

Abstract
Background
Esophageal fibrosis is a complication of eosinophilic esophagitis (EoE) which has been attributed to both subepithelial fibrosis and to epithelial to mesenchymal transition (EMT), a process by which epithelial cells acquire mesenchymal features. Common to both causes of EoE-fibrosis is the notion that granulocyte-derived TGF-β, induces myofibroblast differentiation of the target cell. To date, the role of esophageal epithelial cells as effector cells in esophageal fibrosis has never been explored. Here in, we investigated consequences of cross-talk between esophageal epithelial cells and fibroblasts, and identified profibrotic cytokines which influence the development of EMT in vitro.
Methods and Results
Stimulation of primary fetal esophageal fibroblasts (FEF3) with conditioned media (CEM) from esophageal epithelial cells (EPC2-hTERT), primed FEF3 cells to secrete IL-1β and TNFα, but not TGFβ. To determine whether these cytokines signaled in a paracrine fashion to esophageal epithelial cells, FEF3 cells were stimulated with CEM, followed by transfer of this fibroblast conditioned media (FCM) to EPC2-hTERT cells. Epithelial FCM stimulation increased expression of mesenchymal markers and reduced E-cadherin expression, features of EMT which were TNFα and IL-1β-dependent. Using organotypic culture models, primary EoE epithelial cells exhibited features of EMT compared to non-EoE cells, corresponding to patterns of EMT in native biopsies.
Conclusions
Esophageal epithelial cell and fibroblast cross-talk contributes to esophageal fibrosis. Our results suggest that features of EMT can develop in dependent of TGF-β and granulocytes, which may have important implications in treatment of EoE.
Keywords: Cross-talk, eosinophilic esophagitis, esophageal epithelial cells, fibroblasts, fibrosis, epithelial to mesenchymal transition, cytokines
Introduction
Eosinophilic esophagitis (EoE) is a chronic allergic disease affecting 4 in 10,000 children [1] and adults, characterized by eosinophilic infiltrates of the esophageal mucosa. In older children and adults, the most problematic complication of EoE is the development of esophageal fibrosis leading to dysphagia and esophageal food bolus impactions. The precise etiology of EoE-associated fibrosis remains unknown.
Fibrosis is defined as the inappropriate deposition of extracellular matrix (ECM), leading to deformation of the parenchyma. It is widely believed that stimulation with profibrotic cytokines activates fibroblasts to acquire the activated phenotype of myofibroblasts, morphologic intermediates between fibroblasts and smooth muscle cells which synthesize ECM components including collagen, 3-smooth muscle actin (αSMA), fibronectin, and proteoglycans. Although local fibroblasts are considered to be the most common myofibroblast progenitors, myofibroblasts have also been shown to originate from bone marrow-derived fibrocytes [2–4] and smooth muscle cells [5]. In addition, epithelial cells can acquire a myofibroblast characteristics and lose epithelial cell features [6]via epithelial to mesenchymal transition (EMT) [7]. In EMT, epithelial cells gain contractile and cytoskeleton proteins found in myofibroblasts while losing their characteristic tight junction and adhesion proteins.
Others have recently shown that EoE-associated fibrosis occurs through several mechanisms, including EMT. Aceves et al. showed that esophageal biopsies from EoE patients exhibit increased subepithelial collagen deposition compared to biopsies from control patients and patients with gastroesophageal reflux disease[8] suggesting that activation of fibroblasts within the subepithelium contributes to EoE fibrosis. In contrast, Kagalwalla et al. recently demonstrated that esophageal biopsies from pediatric EoE subjects exhibit features of EMT, characterized by increased expression of the mesenchymal marker vimentin and decreased expression of the epithelial marker cytokeratin within the epithelial compartment [9]. Interestingly, Kagalwalla et al. also observed a correlation between EMT scores and subepithelial fibrosis in pediatric EoE biopsies, indicating that the two processes are not mutually exclusive. In addition, these investigators also showed that features of EMT could be induced in vitro, through stimulation of the HET-1A esophageal epithelial cell line with the profibrotic cytokine, transforming growth factor-β (TGF-β), consistent with findings of Ohashi et al., who also showed that TGF-β stimulation induced EMT in the EPC2-hTERT esophageal epithelial cell line [10].
TGF-β is known as a prototypical profibrotic cytokine in many models of fibrosis[11–13]. Consistent with this notion, both Aceves et al. and Kagalwalla et al. have suggested that TGF-β is necessary for myofibroblast activation in the context of EoE-associated fibrosis. This assumption is supported by the work of others, who have previously shown that TGF-β is produced and released by circulating immune effector cells known to infiltrate the esophageal epithelium in EoE, including mast cells[14] and eosinophils [15].
While TGF-β plays an established role in tissue remodeling, other profibrotic cytokines and soluble mediators can activate fibroblasts and induce ECM production [16]. IL-1β, for example, enhances the effects of TGF-β in the acquisition of the mesenchymal phenotype in human bronchial epithelial cells in vitro[17]. TNF-α has been implicated in the development of EMT in retinal pigment epithelial cells[18], and enhances TGF-β-induced EMT in human alveolar epithelial cells[19]. To date, the potential role for IL-1β and TNF-α in EoE-associated tissue remodeling has not been investigated.
Others have shown that cross-talk between epithelial and mesenchymal cells contributes to remodeling in other model systems [20–22]. Building upon our previous reports that human esophageal epithelial cells function as effector cells in the pathogenesis of esophageal inflammation[23, 24], we hypothesized that esophageal epithelial and mesenchymal cross-talk plays a role in EoE-associated fibrosis. In this study, we show for the first time that esophageal epithelial cells prime esophageal fibroblasts to secrete fibrogenic cytokines IL-1β and TNF-α. Surprisingly, we demonstrate that these cytokines play a role in the development of EMT in vitro, and this can occur in a TGF-β-independent fashion. Using a primary EoE cell line grown in organotypic culture with primary fibroblasts, we further demonstrate that esophageal epithelial cells can function as innate immune effector cells in the context of EoE.
Materials and Methods
Cell lines
Three human esophageal epithelial cell lines, EPC2-hTERT, EPC394, and EPC425, were grown at 37° C in a humidified 5% CO2 incubator, and maintained keratinocyte serum free medium (KSFM, Invitrogen, Grand Island, NY) containing human epidermal growth factor (1ng/mL), bovine pituitary extract (50ug/mL), and penicillin (100 units/mL) and streptomycin (100μg/ml). The EPC2-hTERT cell line is a telomerase-immortalized and nontransformed cell line, whereas the EPC394 and EPC425 cells lines are primary cell lines obtained from an EoE (EPC394) and a non-EoE control (EPC425) patient. Fetal esophageal fibroblasts (FEF3 cells, gift of Hiroshi Nakagawa MD, PhD) and a primary fibroblast cell line (PEF429) from an adolescent patient with EoE, were maintained in Dulbecco’s Minimum Essential Media (DMEM) supplemented with 10% fetal bovine serum (FBS) (GIBCO), and grown at 37° C in a humidified 5% CO2 incubator.
Primary esophageal cell lines
Esophageal biopsies were placed in Hanks BSS buffer, transferred to dispase (BD Biosciences, 50U/mL) for 20 minutes at 37°C, then trypsinized (trypsin-EDTA, GIBCO) at 37° C. Trypsin was inactivated using soybean trypsin inhibitor (SIGMA) and biopsies were gently manually shaken. Samples were poured through a cell strainer and cells were collected in a conical tube. Cells were pelleted by centrifugation at 4 °C for 5 minutes. For epithelial cell isolation, pellets were resuspended in KSFM containing antibiotics and fungizone (1:500) (GIBCO). For fibroblast isolation, pellets were resuspended in DMEM with antibiotics and fungizone (1:500) (GIBCO). Cell suspensions were then seeded in tissue culture plates. Cells were used at passage 2–3.
