-44K; Millipore, Billerica, MA, USA) according to the manufacturer’s instructions. Insulin and C-peptide were analysed by RIA as previously reported [18].
Statistical analysis and calculations
Data are presented as mean±SEM in text and figures. Overall differences among groups were tested using ANOVA, and then individual between-group comparisons were tested using non-parametric methods (Wilcoxon rank sum test). All statistical analyses were performed using SPSS for Windows (version 19.0; SPSS, Chicago, IL, USA), with a two-tailed p value <0.05 considered significant.
Results
Aldosterone deficiency does not prevent HF-feeding-induced obesity
Body weight had increased during HF feeding similarly in the two genotypes at study end (Fig. 1a). Epididymal fat mass increased similarly on the HF diet in the two genotypes (Fig. 1b). Total body fat mass, assessed by NMR, increased to a greater extent in As−/− than wild-type mice on the HF diet (16.3±1.1 vs 13.0±1.3 g, p<0.01; Fig. 1c); results remained significant when analysed as fat percentage per body weight, with an absolute increase of 9.0% body fat in As−/− mice on the HF diet. Muscle mass, assessed by NMR, was similar in the two genotypes on the normal diet and increased on the HF diet in wild-type but not As−/− mice (Fig. 1c).
Fig. 1.

Aldosterone deficiency does not prevent diet-induced obesity. Body weight increased significantly during HF feeding in both genotypes (a). Epididymal fat weight increased similarly in both genotypes (b). Fat mass and free fluid assessed by NMR increased in both genotypes during HF feeding (c, d), although muscle mass increased in wild-type mice but not in As−/− mice during HF feeding (c). White bar, wild-type; grey bar, As−/−; hatched bars, HF diet. *p<0.05, **p≤0.01, ***p≤0.001
Systolic blood pressure was unchanged on the HF diet in wild-type mice, although it was significantly reduced in As−/− + HF mice compared with wild-type + HF (95.5±1.1 vs 100.5± 3.2 mmHg, p<0.01). Aldosterone was below detectable limits in As−/− mice compared with 345.0±59.2 pg/ml in wild-type mice on the normal diet and 356±126 pg/ml on the HF diet (p<0.01 for each vs As−/−). Free fluid assessed by NMR was similar in the two genotypes on the normal diet and increased in both genotypes on the HF diet; the increase in free fluid was significantly attenuated in As−/− mice (Fig. 1d).
Aldosterone deficiency prevents glucose elevation during HF feeding
Fasting glucose was reduced in As−/− mice, and this was not affected by HF feeding (Fig. 2a; 7.5± 1.8 mmol/l in As−/− + HF vs 9.1±1.8 mmol/l in wild-type + HF, p=0.01). HF feeding increased fasting insulin in both genotypes, but to a significantly greater extent in As−/− mice (Fig. 2b; 187.3±101.8 pmol/l in As−/− + HF vs 73.8± 50.8 pmol/l in wild-type + HF, p≤0.01). C-peptide also increased to a greater extent in aldosterone-deficient mice on the HF diet (Fig. 2c), consistent with an effect on insulin secretion.
Fig. 2.
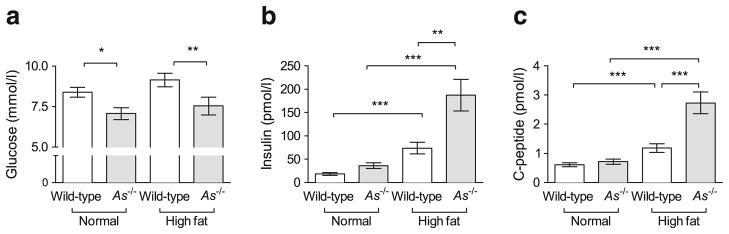
Effect of genetic aldosterone deficiency on fasting glucose and insulin during HF feeding. HF feeding had significantly increased blood glucose after a 5 h morning fast in wild-type, but not in As−/−, mice (a). Fasting plasma insulin had significantly increased in both genotypes but to a greater extent in As−/− (b). Fasting plasma C-peptide data paralleled those of insulin (c). *p<0.05, **p≤0.01, ***p≤0.001
Aldosterone deficiency augments obesity-induced glucose-stimulated insulin secretion in vivo in mice
We assessed insulin secretion during hyperglycaemic clamps by infusing glucose to achieve comparable levels of hyperglycaemia (Fig. 3a). Glucose-stimulated insulin secretion was increased in As−/− mice compared with wild-type mice during normal chow feeding (Fig. 3b), and this effect was augmented during HF feeding (insulin AUC, 4.99±0.88 nmol/l×min in wild-type, 10.28±1.73 nmol/l×min in wild-type + HF, 16.02±2.95 nmol/l×min in As−/−, 30.38±5.34 nmol/l×min in As−/− + HF). Changes in plasma C-peptide concentrations paralleled those of insulin (Fig. 3c), suggesting that increased insulin secretion rather than altered clearance was responsible for differences in concentrations. Histological analysis of pancreatic islet area revealed no significant difference between wild-type and As−/− mice on regular or HF diets.