Conditioned Epithelial Media (CEM) stimulation
Conditioned epithelial media (CEM) was collected from confluent EPC2-hTERT cells grown in complete KSFM, and used to stimulate fibroblast monolayers for 3 and 6 hours. Prior to stimulation of fibroblasts, CEM was supplemented with 10% FBS. Figure 1A shows the schematic of the experimental design. For control conditions, unconditioned complete KSFM was supplemented with 10% FBS. After stimulation with CEM, media was collected for ELISA, and fibroblasts were harvested for RNA isolation.
Figure 1. Conditioned epithelial media (CEM) primes fibroblasts to secrete proinflammatory cytokines.
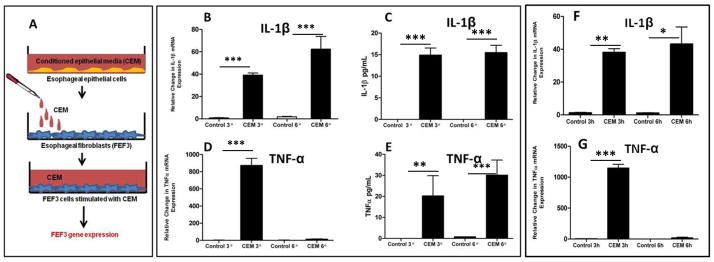
A: Schematic of experimental design: Conditioned epithelial media (CEM) from esophageal epithelial cells (EPC2-hTERT) were transferred to fetal esophageal fibroblasts (FEF3 cells). Media was collected at various time points for quantification of FEF3-secreted cytokines, and FEF3 cells were harvested for analysis of gene expression. B, D: mRNA expression of IL-1β and TNFα by FEF3 cells at various time points after CEM stimulation. C, E: Quantification of IL-1β and TNFα secretion by FEF3 cells at various time points following CEM stimulation. F, G: mRNA expression of IL-1β and TNFα expression by primary esophageal fibroblasts (PEF429) from an adolescent EoE patient following stimulation with CEM for various time points. Data shown are representative of at least 3 individual experiments. Error bars represent standard error. * p<0.05, **p<0.01, ***p<0.001, NS = not significant.
Stimulation of epithelial cells with recombinant cytokines
EPC2-hTERT cells were seeded in 6 well plates at a density of 3 × 105 cells/well one day prior to stimulation. Cells were stimulated in triplicate with combinations of human recombinant TGF-β (R&D Systems, Minneapolis, MN) (10 ng/mL), IL1-β (Sigma, Saint Louis, MO) (10 ng/ml), and TNF-α (R&D) (40 ng/ml). Media, including cytokines, was refreshed weekly, and cells were harvested after 3 weeks of stimulation for RNA isolation.
Fibroblast Conditioned Media (FCM) stimulation
FEF3 cells were first stimulated with CEM. After 6 hours, this fibroblast-conditioned media (FCM) was then used to stimulate fresh monolayers of EPC2-hTERT cells for three weeks. A schematic of the experimental design is shown in Figure 3A. For control conditions, EPC2-hTERT cells were treated for 3 weeks with unconditioned KSFM (+ 10% FBS) which had been applied to FEF3 cells for the same time points. For inhibition studies, cells treated with FCM were also co-treated with infliximab (Remicade) (1ug/mL, gift of Monica Darby), anti-IL1-R (Anakinra)(40ng/mL, R&D), or both. FCM, KSFM, or the inhibitors were refreshed weekly until day 21, when EPC2-hTERT cells were either harvested for RNA or used for immunofluorescence.
Figure 3. Stimulation of esophageal epithelial cells with fibroblast conditioned media (FCM) leads to features of EMT, in an IL-1β and TNFα-dependent fashion.

A: Schematic of experimental design: Following stimulation of FEF3 cells with CEM (2 days), this “fibroblast conditioned media” (FCM) was harvested and transferred to fresh EPC2-hTERT cells. After 3 weeks of FCM stimulation in the presence of absence of competitive inhibitors of IL-1β and/or TNFα, EPC2-hTERT cells were harvested for mRNA isolation, or immunolocalization of mesenchymal/epithelial markers. B, C, D: mRNA expression of E-cadherin, vimentin, and αSMA by EPC2-hTERT cells after stimulation with FCM in the presence or absence of anti-TNFα mAb (Remicade) and/or anti-IL-1R (Anakinra). E, J: Constitutive expression of epithelial E-cadherin (red) and αSMA (green) by EPC2-hTERT cells. Nuclei are counterstained with DAPI (blue). F, K: Loss of E-cadherin expression (red) and enhanced αSMA (green) expression by EPC2-hTERT cells following 3 weeks of stimulation with FCM. G, H: Partial recovery of E-cadherin expression by EPC2-hTERT cells stimulated with FCM and anti-TNFα mAb (Remicade) or anti-IL-1R (Anakinra). L, M: Absence of αSMA in EPC2-hTERT cells treated with FCM in the presence anti-TNFα mAb (Remicade) or anti-IL-1R (Anakinra). I, N: Combination of anti-TNFα mAb (Remicade) and anti-IL-1R (Anakinra) protects EPC2-hTERT cells from effects of FCM stimulation. O, P: Morphology of EPC2-hTERT cells before and after FCM stimulation. Q: Effect of anti-TNFα mAb and anti-IL-1R upon EPC2-hTERT morphology. p-values were calculated based upon comparisons to unstimulated conditions. *p<0.05, **p<0.01, ***p<0.001, NS = not significant.
RNA isolation and quantitative RT-PCR
RNA was isolated using an RNeasy kit (Qiagen, Valencia, CA) according to manufacturer’s recommendations. RNA samples were reverse transcribed using a high-capacity cDNA reverse transcriptase kit (Applied Biosystems, Foster City, CA). Preformulated TaqMan Gene Expression Assays were purchased from Applied Biosystems for human TNF-α, IL-1 β, TGF-β, vimentin, E-cadherin, αSMA, and GAPDH. Quantitative RT-PCR was performed by using Taqman Fast Universal PCR Master Mix kit and reactions were performed in triplicate using 96 well optical plates on a StepOnePlus Real-Time PCR System (Applied Biosystems). GAPDH was used as an endogenous control to normalize the samples using CT method of relative quantification, where CT is the threshold cycle.
Enzyme-linked immunosorbent assay (ELISA)
TNF-α, IL1-β, and TGF-β were quantified in culture supernatants using ELISA (R&D), using manufacturer’s recommendations.
Immunofluorescence
For cell monolayers, esophageal epithelial cells were seeded in glass chamber slides at a density of 3 × 105 cells/chamber. Following 3 weeks of FCM stimulation, cells were fixed and permeabilized using methanol/acetone at −20°C for 10 minutes, followed by incubation in primary antibody [(mouse anti-human E-cadherin (BD Bioscience) (1:200), mouse anti-human αSMA (Sigma) (1:1000)] for 2 hours at 4°C. Secondary antibody [rabbit anti-mouse Dylight (Jackson Immunoresearch Laboratories) (1:600)] was applied for 1 hour at room temperature. Slides, mounted with DAPI mounting media (VECTAshield),were viewed using an Olympus BX51 microscope.
For organotypic culture and patient biopsy slides, sections were re-hydrated and boiled in sodium citrate buffer, then incubated with the primary antibodies [chicken anti-human vimentin (Novus Biologicals, Littleton, CO) 1:5000, E-cadherin 1:200, αSMA 1:1000] at 4°C, followed by secondary anti-chicken antibody (Jackson Immunolaboratories) (1:600)or anti-mouse Dylight antibody (1:600) for 1 hour at room temperature prior to mounting in DAPI mounting media.
Organotypic cell culture (OTC)
OTC models were constructed using previously published methods [25]. Briefly, 5 X 105 esophageal epithelial cells (EPC2-hTERT, primary EoE cell line EPC394, primary non-EoE cell line EPC425) were seeded onto a collagen matrix, containing 7.5 X 104 fetal esophageal fibroblast cells (FEF3). On the fourth day after seeding, epithelial cells were raised to the air-liquid interface and cultured for another 6 days. Cultures were harvested and fixed with 10% neutral buffered formalin and embedded in paraffin. Sections were used for immunofluorescence.
Trichrome staining
Human biopsy slides were deparaffinized, rehydrated, and stained using a Masson trichrome staining protocol[26].
Human subjects
The human subjects protocol was approved by the Institutional Review Board at the Children’s Hospital of Philadelphia. Following informed consent, additional esophageal pinch biopsies were obtained during routine diagnostic esophagogastroduodenoscopy (EGD) for isolation of primary esophageal epithelial or fibroblast cell lines. Consistent with recently published clinical guidelines, the diagnosis of EoE was made histologically by the presence of 15 or more esophageal epithelial eosinophils per high powered field (hpf), hyperplasia of the basal epithelium, and the absence of tissue eosinophilia in the more distal GI tract[27]. All subjects were on high dose PPI therapy for at least 8 weeks prior to biopsy.
Statistical analysis
A two-tailed Student’s t-test was used for analysis of Figure 1, and a one-way ANOVA and post-hoc comparison with Bonferroni was used to analyze Figures 2 and 3. A p value of ≤0.05 was considered to be statistically significant.
Figure 2. Stimulation of esophageal epithelial cell stimulation with IL-1β and TNF-α induces expression of mesenchymal genes and suppresses expression of epithelial markers, features of EMT which are further enhanced by TGF-β.
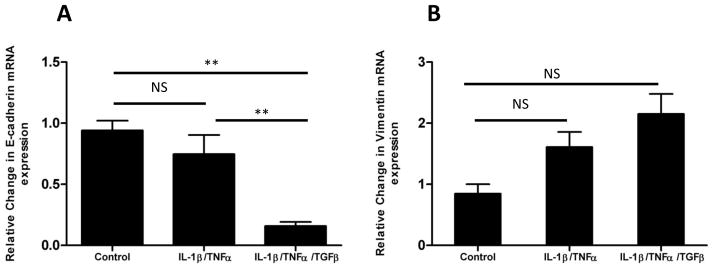
A: mRNA expression of epithelial-specific E-cadherin by EPC2-hTERT cells after 3 weeks of stimulation with combinations of IL-1β, TNFα, and TGF-β. B: Expression of mesenchymal marker vimentin by EPC2-hTERT cells after three weeks of dual cytokine (IL-1β/TNFα) or triple cytokine (IL-1β/TNFα/TGF-β) stimulation. Data shown are representative of at least 3 individual experiments. p-values were calculated based upon comparisons to unstimulated conditions. *p<0.05, **p<0.01, NS = not significant.
Results
1) Conditioned esophageal epithelial media primes esophageal fibroblasts to secrete IL-1β, TNF-α, but not TGF-β
As a first step in investigating esophageal epithelial and mesenchymal cross-talk, we determined whether esophageal fibroblasts could sense factors released by esophageal epithelial cells in vitro. We investigated cross-talk using the immortalized nontransformed EPC2-hTERT esophageal epithelial cells, and the primary fetal esophageal fibroblast (FEF3) cell line. EPC2-hTERT cells exhibit a normal karyotype, do not undergo a slow-growth phase, and have been routinely used through 200 passage days (PD) by others [28]. EPC2-hTERT cells in this study were used between 30–50 PD. CEM from confluent EPC2-hTERT cells was used to stimulate confluent FEF3 cells for various time points. Unconditioned, fresh KSFM was used for control conditions. A schematic of the experimental design is shown in Figure 1A. To analyze the pro-fibrotic response, we quantified FEF3 mRNA expression and secretion of IL-1β, TNF-α, and TGF-β in response to CEM stimulation. Fibroblast mRNA expression of IL-1β peaked at the 6 hour time point following stimulation, with corresponding protein secretion sustained at both the 3 and 6 hour time points (Figures 1B, 1C). Robust mRNA expression of TNF-α was detected at 3 hours following CEM stimulation, with protein secretion sustained at 3 and 6 hours post-stimulation (Figures 1D,1E). Protein concentrations of TNF-α and IL-1β remained unchanged through 5 days(data not shown). Notably, there was no detectable TGF-β in the CEM, nor did CEM induce any mRNA expression or protein secretion of TGF-β from stimulated FEF3 cells (data not shown).
Fetal-derived fibroblasts, including FEF3 cells, may have distinct functional differences from mature fibroblasts[29–31]To control for this possibility, CEM was also used to stimulate primary esophageal fibroblasts isolated from an adolescent patient with EoE (PEF429), and CEM-induced mRNA expression of IL-1β, TNF-α, and TGF-β were quantified. The clinical characteristics of the EoE subject from which the esophageal fibroblasts were acquired are shown in Table 1. PEF429 response to CEM paralleled that of FEF3 cells, with significant induction in IL-1β (Figure 1F) and TNF-α (Figure 1G), but not TGF-β (not shown). Based upon the similarities in response to CEM between the two fibroblast cell lines, the remainder of experiments in this study were performed using the FEF3 cell line.
Table 1.
Clinical characteristics of subjects 394 and 425, which were used to generate primary esophageal epithelial cell lines EPC394 and EPC425. Esophageal biopsies from subject 429 were used to generate the primary esophageal fibroblast cell line PEF429.
| Subject ID | 394 | 425 | 429 |
|---|---|---|---|
| Age (Years) | 11 | 12 | 13 |
| Gender | Male | Male | Female |
| Symptoms | Dysphagia | Heartburn, abdominal pain | Dysphagia |
| Medications | Lansoprazole, allergy shots | Omeprazole, cetirizine | Omeprazole |
| # Eosinophils per hpf | 37 | 0 | 15 |
| Clinical Diagnosis | EoE | Abdominal pain | EoE |
| Esophageal Cell line | Epithelial (EPC394) | Epithelial (EPC425) | Fibroblast (PEF429) |
2) Exposure of esophageal epithelial cells to pro-fibrogenic cytokinesIL-1β, TNF-α, and TGF-β leads to features of EMT in vitro
We hypothesized that CEM-induced production of fibroblast-derived cytokines IL-1β and TNF-α might exert pro-fibrogenic effects upon esophageal epithelial cells. To recapitulate this hypothesized paracrine signaling pathway in vitro, we cultured EPC2-hTERT cells in the presence of recombinant human IL-1β and TNF-α, and quantified mRNA expression of the epithelial-specific marker E-cadherin and the mesenchymal marker vimentin after three weeks in culture. The three week time point was chosen based upon the findings of Ohashi et al. who previously demonstrated that EPC2-hTERT cells undergo maximal TGF-β-induced transition to spindle-like morphology after 21 days of continuous cytokine exposure in vitro[10]. Although fibroblast expression and secretion of TGF-β was not detected in our model system, we hypothesized that exogenous TGF-β might further enhance the pro-fibrogenic effects of IL-1β and TNFα upon EPC2-hTERT cells.