Fig. 3.
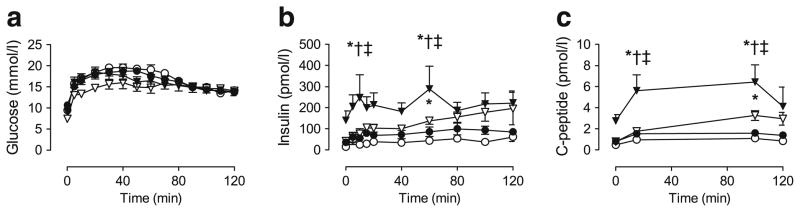
Glucose-stimulated insulin secretion is increased in aldosterone-deficient mice during normal chow and HF feeding. During hyperglycaemic clamp conditions, glucose was infused to achieve equivalent hyperglycaemia in normal-chow-fed wild-type (white circles) and aldosterone-deficient (white triangles) mice and HF-fed wild-type (black circles) and As−/− mice (black triangles) (a). Plasma insulin (b) and C-peptide (c) concentrations were significantly higher in As−/− mice during either normal chow or HF feeding than in wild-type. *p≤0.05 vs wild-type; †p≤0.05 vs wild-type + HF; ‡p≤0.05 vs As−/−
Aldosterone deficiency does not prevent insulin resistance induced by HF feeding
We assessed insulin sensitivity using the hyperinsulinaemic clamp. Because fasting plasma insulin concentrations differed significantly among genotype and diet groups and can confound assessment of insulin sensitivity, we modified the hyperinsulinaemic clamp by infusing somatostatin to suppress endogenous insulin secretion. A similar approach has been used in humans and rats to achieve equal insulin concentrations during exogenous insulin administration [22, 23]. At initiation of the clamp studies (Fig. 4a, b, Basal), plasma C-peptide and insulin concentrations varied significantly among groups. After somatostatin infusion, plasma C-peptide decreased markedly (Fig. 4a, End), indicating suppression of endogenous insulin secretion. Plasma insulin at steady-state was equivalent in the two genotype groups (Fig. 4b, End), and higher in HF mice regardless of genotype. A similar level of euglycaemia was maintained near 6.9 mmol/l (125 mg/dl) (Fig. 4c). HF feeding reduced overall insulin sensitivity significantly and similarly in both genotypes, as demonstrated by the reduction in GIR needed to maintain normoglycaemia (Fig. 4d). Results were unaffected after adjustment of GIR for lean body mass. Tissue-specific glucose (2-DG) uptake was also significantly reduced in wild-type adipose and heart tissues during HF feeding and was not affected by genetic aldosterone deficiency (Table 1). These data suggest that aldosterone deficiency did not prevent obesity-induced insulin resistance, but preserved normoglycaemia because of increased beta cell responsiveness.
Fig. 4.

HF feeding produces insulin resistance independently of aldosterone. Somatostatin-modified hyperinsulinaemic–euglycaemic clamps were used to abolish baseline differences in insulin secretion between wild-type and As−/− mice during HF feeding. Before the clamp, plasma C-peptide (a, basal) and insulin (b, basal) varied among groups at basal time points (white circle, normal chow wild-type; white triangle, normal chow As−/−; black circle, HF-fed wild-type; black triangle, HF-fed As−/−). Somatostatin suppressed endogenous insulin secretion, demonstrated by C-peptide in all groups (a, end), and equivalent plasma insulin was achieved between genotypes (b, End). Blood glucose was maintained at 6.9 mmol/l (125 mg/dl) (c). HF feeding significantly decreased insulin sensitivity in both wild-type and As−/− mice, demonstrated by the GIR required to maintain euglycaemia (d). ***p≤0.001
Table 1.
Tissue-specific glucose uptake (Rg) during hyperinsulinaemic–euglycaemic clamp
| Group | Tissue Rg (μgmin−1 [g tissue] −1)
|
|||||
|---|---|---|---|---|---|---|
| Soleus | Gastrocnemius | Vastus lateralis | Adipose | Diaphragm | Heart | |
| Wild-type | 135.6±55.9 | 16.8±3.3 | 15.3±2.3 | 9.7±1.3 | 84.3±8.8 | 432.7±55 |
| As−/− | 65.9±16 | 14.3±7.4 | 9.4±4 | 6.3±1.3** | 122.4±56.7 | 176.9±54** |
| Wild-type + HF | 59.4±10.2 | 9.3±1.6 | 9.7±2.5 | 3.9±0.6** | 90.4±22.5 | 216.5±50.6** |
| As−/− + HF | 68.2±48.1 | 12.8±5 | 13.6±6.1 | 4.4±0.8** | 130.3±32.5 | 283.2±85.2* |
Values are mean±SEM
p≤0.05 vs wild-type;
p<0.01 vs wild-type
Aldosterone deficiency attenuates HF-feeding-induced hepatic steatosis
HF feeding increased hepatic fat deposition in wild-type mice, as assessed by Oil Red O positivity, and this effect was attenuated in As−/− mice (Fig. 5a). Hepatic triacylglycerol content was similar in the two genotype groups during normal chow feeding and increased significantly in both genotypes during HF feeding. The effect of HF feeding on hepatic triacylglycerol content was attenuated in As−/− mice (Fig. 5b, 33.3±3.4 mg/g tissue in As−/− + HF vs 44.9±2.2 mg/g tissue in wild-type + HF, p<0.05). Muscle triacylglycerol content tended to increase in wild-type mice on the HF diet (7.2±0.7 vs 9.7±1.1 mg/g tissue on normal and HF diet, respectively) and was similar in As−/− mice (9.2±0.8 vs 10.1±0.6 mg/g tissue). These data show that aldosterone deficiency improved HF-feeding-induced hepatic steatosis despite a similar degree of whole-animal insulin resistance.
Fig. 5.
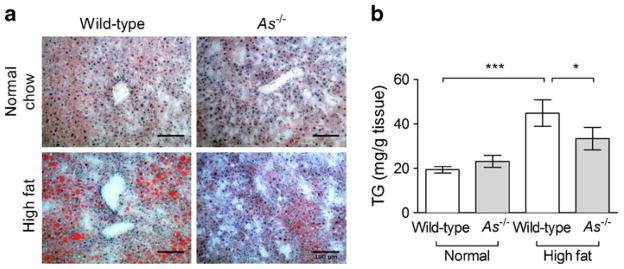
Aldosterone deficiency attenuates obesity-induced hepatic steatosis. Fat deposition increased during HF feeding in wild-type liver, but appeared to be less in As−/− mice (a, Oil Red O staining; scale bar=100 μm). Liver triacylglycerol (TG) content increased significantly during HF feeding in wild-type mice, but was attenuated in As−/− mice (b). *p<0.05, ***p≤0.001
Aldosterone deficiency prevents adipokine dysfunction and macrophage infiltration during HF feeding
Plasma adiponectin increased during HF feeding in wild-type and As−/− mice, but to a greater extent in As−/− mice (17.3±1.8 vs 11.1± 1.2 μg/ml, p<0.001, Fig. 6a). Analysis by non-denaturing electrophoresis demonstrated that the biologically active high molecular weight (HMW) isoform was selectively increased in HF-fed As−/− mice (Fig. 6b). Consistent with increased adiposity, plasma leptin concentrations also increased during HF feeding and to a greater extent in As−/− mice (Fig. 6c). Plasma TNF-α, which is typically suppressed by adiponectin, also tended to decrease during HF feeding in As−/− mice compared with wild-type (0.03±0.04 ng/ml in As−/− + HF vs 1.9±1.4 ng/ml in wild-type + HF; p=0.08), suggesting a physiological effect of increased HMW adiponectin. Adipocyte size increased during HF feeding to a similar extent in the two genotypes (Fig. 7a, b), and there was no difference in adipocyte size distribution between genotypes. Infiltration of F4/80-positive macrophages increased markedly in HF-fed wild-type mice, and this was attenuated in As−/− mice (Fig. 7a, c). Likewise, crown-like structures were increased during HF feeding in both wild-type (1.4±0.9 vs 12.7±3.9 per ten high-power fields; p=0.03) and As−/− (0.3±0.17 vs 8.2±1.9 per ten high-power fields; p<0.001) mice.
Fig. 6.
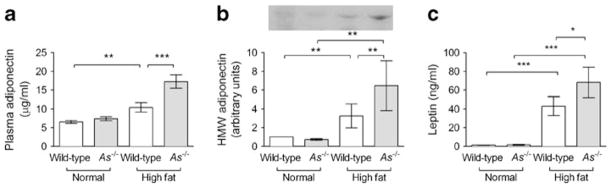
Obesity-induced adipokine dysfunction is prevented in aldosterone-deficient mice. Plasma adiponectin increased during HF feeding to a greater extent in As−/− than wild-type mice (a). Quantification of adiponectin oligomers revealed a selective increase in HMW adiponectin during HF feeding, which increased to a greater extent in As−/− mice. A representative western blot of plasma HMW adiponectin oligomers is shown (b). Plasma leptin increased significantly during HF feeding, to a greater extent in As−/− than wild-type mice (c). *p<0.05, **p≤0.01, ***p≤0.001
Fig. 7.