Though not statistically significant, the expression of the epithelial marker E-cadherin was modestly reduced by IL-1β/TNF-α stimulation. This effect was enhanced by the addition of TGF-β, which led to a significant reduction in E-cadherin expression(Figure 2A). Notably, 3 week stimulation with TGF-β alone led to a reduction in E-cadherin expression, though not statistically significant (data not shown). Though the effects of these pro-fibrotic cytokines upon vimentin expression did not reach statistical significance, expression of this mesenchymal marker trended upward following IL-1β/TNF-α stimulation, and was further increased following the addition of exogenous TGF-β, suggestive of EMT (Figure 2B).
3) Esophageal epithelial exposure to fibroblast conditioned media (FCM) leads to features of EMT in a TNF-α and IL-1β-dependent fashion
To further interrogate epithelial-fibroblast cross-talk, we stimulated EPC2-hTERT cells with media harvested from CEM-stimulated FEF3 cells, which was designated as “fibroblast conditioned media” (FCM). Unconditioned cell culture media not previously in contact with epithelial cells, was applied to FEF3 cells for control conditions. A schematic of the experimental design is shown in Figure 3A. To determine the role of IL-1β and TNF-α in the development of FCM-induced EMT, competitive inhibition experiments were performed in the presence of combinations of anti-TNF-α (Remicade) and anti-IL-1R (Anakinra)[32, 33]. Competitive inhibition of TGF-β signaling was not performed based upon the absence of TGF-β mRNA expression in FCM-stimulated esophageal epithelial cells (data not shown).
Following 3 weeks of culture in FCM, the mRNA expression of E-cadherin was significantly reduced (Figure 3B). This effect was not reversed with competitive inhibition using either anti-TNF-α or anti-IL-1R alone. Remarkably, however, combined anti-TNF-α andanti-IL-1R almost completely rescued epithelial cells from the FCM-induced suppression of E-cadherin. This pattern of expression was also evident using immunofluorescent staining for E-cadherin (Figures 3E–I).
Consistent with EMT, FCM stimulation also enhanced the expression of mesenchymal genes. FCM significantly induced the mRNA expression of vimentin, an effect which was reversible through co-inhibition of TNF-α and IL-1 signaling. Similar to E-cadherin, the effect of FCM upon vimentin expression was not affected by either inhibitor alone (Figure 3C). Though not statistically significant, the effect of FCM stimulation upon αSMA mirrored that of vimentin (Figure 3D). Immunostaining for αSMA demonstrated enhanced expression of this mesenchymal marker in FCM-stimulated cells, which appeared to be rescued by anti-TNF-α and anti-IL-1R, both alone and in combination (Figures 3J–N). Unexpectedly, although anti-TNF-α enhanced the mRNA expression of αSMA, this was not reflected in immunofluorescent staining for αSMA (Figure 3L).
Cell morphology was also altered during the three weeks of FCM exposure. In contrast to control cells, FCM-stimulated epithelial cells developed elongated, spindle-like morphology. Competitive inhibition of TNF-α and IL-1R rescued epithelial cells from these morphologic changes (Figure 3O–Q).
4) Primary EoE esophageal epithelial cells exhibit features of EMT when grown in organotypic cell culture
To explore epithelial and mesenchymal cross-talk within physiologic context, we used primary esophageal epithelial cell lines grown in organotypic cell culture (OTC) models. In the organotypic model, esophageal epithelial cells and fibroblasts grow in physiologic context, where direct cell-cell contact is maintained for over 2 weeks. We hypothesized that OTC-cultured primary esophageal epithelial cells derived from an EoE subject (EPC394) would exhibit enhanced features of EMT, compared to both a non-EoE subject (EPC425) and the EPC2-hTERT cell line. The clinical characteristics of the EoE and control subject are outlined in Table 1.
Using previously published methods, three OTC models were constructed using esophageal epithelial cells (EPC2-hTERT, EoE-EPC394, and non-EoE-EPC425) seeded on FEF3 cells embedded in a collagen matrix. Following differentiation and stratification, cultures were harvested for immunolocalization of E-cadherin, αSMA, and vimentin. Consistent with our findings in cell monolayers, we observed a modest decrease in E-cadherin expression in the EoE cell line, along with increased expression of mesenchymal markers αSMA and vimentin. In contrast, the non-EoE control EPC425 cell line exhibited similar expression of both epithelial and mesenchymal markers compared to the EPC2-hTERT cell line (Figure 4).
Figure 4. Primary esophageal epithelial cells from an EoE subject exhibit fibrogenic behavior compared to non-EoE control when grown in organotypic cell culture (OTC).

Primary esophageal epithelial cells (passage 3) were harvested from subjects 394 (EoE) and 425 (non-EoE), and seeded onto a matrix of FEF3 cells within a collagen matrix. OTC was also constructed using EPC2-hTERT cells. Following epithelial differentiation and stratification, OTC were harvested for immunolocalization of EMT markers. A, B, C: Expression of mesenchymal marker vimentin (yellow) in EPC2-hTERT, EPC394 (EoE) and EPC425 (non-EoE) OTC. D, E, F: E-cadherin expression (red) in EPC2-hTERT, EPC394 (EoE) and EPC425 (non-EoE) grown in OTC. G, H, I: Expression of α-SMA (green) in OTC constructed using EPC2-hTERT, EPC394, and EPC425 cell lines. In all sections, nuclei are counterstained with DAPI (blue). Epithelial (Epi) and subepithelial (Sub) compartments are labeled. Images shown are at 200X magnification.
5) Validation of fibrosis and EMT in vivo in EoE
In order to validate our in vitro organotypic findings, we evaluated the esophageal biopsy samples from the EoE and non EoE subjects from which the EoE and non-EoE primary epithelial cell lines were derived. Trichrome staining revealed that the EoE subject not only exhibited densely packed collagen within the subepithelial compartment, but also had extension of collagen deposition into the papillae (Figure 5A). In contrast, loose collagen fibrils were seen in the subepithlial compartment of the non-EoE subject (Figure 5E). Similar to the findings described by Kagalwalla et al.[9] markers of EMT were detected using immunofluorescence in biopsies from the EoE subject (decreased E-cadherin, increased αSMA and vimentin, Figures 5B–D) compared to the non-EoE subject (Figures 5F–H).
Figure 5. Expression of subepithelial collagen and mesenchymal/epithelial markers in native biopsies from EoE and control subjects used for primary esophageal epithelial cell lines.
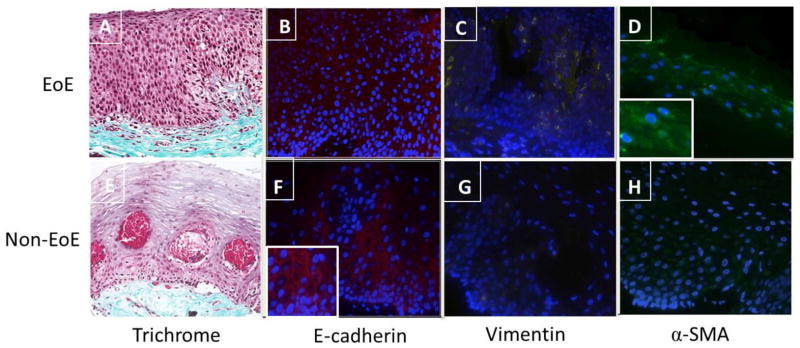
A, E: Trichrome stain of esophageal biopsy from subjects 394 and 425 shows differential subepithelial collagen deposition (blue) in EoE (394) and non EoE (425) subjects. B, F: Reduced epithelial E-cadherin (red) expression in EoE subject compared to normal control. Figure Finset shows magnified detail of E-cadherin in the normal control. C, D: Expression of mesenchymal marker vimentin (yellow) and α-SMA (green) in EoE subject. Figure D inset shows magnified detail of α-SMA expression in the EoE biopsy sample. G, H: Vimentin and α-SMA expression in control subject biopsy. Images are shown at 200X magnification.