Aldosterone deficiency attenuated HF-feeding-induced adipose tissue macrophage infiltration but not adipocyte hypertrophy. HF feeding increased F4/80 positivity in wild-type mouse epididymal fat tissues but appeared to be less in As−/− mice (a, diaminobenzidine staining; scale bar 100 μm). Adipocyte size increased significantly and similarly in wild-type and As−/− mice during HF feeding (b). HF feeding markedly increased the percentage of F4/80-positive macrophages within the adipose tissue in wild-type mice, but this effect was attenuated in As−/− mice (c). **p≤0.01, ***p≤0.001
Discussion
Obesity is associated with inappropriately elevated plasma aldosterone, and MR antagonism attenuates obesity-related hypertension and adipose dysfunction [5]. Other steroids can also bind to and activate the MR, and the effect of decreasing aldosterone on insulin resistance, insulin secretion and adipose tissue inflammation has not been previously explored. Although glucose-stimulated insulin secretion is increased in As−/− mice [18], whether this response is beneficial or maladaptive under conditions of metabolic stress is uncertain. In the present study, genetic aldosterone deficiency significantly attenuated or prevented obesity-related beta cell dysfunction, hyperglycaemia, hepatic steatosis and adipose inflammation. Surprisingly, these beneficial effects were observed despite the development of diet-induced weight gain and insulin resistance.
We previously demonstrated that endogenous aldosterone impairs glucose-stimulated insulin secretion in vivo and that exogenous aldosterone impairs insulin secretion in isolated murine islets and MIN6 cells [18]. Clinical studies have shown that modest or severe hyperaldosteronism, due to either idiopathic or adrenal adenoma, is associated with impaired insulin secretion, suggesting that these findings in mice are directly relevant in humans [16, 17]. The present study demonstrates that genetic aldosterone deficiency and increased insulin secretion attenuates hyperglycaemia and hepatic steatosis during HF-feeding-induced obesity. This protection occurred despite a similar weight gain on the HF diet, and even an apparent increase in relative fat accumulation and no change in adipocyte size in As−/− mice.
In other genetic mouse models, insulin hypersecretion is associated with improved glucose metabolism [24, 25]. The majority of identified genes associated with susceptibility to type 2 diabetes affect insulin processing or secretory pathways [15, 26]. Polymorphisms in the aldosterone synthase gene (CYP11B2) are also associated with development of the metabolic syndrome and type 2 diabetes [27–29], although a link to insulin secretion has not been investigated. Genetic or obesity-induced variation in adrenal aldosterone production may affect insulin secretion in humans, and further studies are warranted to investigate this relationship.
Adrenal aldosterone secretion is also associated with obesity. Aldosterone concentration correlates strongly with measures of fat mass in humans [1, 2], and decreases after diet-induced weight loss [30, 31]. Aldosterone is similarly raised in rodent models of obesity such as db/db mice and SHR/NDmc corpulent rats [11, 32]. In other studies, as in ours, aldosterone is not increased, but is not appropriately suppressed by the sodium and fluid accumulation that occurs on an HF diet [33]. Adipocyte-derived factors such as oxidised fatty acids stimulate aldosterone secretion independently of angiotensin II in vitro, possibly explaining this association [34, 35], and recent studies suggest that adipocytes may be capable of aldosterone production [36]. Aldosterone may also promote adiposity by inducing terminal adipocyte differentiation via the MR [37]. It has therefore been proposed that aldosterone excess contributes to fat expansion. Our data do not support an essential role of aldosterone in diet-induced obesity. It may affect fat distribution by increasing visceral fat relative to peripheral fat, although we could not definitively address this in the present study. Adipose tissue produces relatively little 11β-hydroxysteroid dehydrogenase type 2 (11βHSD2), which inactivates cortisol and prevents MR activation [37]. Because glucocorticoids promote visceral fat accumulation [38, 39], an imbalance between aldosterone and glucocorticoids could alter fat partitioning. Furthermore, adipose 11βHSD1 may contribute to local cortisol metabolism and affect insulin sensitivity [40]. Our data show that adipose tissue expansion is clearly aldosterone-independent, although we cannot exclude a contribution of the MR.
In obesity, adipose tissue appearance and function is altered, characterised by macrophage infiltration, crown-like structures and altered adipokine secretion [41]. Aldosterone decreases adiponectin secretion and increases proinflammatory gene expression in 3T3 cells via MR activation in vitro [11, 42]. MR activation also reduces adipose tissue inflammatory gene expression in the adipose tissue of obese rodents [11]. Glucocorticoids also impair adipokine gene expression via the MR in 3T3 cells [43], and circulate at a much higher concentration in vivo, raising the possibility that endogenous glucocorticoids may alter adipokines even in the absence of aldosterone. In the present study, aldosterone-deficient mice fed an HF diet have increased circulating adiponectin and less macrophage adipose infiltration and hepatic triacylglycerol accumulation. These findings are consistent with the emerging hypothesis that certain adipose tissues can serve as a fatty acid depot and prevent ectopic fat accumulation in visceral tissues [44]. However, it is of interest that improvement in these adipokines and adipose tissue histological appearance was not associated with preserved insulin sensitivity. Therefore, obesity-associated insulin resistance may be independent of elevated TNF-α, hypoadiponectinaemia and adipose tissue inflammation. Our results demonstrate that endogenous aldosterone contributes to obesity-induced adipokine dysfunction and adipose tissue macrophage infiltration in vivo.
Exogenous aldosterone induces insulin resistance via the MR [5, 11, 45]. Plasma aldosterone also predicts the development of insulin resistance in humans [46]. Because obesity is associated with inappropriately increased aldosterone and MR activation, we hypothesised that aldosterone deficiency would prevent HF-feeding-induced insulin resistance. Surprisingly, As−/− mice were not protected against obesity-induced insulin resistance, assessed by both whole-organism and tissue-specific measurements. Ligands other than aldosterone may contribute to insulin resistance via the MR. Glucocorticoids bind the MR with similar affinity to aldosterone and impair skeletal muscle insulin receptor signalling [47]. However, corticosterone concentrations were similar in wild-type and As−/− mice [18]. Tissue-specific overproduction of 11βHSD1 within adipose tissue or skeletal muscle increases local glucocorticoids and similarly induces insulin resistance [39, 48]. The relatively low abundance of 11βHSD2 in skeletal muscle allows glucocorticoids to activate the MR, and may contribute to glucocorticoid-induced insulin resistance.
The use of somatostatin to equalise insulin during clamp studies in mice represents a technical advance that can be used to investigate differences in insulin sensitivity despite marked differences in basal insulin concentrations. The HOMA has not been validated in rodents [49], so we used the gold standard hyperinsulinaemic–euglycaemic clamp to measure insulin resistance [19]. Differences in insulin levels would violate a major assumption of the clamp, so we equalised insulin concentrations by infusing somatostatin and glucagon. Endogenous insulin secretion was entirely suppressed, as documented by C-peptide, enabling matched insulin concentrations between genotypes. The remaining difference in insulin concentrations between diet groups is probably due to weight-based insulin dosing. Although HF-fed animals were exposed to a slightly higher insulin concentration, this would bias towards the null hypothesis in comparisons of HF and regular chow groups. Therefore, our results suggest that the HF diet induced a greater degree of insulin resistance than the GIR alone suggests. Future refinement of the protocol by adjusting the insulin dose by lean body mass may result in more comparable insulin levels among animals with greatly differing weights.
Aldosterone synthase inhibitors are being developed for clinical use and effectively reduce circulating aldosterone and blood pressure in hypertensive patients [50]. Our findings suggest that these agents may provide beneficial metabolic effects in humans beyond blood pressure reduction, and further studies are needed in other prediabetes models and in clinical trials.
Acknowledgments
We thank N. Brown (Vanderbilt Medical Center), for reading the manuscript, and T. Ansari and C. Malabanan (Vanderbilt MMPC), for their excellent technical expertise during clamp studies. This work was presented in part at the 2011 AHA Council for High Blood Pressure Research.
Funding This work was supported by grants from the NIH (DK081662, HL060906, DK58404 and DK038226-24), and the Vanderbilt Mouse Metabolic Phenotyping Center (DK59637). Steroid hormone assays were performed by the Vanderbilt Diabetes Research and Training Center Hormone Assay Core Lab (DK20593).
Abbreviations
- As−/−
Aldosterone synthase-deficient
- 2-DG
2-Deoxyglucose
- GIR
Glucose infusion rate
- HF
High fat
- HM
High molecular weight
- 11βHSD
11β-Hydroxysteroid dehydrogenase
- MR
Mineralocorticoid receptor
- NMR
Nuclear magnetic resonance
- RAAS
Renin–angiotensin–aldosterone system
Footnotes
Duality of interest All other authors report that there is no duality of interest associated with this manuscript.
Contribution statement All authors contributed to experimental design, researched data, wrote the manuscript, contributed to discussion, and approved the final version. JML is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Contributor Information
P. Luo, Division of Clinical Pharmacology, Department of Medicine, Vanderbilt University Medical Center, 2200 Pierce Avenue, 560 RRB, Nashville, TN 37232-6602, USA. Huangshi Central Hospital, Huangshi, Hubei Province, People’s Republic of China
A. Dematteo, Division of Clinical Pharmacology, Department of Medicine, Vanderbilt University Medical Center, 2200 Pierce Avenue, 560 RRB, Nashville, TN 37232-6602, USA
Z. Wang, Division of Clinical Pharmacology, Department of Medicine, Vanderbilt University Medical Center, 2200 Pierce Avenue, 560 RRB, Nashville, TN 37232-6602, USA