Discussion
In this study, we show for the first time that cross-talk between esophageal epithelial cells and esophageal fibroblasts leads to features of EMT in vitro. We demonstrate that two cytokines previously implicated in other models of cross-talk and fibrosis, IL-1β and TNF-α, may play an inciting role in the development of EMT. Our results support the recent report by Kagalwalla et al, which demonstrated that EMT occurs in the esophageal epithelium of EoE subjects[9]. Importantly, however, we now demonstrate that some of the cardinal features of EMT, acquisition of mesenchymal markers and loss of epithelial markers, can occur in a TGF-β independent fashion. Our in vitro organotypic model further corroborates our hypothesis of epithelial-mesenchymal cross-talk, and demonstrates that some features of EMT can occur in the absence of immune cells, tissue injury, or chronic inflammation.
In EoE, TGF-β has been suggested as a primary effector of fibrosis, supported by immunostaining for TGF-β [9] and its signaling molecule phospho-Smad 2/3 [8] in esophageal biopsies of EoE patients. Previous reports demonstrate that granulocyte populations which infiltrate the esophageal mucosa, including mast cells [34]and eosinophils[15] secrete TGF-β, further supporting the notion that this cytokine may play a role in EoE fibrogenesis. The role of TGF-β in myofibroblast development [7, 13] and fibrogenesis [2, 11, 35] has been very well-characterized in other model systems. Interestingly, eosinophils, when co-cultured with fibroblasts, have also been shown to activate fibroblasts by releasing both TGF-α and IL1-α [36].
To our knowledge, this is the first study which looks beyond eosinophil and mast cell-derived cytokines as the major driving force behind tissue remodeling in EoE. In our granulocyte-free model, TGF-β is not secreted by the epithelium or by epithelial-primed fibroblasts. Our results contrast with the findings of others who have previously shown that epithelial-derived TGF-β contributes to the development of EMT via autocrine signaling[37, 38]. In the absence of the inflammatory triggers and granulocytes important to EoE pathogenesis, the possibility that autocrine TGF-β signaling contributes to EoE-associated tissue remodeling cannot be excluded (Figure 6).
Figure 6. Proposed mechanism of esophageal epithelial-mesenchymal cross talk and EMT.
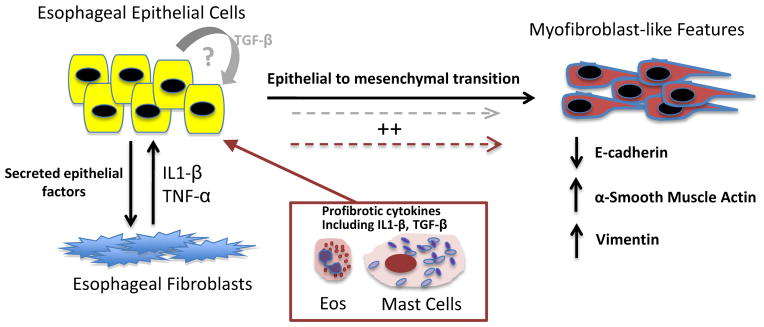
Together, our model suggests that in a genetically predisposed host, factors secreted by esophageal epithelial cells prime esophageal fibroblasts to secrete cytokines including IL-1β and TNF-α. Fibroblast-derived cytokines IL-1β and TNF-α, then stimulate adjacent esophageal epithelial cells to lose epithelial markers (E-cadherin) and gain expression of mesenchymal markers including vimentin and α-SMA. Though our in vitro model suggests that while these features of EMT can occur in the absence of TGF-β, granulocyte-derived TGF-β and IL-1β (maroon box and arrows)and potentially autocrine signaling by epithelial-derived TGF-β (gray arrows),may further enhance the development of EMT in vivo.
Notably, the effect of IL-1β and TNF-α stimulation upon the development of EMT in EPC2-hTERT cells was enhanced by the addition of TGF-β through suppression of epithelial E-cadherin and induction of mesenchymal vimentin expression. This supports previous findings that TGF-β alone is insufficient to induce fibrosis in specific model systems. In a murine model of systemic sclerosis, Mori et al. found that skin fibrosis was induced only when mice were injected with both connective tissue growth factor (CTGF) and TGF-β [39]. Fattough et al found that allergic airway remodeling can occur independently from TGF-β and may depend on IL-13 and other eosinophil derived factors[40]. Overall, our findings may suggest a pathway by which, in a genetically predisposed individual, esophageal epithelial and mesenchymal cross talk participates in the pathogenesis of EMT. The addition of environmental triggers including diet[41] and pollen[42, 43], may lead to the infiltration and activation of innate granulocyte populations (eosinophils, mast cells) which secrete TGF-β and IL1β [36], synergistically enhancing tissue remodeling in EoE(Figure 6).
Unexpectedly, FCM was a more potent inducer of EMT (Figure 3) compared to the combined effect of recombinant cytokines (Figure 2). Although our reductionist approach suggests an important and novel role for IL-1β and TNF-α in FCM-induced EMT, this observation clearly suggests that other soluble mediators play a role in our cell culture model of EMT. Some candidates for future studies include growth factors (including insulin-like growth factor I, epidermal growth factor, basic fibroblast growth factor, platelet-derived growth factor) and cytokines (IL-1, IL-4, IL-6, IL-13, and IL-21) known to activate fibroblasts in other models [16, 44]. Alternatively, the role of Notch signaling in epithelial-mesenchymal cross talk may be explored, as Notch signaling has been previously shown to play a role in EMT in both TGF-β-dependent [45, 46] and TGF-β independent [47] model systems.
The role of IL-1β and TNF-α as profibrotic mediators has been reported in other models. IL-1β has been implicated in pancreatic fibrosis and liver fibrosis. Shen et al. showed that intraperitoneal injections of anti-IL1-R in mice with chronic pancreatitis attenuated pancreatic fibrosis. [48] Furthermore IL-1β −/− mice fed a high-fat diet are protected against steatohepatitis and liver fibrosis compared to wild type controls[49]. Likewise, TNF-α has been implicated in skin EMT [50]. Interestingly many studies suggest that both of these cytokines require TGF-β as a co-stimulant in order to induce fibrosis[51, 52].
Although the use of the FEF3 fetal fibroblast cell line has been validated in organotypic models of esophageal cancer [53, 54], a potential weakness of our study is the exclusive use this fetal cell line. Indeed, it has been proposed that fetal and adult fibroblasts have differential migratory abilities [29] and different responses to TGF-β [31]. In addition, variations in fibroblast phenotype and activation state are known to influence the invasive behavior of the adjacent epithelium in cell culture models of esophageal cancer [55]. However, functional comparisons of esophageal fibroblasts in pediatric EoE have not been reported. Though our results show that primary esophageal fibroblasts from a single EoE patient have similar innate immune responsiveness to CEM compared to the fetal FEF3 cell line (Figures 1F, 1G) future studies will investigate interactions between esophageal epithelial cells from EoE subjects with esophageal fibroblasts from age-matched controls.
Kagalwalla’s study showed that treatment of EoE with dietary restriction or topical corticosteroids (TC) reduced tissue eosinophil load and EMT scores, suggesting that therapies which reduce eosinophil counts ameliorate EoE by reversing EMT. While the precise mechanisms by which dietary restriction and TC improve EoE esophageal inflammation are unknown, it is likely that their anti-inflammatory effects involve esophageal epithelial immune responses. In asthma, the effects of budesonide upon bronchial epithelial cells have been well-described[56–58], and the efficacy of topical corticosteroid therapy in EoE further supports a role for esophageal epithelial cells in EoE pathogenesis. Interestingly, Mulder et al. showed that esophageal epithelial cells can internalize, process, and present ovalbumin to activated T-cells, implicating esophageal epithelial cells as nonprofessional antigen presenting cells in diet-triggered EoE[59]. In the current study, our results now suggest an additional role for esophageal epithelial cells as profibrogenic effector cells in EoE fibrosis. Continued studies using additional primary esophageal epithelial and fibroblast cell lines will be important to further elucidate signaling mechanisms involved in pathogenesis of this complex disease.
Highlights.
Cytokines IL-1β and TNFα play a role in the development of esophageal EMT in vitro.
Features of EMT can occur in the absence of TGF-β in cultured human esophageal epithelial cells.
Esophageal epithelial cells act as effector cells in EoE-associated tissue remodeling in vitro.
Acknowledgments
Funding Sources:
NIH RO1DK087789 (to M.L.W.)
American Partnership for Eosinophilic Disorders (to M.L.W.)
Department of Defense A-16809.2 (to J.M.S.)
NIH/NIDDK P30 Center for Molecular Studies in Digestive and Liver Diseases (P30-DK050306) (to M.L.W.)
5 T32 HD 43021-9 (to A.B.M.)
Abbreviations
- EoE
eosinophilic esophagitis
- EMT
epithelial to mesenchymal transition
- IL-1β
interleukin 1-beta
- TNFα
tumor necrosis factor alpha
- TGFβ
transforming growth factor beta
- αSMA
alpha smooth muscle actin
- CEM
conditioned epithelial media
- FCM
fibroblast conditioned media
- OTC
organotypic cell culture
Footnotes
Conflicts of Interest: None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Noel RJ, Putnam PE, Rothenberg ME. Eosinophilic esophagitis. The New England journal of medicine. 2004;351(9):940–1. doi: 10.1056/NEJM200408263510924. [DOI] [PubMed] [Google Scholar]
- 2.Scholten D, Reichart D, Paik YH, Lindert J, Bhattacharya J, Glass CK, Brenner DA, Kisseleva T. Migration of fibrocytes in fibrogenic liver injury. Am J Pathol. 2011;179(1):189–98. doi: 10.1016/j.ajpath.2011.03.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Keeley EC, Mehrad B, Strieter RM. The role of fibrocytes in fibrotic diseases of the lungs and heart. Fibrogenesis & tissue repair. 2011;4:2. doi: 10.1186/1755-1536-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wada T, Sakai N, Sakai Y, Matsushima K, Kaneko S, Furuichi K. Involvement of bone-marrow-derived cells in kidney fibrosis. Clin Exp Nephrol. 2011;15(1):8–13. doi: 10.1007/s10157-010-0372-2. [DOI] [PubMed] [Google Scholar]
- 5.Hao H, Gabbiani G, Camenzind E, Bacchetta M, Virmani R, Bochaton-Piallat ML. Phenotypic modulation of intima and media smooth muscle cells in fatal cases of coronary artery lesion. Arterioscler Thromb Vase Biol. 2006;26(2):326–32. doi: 10.1161/01.ATV.0000199393.74656.4c. [DOI] [PubMed] [Google Scholar]
- 6.Kalluri R, Neilson EG. Epithelial-mesenchymal transition and its implications for fibrosis. The journal of Clinical Investigation. 2003;112(12):1776–84. doi: 10.1172/JCI20530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hinz B, Phan SH, Thannickal VJ, Galli A, Bochaton-Piallat ML, Gabbiani G. The myofibroblast: one function, multiple origins. Am J Pathol. 2007;170(6):1807–16. doi: 10.2353/ajpath.2007.070112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aceves SS, Newbury RO, Dohil R, Bastian JF, Broide DH. Esophageal remodeling in pediatric eosinophilic esophagitis. The Journal of allergy and clinical immunology. 2007;119(1):206–12. doi: 10.1016/j.jaci.2006.10.016. [DOI] [PubMed] [Google Scholar]
- 9.Kagalwalla AF, Akhtar N, Woodruff SA, Rea BA, Masterson JC, Mukkada V, Parashette KR, Du J, Fillon S, Protheroe CA, Lee JJ, Amsden K, Melin-Aldana H, Capocelli KE, Furuta GT, Ackerman SJ. Eosinophilic esophagitis: Epithelial mesenchymal transition contributes to esophageal remodeling and reverses with treatment. J Allergy Clin Immunol. 2012;129(5):1387–1396. e7. doi: 10.1016/j.jaci.2012.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ohashi S, Natsuizaka M, Wong GS, Michaylira CZ, Grugan KD, Stairs DB, Kalabis J, Vega ME, Kalman RA, Nakagawa M, Klein-Szanto AJ, Herlyn M, Diehl JA, Rustgi AK, Nakagawa H. Epidermal growth factor receptor and mutant p53 expand an esophageal cellular subpopulation capable of epithelial-to-mesenchymal transition through ZEB transcription factors. Cancer research. 2010;70(10):4174–84. doi: 10.1158/0008-5472.CAN-09-4614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Balestrini JL, Chaudhry S, Sarrazy V, Koehler A, Hinz B. The mechanical memory of lung myofibroblasts. Integr Biol (Camb) 2012;4(4):410–21. doi: 10.1039/c2ib00149g. [DOI] [PubMed] [Google Scholar]
- 12.Barrientos S, Stojadinovic O, Golinko MS, Brem H, Tomic-Canic M. Growth factors and cytokines in wound healing. Wound Repair Regen. 2008;16(5):585–601. doi: 10.1111/j.1524-475X.2008.00410.x. [DOI] [PubMed] [Google Scholar]
- 13.Hinz B. Tissue stiffness, latent TGF-beta1 activation, and mechanical signal transduction: implications for the pathogenesis and treatment of fibrosis. Curr Rheumatol Rep. 2009;11(2):120–6. doi: 10.1007/s11926-009-0017-1. [DOI] [PubMed] [Google Scholar]
- 14.Aceves SS, Chen D, Newbury RO, Dohil R, Bastian JF, Broide DH. Mast cells infiltrate the esophageal smooth muscle in patients with eosinophilic esophagitis, express TGF-beta1, and increase esophageal smooth muscle contraction. The Journal of allergy and clinical immunology. 2010;126(6):1198–204. e4. doi: 10.1016/j.jaci.2010.08.050. [DOI] [PubMed] [Google Scholar]
- 15.Minshall EM, Leung DY, Martin RJ, Song YL, Cameron L, Ernst P, Hamid Q. Eosinophil-associated TGF-beta1 mRNA expression and airways fibrosis in bronchial asthma. Am J Respir Cell Mol Biol. 1997;17(3):326–33. doi: 10.1165/ajrcmb.17.3.2733. [DOI] [PubMed] [Google Scholar]