L. Zhu, Division of Endocrinology and Diabetes, Department of Medicine, Vanderbilt University School of Medicine, Nashville, TN, USA. Department of Veterans Affairs, Nashville, TN, USA
A. Wang, Eastern Virginia Medical School, Norfolk, VA, USA
H.-S. Kim, Departments of Pathology and Laboratory Medicine, University of North Carolina, Chapel Hill, NC, USA
A. Pozzi, Department of Veterans Affairs, Nashville, TN, USA. Division of Nephrology and Hypertension, Department of Medicine, Vanderbilt University School of Medicine, Nashville, TN, USA
J. M. Stafford, Division of Endocrinology and Diabetes, Department of Medicine, Vanderbilt University School of Medicine, Nashville, TN, USA. Department of Veterans Affairs, Nashville, TN, USA
J. M. Luther, Email: James.Luther@Vanderbilt.edu, Division of Clinical Pharmacology, Department of Medicine, Vanderbilt University Medical Center, 2200 Pierce Avenue, 560 RRB, Nashville, TN 37232-6602, USA. Division of Nephrology and Hypertension, Department of Medicine, Vanderbilt University School of Medicine, Nashville, TN, USA
References
- 1.Bentley-Lewis R, Adler GK, Perlstein T, et al. Body mass index predicts aldosterone production in normotensive adults on a high-salt diet. J Clin Endocrinol Metab. 2007;92:4472–4475. doi: 10.1210/jc.2007-1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goodfriend TL, Kelley DE, Goodpaster BH, Winters SJ. Visceral obesity and insulin resistance are associated with plasma aldosterone levels in women. Obes Res. 1999;7:355–362. doi: 10.1002/j.1550-8528.1999.tb00418.x. [DOI] [PubMed] [Google Scholar]
- 3.Vasan RS, Evans JC, Larson MG, et al. Serum aldosterone and the incidence of hypertension in nonhypertensive persons. N Engl J Med. 2004;351:33–41. doi: 10.1056/NEJMoa033263. [DOI] [PubMed] [Google Scholar]
- 4.Ingelsson E, Pencina MJ, Tofler GH, et al. Multimarker approach to evaluate the incidence of the metabolic syndrome and longitudinal changes in metabolic risk factors: the Framingham Offspring Study. Circulation. 2007;116:984–992. doi: 10.1161/CIRCULATIONAHA.107.708537. [DOI] [PubMed] [Google Scholar]
- 5.Sowers JR, Whaley-Connell A, Epstein M. Narrative review: the emerging clinical implications of the role of aldosterone in the metabolic syndrome and resistant hypertension. Ann Intern Med. 2009;150:776–783. doi: 10.7326/0003-4819-150-11-200906020-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Briet M, Schiffrin EL. The role of aldosterone in the metabolic syndrome. Curr Hypertens Rep. 2011;13:163–172. doi: 10.1007/s11906-011-0182-2. [DOI] [PubMed] [Google Scholar]
- 7.Bosch J, Yusuf S, Gerstein HC, et al. Effect of ramipril on the incidence of diabetes. N Engl J Med. 2006;355:1551–1562. doi: 10.1056/NEJMoa065061. [DOI] [PubMed] [Google Scholar]
- 8.McMurray JJ, Holman RR, Haffner SM, et al. Effect of valsartan on the incidence of diabetes and cardiovascular events. N Engl J Med. 2010;362:1477–1490. doi: 10.1056/NEJMoa1001121. [DOI] [PubMed] [Google Scholar]
- 9.Kahn SE, Hull RL, Utzschneider KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature. 2006;444:840–846. doi: 10.1038/nature05482. [DOI] [PubMed] [Google Scholar]
- 10.Weyer C, Bogardus C, Mott DM, Pratley RE. The natural history of insulin secretory dysfunction and insulin resistance in the pathogenesis of type 2 diabetes mellitus. J Clin Invest. 1999;104:787–794. doi: 10.1172/JCI7231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guo C, Ricchiuti V, Lian BQ, et al. Mineralocorticoid receptor blockade reverses obesity-related changes in expression of adiponectin, peroxisome proliferator-activated receptor-gamma, and proinflammatory adipokines. Circulation. 2008;117:2253–2261. doi: 10.1161/CIRCULATIONAHA.107.748640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lastra G, Whaley-Connell A, Manrique C, et al. Low-dose spironolactone reduces reactive oxygen species generation and improves insulin-stimulated glucose transport in skeletal muscle in the TG(mRen2)27 rat. Am J Physiol Endocrinol Metab. 2008;295:E110–E116. doi: 10.1152/ajpendo.00258.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Festa A, Williams K, Hanley AJ, Haffner SM. Beta-cell dysfunction in subjects with impaired glucose tolerance and early type 2 diabetes: comparison of surrogate markers with first-phase insulin secretion from an intravenous glucose tolerance test. Diabetes. 2008;57:1638–1644. doi: 10.2337/db07-0954. [DOI] [PubMed] [Google Scholar]