- 16.Fiocchi C, Lund PK. Themes in fibrosis and gastrointestinal inflammation. Am J Physiol Gastrointest Liver Physiol. 2011;300(5):G677–83. doi: 10.1152/ajpgi.00104.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Doerner AM, Zuraw BL. TGF-beta1 induced epithelial to mesenchymal transition (EMT) in human bronchial epithelial cells is enhanced by IL-1beta but not abrogated by corticosteroids. Respir Res. 2009;10:100. doi: 10.1186/1465-9921-10-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Takahashi E, Nagano O, Ishimoto T, Yae T, Suzuki Y, Shinoda T, Nakamura S, Niwa S, Ikeda S, Koga H, Tanihara H, Saya H. Tumor necrosis factor-alpha regulates transforming growth factor-beta-dependent epithelial-mesenchymal transition by promoting hyaluronan-CD44-moesin interaction. J Biol Chem. 2010;285(6):4060–73. doi: 10.1074/jbc.M109.056523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yamauchi Y, Kohyama T, Takizawa H, Kamitani S, Desaki M, Takami K, Kawasaki S, Kato J, Nagase T. Tumor necrosis factor-alpha enhances both epithelial-mesenchymal transition and cell contraction induced in A549 human alveolar epithelial cells by transforming growth factor-beta1. Exp Lung Res. 2010;36(1):12–24. doi: 10.3109/01902140903042589. [DOI] [PubMed] [Google Scholar]
- 20.Selman M, Pardo A. Idiopathic pulmonary fibrosis: an epithelial/fibroblastic cross-talk disorder. Respir Res. 2002;3:3. doi: 10.1186/rr175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chapman HA. Epithelial-mesenchymal interactions in pulmonary fibrosis. Annu Rev Physiol. 2011;73:413–35. doi: 10.1146/annurev-physiol-012110-142225. [DOI] [PubMed] [Google Scholar]
- 22.Davies DE. The role of the epithelium in airway remodeling in asthma. Proc Am Thorac Soc. 2009;6(8):678–82. doi: 10.1513/pats.200907-067DP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lim DM, Narasimhan S, Michaylira CZ, Wang ML. TLR3-mediated NF-{kappa}B signaling in human esophageal epithelial cells. Am J Physiol Gastrointest Liver Physiol. 2009;297(6):G1172–1180. doi: 10.1152/ajpgi.00065.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lim DM, Wang ML. Toll-like receptor 3 signaling enables human esophageal epithelial cells to sense endogenous danger signals released by necrotic cells. Am J Physiol Gastrointest Liver Physiol. 2011;301(1):G91–9. doi: 10.1152/ajpgi.00471.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kalabis J, Wong GS, Vega ME, Natsuizaka M, Robertson ES, Herlyn M, Nakagawa H, Rustgi AK. Isolation and characterization of mouse and human esophageal epithelial cells in 3D organotypic culture. Nature protocols. 2012;7(2):235–46. doi: 10.1038/nprot.2011.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carson FL. Histotechnology: A Self-instructional text. 3. American Society for clinical Pathology Press; 2009. [Google Scholar]
- 27.Liacouras CA, Furuta GT, Hirano I, Atkins D, Attwood SE, Bonis PA, Burks AW, Chehade M, Collins MH, Dellon ES, Dohil R, Falk GW, Gonsalves N, Gupta SK, Katzka DA, Lucendo AJ, Markowitz JE, Noel RJ, Odze RD, Putnam PE, Richter JE, Romero Y, Ruchelli E, Sampson HA, Schoepfer A, Shaheen NJ, Sicherer SH, Spechler S, Spergel JM, Straumann A, Wershil BK, Rothenberg ME, Aceves SS. Eosinophilic esophagitis: updated consensus recommendations for children and adults. The Journal of allergy and clinical immunology. 2011;128(1):3–20. e6. doi: 10.1016/j.jaci.2011.02.040. quiz 21–2. [DOI] [PubMed] [Google Scholar]
- 28.Harada H, Nakagawa H, Oyama K, Takaoka M, Andl CD, Jacobmeier B, von Werder A, Enders GH, Opitz OG, Rustgi AK. Telomerase induces immortalization of human esophageal keratinocytes without p16INK4a inactivation. Molecular Cancer Research: MCR. 2003;1(10):729–38. [PubMed] [Google Scholar]
- 29.Ellis I, Banyard J, Schor SL. Differential response of fetal and adult fibroblasts to cytokines: cell migration and hyaluronan synthesis. Development. 1997;124(8):1593–600. doi: 10.1242/dev.124.8.1593. [DOI] [PubMed] [Google Scholar]
- 30.Ellis IR, Schor SL. Differential effects of TGF-beta1 on hyaluronan synthesis by fetal and adult skin fibroblasts: implications for cell migration and wound healing. Exp Cell Res. 1996;228(2):326–33. doi: 10.1006/excr.1996.0332. [DOI] [PubMed] [Google Scholar]
- 31.Ellis IR, Schor SL. Differential motogenic and biosynthetic response of fetal and adult skin fibroblasts to TGF-beta isoforms. Cytokine. 1998;10(4):281–9. doi: 10.1006/cyto.1997.0294. [DOI] [PubMed] [Google Scholar]
- 32.Juuti-Uusitalo K, Klunder LJ, Sjollema KA, Mackovicova K, Ohgaki R, Hoekstra D, Dekker J, van Ijzendoorn SC. Differential effects of TNF (TNFSF2) and IFN-gamma on intestinal epithelial cell morphogenesis and barrier function in three-dimensional culture. PloS one. 2011;6(8):e22967. doi: 10.1371/journal.pone.0022967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Long-Smith CM, Collins L, Toulouse A, Sullivan AM, Nolan YM. Interleukin-1beta contributes to dopaminergic neuronal death induced by lipopolysaccharide-stimulated rat glia in vitro. Journal of neuroimmunology. 2010;226(1–2):20–6. doi: 10.1016/j.jneuroim.2010.05.030. [DOI] [PubMed] [Google Scholar]
- 34.Aceves SS, Chen D, Newbury RO, Dohil R, Bastian JF, Broide DH. Mast cells infiltrate the esophageal smooth muscle in patients with eosinophilic esophagitis, express TGF-beta1, and increase esophageal smooth muscle contraction. J Allergy Clin Immunol. 2010;126(6):1198–204. e4. doi: 10.1016/j.jaci.2010.08.050. [DOI] [PubMed] [Google Scholar]
- 35.Kisseleva T, Brenner DA. Fibrogenesis of parenchymal organs. Proc Am Thorac Soc. 2008;5(3):338–42. doi: 10.1513/pats.200711-168DR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gomes I, Mathur SK, Espenshade BM, Mori Y, Varga J, Ackerman SJ. Eosinophil-fibroblast interactions induce fibroblast IL-6 secretion and extracellular matrix gene expression: implications in fibrogenesis. The Journal of allergy and clinical immunology. 2005;116(4):796–804. doi: 10.1016/j.jaci.2005.06.031. [DOI] [PubMed] [Google Scholar]
- 37.Ito J, Harada N, Nagashima O, Makino F, Usui Y, Yagita H, Okumura K, Dorscheid DR, Atsuta R, Akiba H, Takahashi K. Wound-induced TGF-beta1 and TGF-beta2 enhance airway epithelial repair via HB-EGF and TGF-alpha. Biochemical and Biophysical Research Communications. 2011;412(1):109–14. doi: 10.1016/j.bbrc.2011.07.054. [DOI] [PubMed] [Google Scholar]
- 38.Gregory PA, Bracken CP, Smith E, Bert AG, Wright JA, Roslan S, Morris M, Wyatt L, Farshid G, Lim YY, Lindeman GJ, Shannon MF, Drew PA, Khew-Goodall Y, Goodall GJ. An autocrine TGF-beta/ZEB/miR-200 signaling network regulates establishment and maintenance of epithelial-mesenchymal transition. Molecular Biology of the Cell. 2011;22(10):1686–98. doi: 10.1091/mbc.E11-02-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mori T, Kawara S, Shinozaki M, Hayashi N, Kakinuma T, Igarashi A, Takigawa M, Nakanishi T, Takehara K. Role and interaction of connective tissue growth factor with transforming growth factor-beta in persistent fibrosis: A mouse fibrosis model. Journal of Cellular Physiology. 1999;181(1):153–9. doi: 10.1002/(SICI)1097-4652(199910)181:1<153::AID-JCP16>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 40.Fattouh R, Jordana M. TGF-beta, eosinophils and IL-13 in allergic airway remodeling: a critical appraisal with therapeutic considerations. Inflammation & allergy drug targets. 2008;7(4):224–36. doi: 10.2174/187152808786848388. [DOI] [PubMed] [Google Scholar]