- 14.Jensen CC, Cnop M, Hull RL, Fujimoto WY, Kahn SE. Beta-cell function is a major contributor to oral glucose tolerance in high-risk relatives of four ethnic groups in the U.S. Diabetes. 2002;51:2170–2178. doi: 10.2337/diabetes.51.7.2170. [DOI] [PubMed] [Google Scholar]
- 15.Billings LK, Florez JC. The genetics of type 2 diabetes: what have we learned from GWAS? Ann N Y Acad Sci. 2010;1212:59–77. doi: 10.1111/j.1749-6632.2010.05838.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mosso LM, Carvajal CA, Maiz A, et al. A possible association between primary aldosteronism and a lower beta-cell function. J Hypertens. 2007;25:2125–2130. doi: 10.1097/HJH.0b013e3282861fa4. [DOI] [PubMed] [Google Scholar]
- 17.Conn JW. Hypertension, the potassium ion and impaired carbohydrate tolerance. N Engl J Med. 1965;273:1135–1143. doi: 10.1056/NEJM196511182732106. [DOI] [PubMed] [Google Scholar]
- 18.Luther JM, Luo P, Kreger MT, et al. Aldosterone decreases glucose-stimulated insulin secretion in vivo in mice and in murine islets. Diabetologia. 2011;54:2152–2163. doi: 10.1007/s00125-011-2158-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ayala JE, Bracy DP, McGuinness OP, Wasserman DH. Considerations in the design of hyperinsulinemic-euglycemic clamps in the conscious mouse. Diabetes. 2006;55:390–397. doi: 10.2337/diabetes.55.02.06.db05-0686. [DOI] [PubMed] [Google Scholar]
- 20.Bobbert T, Rochlitz H, Wegewitz U, et al. Changes of adiponectin oligomer composition by moderate weight reduction. Diabetes. 2005;54:2712–2719. doi: 10.2337/diabetes.54.9.2712. [DOI] [PubMed] [Google Scholar]
- 21.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW., Jr Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112:1796–1808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu K, Cappel D, Martinez M, Stafford JM. Impaired-inactivation of FoxO1 contributes to glucose-mediated increases in serum very low-density lipoprotein. Endocrinology. 2010;151:3566–3576. doi: 10.1210/en.2010-0204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meneilly GS, McIntosh CH, Pederson RA, et al. Effect of glucagon-like peptide 1 on non-insulin-mediated glucose uptake in the elderly patient with diabetes. Diabetes Care. 2001;24:1951–1956. doi: 10.2337/diacare.24.11.1951. [DOI] [PubMed] [Google Scholar]
- 24.Turban S, Liu X, Ramage L, et al. Optimal elevation of beta-cell 11beta-hydroxysteroid dehydrogenase type 1 is a compensatory mechanism that prevents high-fat diet-induced beta-cell failure. Diabetes. 2012;61:642–652. doi: 10.2337/db11-1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shafrir E, Ziv E, Mosthaf L. Nutritionally induced insulin resistance and receptor defect leading to beta-cell failure in animal models. Ann N Y Acad Sci. 1999;892:223–246. doi: 10.1111/j.1749-6632.1999.tb07798.x. [DOI] [PubMed] [Google Scholar]
- 26.Ingelsson E, Langenberg C, Hivert MF, et al. Detailed physiologic characterization reveals diverse mechanisms for novel genetic Loci regulating glucose and insulin metabolism in humans. Diabetes. 2010;59:1266–1275. doi: 10.2337/db09-1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bellili NM, Foucan L, Fumeron F, et al. Associations of the -344 T>C and the 3097 G>A polymorphisms of CYP11B2 gene with hypertension, type 2 diabetes, and metabolic syndrome in a French population. Am J Hypertens. 2010;23:660–667. doi: 10.1038/ajh.2010.44. [DOI] [PubMed] [Google Scholar]
- 28.Ranade K, Wu KD, Risch N, et al. Genetic variation in aldosterone synthase predicts plasma glucose levels. Proc Natl Acad Sci U S A. 2001;98:13219–13224. doi: 10.1073/pnas.221467098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Russo P, Lauria F, Loguercio M, et al. 344C/T Variant in the promoter of the aldosterone synthase gene (CYP11B2) is associated with metabolic syndrome in men. Am J Hypertens. 2007;20:218–222. doi: 10.1016/j.amjhyper.2006.07.012. [DOI] [PubMed] [Google Scholar]
- 30.Ho JT, Keogh JB, Bornstein SR, et al. Moderate weight loss reduces renin and aldosterone but does not influence basal or stimulated pituitary-adrenal axis function. Horm Metab Res. 2007;39:694–699. doi: 10.1055/s-2007-985354. [DOI] [PubMed] [Google Scholar]
- 31.Tuck ML, Sowers J, Dornfeld L, Kledzik G, Maxwell M. The effect of weight reduction on blood pressure, plasma renin activity, and plasma aldosterone levels in obese patients. N Engl J Med. 1981;304:930–933. doi: 10.1056/NEJM198104163041602. [DOI] [PubMed] [Google Scholar]
- 32.Nagase M, Yoshida S, Shibata S, et al. Enhanced aldosterone signaling in the early nephropathy of rats with metabolic syndrome: possible contribution of fat-derived factors. J Am Soc Nephrol. 2006;17:3438–3446. doi: 10.1681/ASN.2006080944. [DOI] [PubMed] [Google Scholar]