- 41.Kagalwalla AF, Sentongo TA, Ritz S, Hess T, Nelson SP, Emerick KM, Melin-Aldana H, Li BUK. Effect of six-food elimination diet on clinical and histologic outcomes in eosinophilic esophagitis. Clinical gastroenterology and hepatology: the official clinical practice journal of the American Gastroenterological Association. 2006;4(9):1097–102. doi: 10.1016/j.cgh.2006.05.026. [DOI] [PubMed] [Google Scholar]
- 42.Fogg MI, Ruchelli E, Spergel JM. Pollen and eosinophilic esophagitis. The Journal of allergy and clinical immunology. 2003;112(4):796–7. doi: 10.1016/s0091-6749(03)01715-9. [DOI] [PubMed] [Google Scholar]
- 43.Spergel JM. Eosinophilic oesophagitis and pollen. Clinical and experimental allergy: journal of the British Society for Allergy and Clinical Immunology. 2005;35(11):1421–2. doi: 10.1111/j.1365-2222.2005.02372.x. [DOI] [PubMed] [Google Scholar]
- 44.Wick G, Backovic A, Rabensteiner E, Plank N, Schwentner C, Sgonc R. The immunology of fibrosis: innate and adaptive responses. Trends in Immunology. 2010;31(3):110–9. doi: 10.1016/j.it.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Aoyagi-Ikeda K, Maeno T, Matsui H, Ueno M, Hara K, Aoki Y, Aoki F, Shimizu T, Doi H, Kawai-Kowase K, Iso T, Suga T, Arai M, Kurabayashi M. Notch induces myofibroblast differentiation of alveolar epithelial cells via transforming growth factor-{beta}-Smad3 pathway. American journal of respiratory cell and molecular biology. 2011;45(1):136–44. doi: 10.1165/rcmb.2010-0140oc. [DOI] [PubMed] [Google Scholar]
- 46.Matsuno Y, Coelho AL, Jarai G, Westwick J, Hogaboam CM. Notch signaling mediates TGF-beta1-induced epithelial-mesenchymal transition through the induction of Snai1. The international journal of biochemistry & cell biology. 2012;44(5):776–89. doi: 10.1016/j.biocel.2012.01.021. [DOI] [PubMed] [Google Scholar]
- 47.Namba T, Tanaka KI, Ito Y, Hoshino T, Matoyama M, Yamakawa N, Isohama Y, Azuma A, Mizushima T. Induction of EMT-like phenotypes by an active metabolite of leflunomide and its contribution to pulmonary fibrosis. Cell death and differentiation. 2010;17(12):1882–95. doi: 10.1038/cdd.2010.64. [DOI] [PubMed] [Google Scholar]
- 48.Shen J, Gao J, Zhang J, Xiang D, Wang X, Qian L, Yang L, Zhu S, Wu M, Yu Y, Han W. Recombinant human interleukin-1 receptor antagonist (rhIL-1Ra) attenuates caerulein-induced chronic pancreatitis in mice. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie. 2012;66(2):83–8. doi: 10.1016/j.biopha.2011.11.017. [DOI] [PubMed] [Google Scholar]
- 49.Kamari Y, Shaish A, Vax E, Shemesh S, Kandel-Kfir M, Arbel Y, Olteanu S, Barshack I, Dotan S, Voronov E, Dinarello CA, Apte RN, Harats D. Lack of interleukin-1alpha or interleukin-1beta inhibits transformation of steatosis to steatohepatitis and liver fibrosis in hypercholesterolemic mice. Journal of hepatology. 2011;55(5):1086–94. doi: 10.1016/j.jhep.2011.01.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yan C, Grimm WA, Garner WL, Qin L, Travis T, Tan N, Han YP. Epithelial to mesenchymal transition in human skin wound healing is induced by tumor necrosis factor-alpha through bone morphogenic protein-2. The American journal of pathology. 2010;176(5):2247–58. doi: 10.2353/ajpath.2010.090048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Camara J, Jarai G. Epithelial-mesenchymal transition in primary human bronchial epithelial cells is Smad-dependent and enhanced by fibronectin and TNF-alpha. Fibrogenesis & tissue repair. 2010;3(1):2. doi: 10.1186/1755-1536-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Doerner AM, Zuraw BL. TGF-beta1 induced epithelial to mesenchymal transition (EMT) in human bronchial epithelial cells is enhanced by IL-1beta but not abrogated by corticosteroids. Respiratory research. 2009;10:100. doi: 10.1186/1465-9921-10-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Okawa T, Michaylira CZ, Kalabis J, Stairs DB, Nakagawa H, Andl CD, Johnstone CN, Klein-Szanto AJ, El-Deiry WS, Cukierman E, Herlyn M, Rustgi AK. The functional interplay between EGFR overexpression, hTERT activation, and p53 mutation in esophageal epithelial cells with activation of stromal fibroblasts induces tumor development, invasion, and differentiation. Genes & development. 2007;21(21):2788–803. doi: 10.1101/gad.1544507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Michaylira CZ, Wong GS, Miller CG, Gutierrez CM, Nakagawa H, Hammond R, Klein-Szanto AJ, Lee JS, Kim SB, Herlyn M, Diehl JA, Gimotty P, Rustgi AK. Periostin, a cell adhesion molecule, facilitates invasion in the tumor microenvironment and annotates a novel tumor-invasive signature in esophageal cancer. Cancer Research. 2010;70(13):5281–92. doi: 10.1158/0008-5472.CAN-10-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Grugan KD, Miller CG, Yao Y, Michaylira CZ, Ohashi S, Klein-Szanto AJ, Diehl JA, Herlyn M, Han M, Nakagawa H, Rustgi AK. Fibroblast-secreted hepatocyte growth factor plays a functional role in esophageal squamous cell carcinoma invasion. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(24):11026–31. doi: 10.1073/pnas.0914295107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gallelli L, Pelaia G, Fratto D, Muto V, Falcone D, Vatrella A, Curto LS, Renda T, Busceti MT, Liberto MC, Savino R, Cazzola M, Marsico SA, Maselli R. Effects of budesonide on P38 MAPK activation, apoptosis and IL-8 secretion, induced by TNF-alpha and Haemophilus influenzae in human bronchial epithelial cells. International journal of immunopathology and pharmacology. 2010;23(2):471–9. doi: 10.1177/039463201002300209. [DOI] [PubMed] [Google Scholar]
- 57.Huang YC, Leyko B, Frieri M. Effects of omalizumab and budesonide on markers of inflammation in human bronchial epithelial cells. Annals of allergy, asthma & immunology: official publication of the American College of Allergy, Asthma, & Immunology. 2005;95(5):443–51. doi: 10.1016/S1081-1206(10)61170-2. [DOI] [PubMed] [Google Scholar]
- 58.Strandberg K, Palmberg L, Larsson K. Effect of budesonide and formoterol on IL-6 and IL-8 release from primary bronchial epithelial cells. The Journal of asthma: official journal of the Association for the Care of Asthma. 2008;45(3):201–3. doi: 10.1080/02770900801890372. [DOI] [PubMed] [Google Scholar]
- 59.Mulder DJ, Pooni A, Mak N, Hurlbut DJ, Basta S, Justinich CJ. Antigen presentation and MHC class II expression by human esophageal epithelial cells: role in eosinophilic esophagitis. The American journal of pathology. 2011;178(2):744–53. doi: 10.1016/j.ajpath.2010.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]