- 33.Hall JE, Brands MW, Dixon WN, Smith MJ., Jr Obesity-induced hypertension. Renal function and systemic hemodynamics. Hypertension. 1993;22:292–299. doi: 10.1161/01.hyp.22.3.292. [DOI] [PubMed] [Google Scholar]
- 34.Goodfriend TL, Ball DL, Egan BM, Campbell WB, Nithipatikom K. Epoxy-keto derivative of linoleic acid stimulates aldosterone secretion. Hypertension. 2004;43:358–363. doi: 10.1161/01.HYP.0000113294.06704.64. [DOI] [PubMed] [Google Scholar]
- 35.Ehrhart-Bornstein M, Lamounier-Zepter V, Schraven A, et al. Human adipocytes secrete mineralocorticoid-releasing factors. Proc Natl Acad Sci U S A. 2003;100:14211–14216. doi: 10.1073/pnas.2336140100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Briones AM, Cat AN, Callera GE, et al. Adipocytes produce aldosterone through calcineurin-dependent signaling pathways: implications in diabetes mellitus-associated obesity and vascular dysfunction. Hypertension. 2012;59:1069–1078. doi: 10.1161/HYPERTENSIONAHA.111.190223. [DOI] [PubMed] [Google Scholar]
- 37.Marzolla V, Armani A, Zennaro MC, et al. The role of the mineralocorticoid receptor in adipocyte biology and fat metabolism. Mol Cell Endocrinol. 2012;350:281–288. doi: 10.1016/j.mce.2011.09.011. [DOI] [PubMed] [Google Scholar]
- 38.Geer EB, Shen W, Strohmayer E, Post KD, Freda PU. Body composition and cardiovascular risk markers after remission of Cushing’s disease: a prospective study using whole-body MRI. J Clin Endocrinol Metab. 2012;97:1702–1711. doi: 10.1210/jc.2011-3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Masuzaki H, Paterson J, Shinyama H, et al. A transgenic model of visceral obesity and the metabolic syndrome. Science. 2001;294:2166–2170. doi: 10.1126/science.1066285. [DOI] [PubMed] [Google Scholar]
- 40.Hughes KA, Manolopoulos KN, Iqbal J, et al. Recycling between cortisol and cortisone in human splanchnic, subcutaneous adipose, and skeletal muscle tissues in vivo. Diabetes. 2012;61:1357–1364. doi: 10.2337/db11-1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sun K, Kusminski CM, Scherer PE. Adipose tissue remodeling and obesity. J Clin Invest. 2011;121:2094–2101. doi: 10.1172/JCI45887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li P, Zhang XN, Pan CM, et al. Aldosterone perturbs adiponectin and PAI-1 expression and secretion in 3T3-L1 adipocytes. Horm Metab Res. 2011;43:464–469. doi: 10.1055/s-0031-1277226. [DOI] [PubMed] [Google Scholar]
- 43.Hirata A, Maeda N, Nakatsuji H, et al. Contribution of glucocorticoid-mineralocorticoid receptor pathway on the obesity-related adipocyte dysfunction. Biochem Biophys Res Commun. 2012;419:182–187. doi: 10.1016/j.bbrc.2012.01.139. [DOI] [PubMed] [Google Scholar]
- 44.Kim JY, van de Wall E, Laplante M, et al. Obesity-associated improvements in metabolic profile through expansion of adipose tissue. J Clin Invest. 2007;117:2621–2637. doi: 10.1172/JCI31021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wada T, Kenmochi H, Miyashita Y, et al. Spironolactone improves glucose and lipid metabolism by ameliorating hepatic steatosis and inflammation and suppressing enhanced gluconeogenesis induced by high-fat and high-fructose diet. Endocrinology. 2010;151:2040–2049. doi: 10.1210/en.2009-0869. [DOI] [PubMed] [Google Scholar]
- 46.Kumagai E, Adachi H, Jacobs DR, Jr, et al. Plasma aldosterone levels and development of insulin resistance: prospective study in a general population. Hypertension. 2011;58:1043–1048. doi: 10.1161/HYPERTENSIONAHA.111.180521. [DOI] [PubMed] [Google Scholar]
- 47.Gathercole LL, Stewart PM. Targeting the pre-receptor metabolism of cortisol as a novel therapy in obesity and diabetes. J Steroid Biochem Mol Biol. 2010;122:21–27. doi: 10.1016/j.jsbmb.2010.03.060. [DOI] [PubMed] [Google Scholar]
- 48.Morgan SA, Sherlock M, Gathercole LL, et al. 11beta-hydroxysteroid dehydrogenase type 1 regulates glucocorticoid-induced insulin resistance in skeletal muscle. Diabetes. 2009;58:2506–2515. doi: 10.2337/db09-0525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wallace TM, Levy JC, Matthews DR. Use and abuse of HOMA modeling. Diabetes Care. 2004;27:1487–1495. doi: 10.2337/diacare.27.6.1487. [DOI] [PubMed] [Google Scholar]
- 50.Calhoun DA, White WB, Krum H, et al. Effects of a novel aldosterone synthase inhibitor for treatment of primary hypertension: results of a randomized, double-blind, placebo- and active-controlled phase 2 trial. Circulation. 2011;124:1945–1955. doi: 10.1161/CIRCULATIONAHA.111.029892. [DOI] [PubMed] [Google Scholar]