

Abstract
Glial reaction is a common feature of neurodegenerative diseases. Recent studies have suggested that reactive astrocytes gain neurotoxic properties, but exactly how reactive astrocytes contribute to neurotoxicity remains to be determined. Here, we identify lipocalin 2 (lcn2) as an inducible factor that is secreted by reactive astrocytes and that is selectively toxic to neurons. We show that lcn2 is induced in reactive astrocytes in transgenic rats with neuronal expression of mutant human TAR DNA-binding protein 43 (TDP-43) or RNA-binding protein fused in sarcoma (FUS). Therefore, lcn2 is induced in activated astrocytes in response to neurodegeneration, but its induction is independent of TDP-43 or FUS expression in astrocytes. We found that synthetic lcn2 is cytotoxic to primary neurons in a dose-dependent manner, but is innocuous to astrocytes, microglia, and oligodendrocytes. Lcn2 toxicity is increased in neurons that express a disease gene, such as mutant FUS or TDP-43. Conditioned medium from rat brain slice cultures with neuronal expression of mutant TDP-43 contains abundant lcn2 and is toxic to primary neurons as well as neurons in cultured brain slice from WT rats. Partial depletion of lcn2 by immunoprecipitation reduced conditioned medium-mediated neurotoxicity. Our data indicate that reactive astrocytes secrete lcn2, which is a potent neurotoxic mediator.
Keywords: amyotrophic lateral sclerosis, astrocytosis
Glial reaction is a common feature of neurodegenerative diseases, including amyotrophic lateral sclerosis (ALS), frontotemporal lobar degeneration (FTLD), Huntington disease, Parkinson disease, and Alzheimer’s disease. Astrocytes and microglia become reactive during neurodegenerative processes (1, 2), and activated astrocytes may exhibit differential expression of astrocytic receptors, transporters, and transmitters; metabolic changes; and altered synthesis and release of proteins, chemokines, and cytokines (3–6). Controlled activation of astrocytes is considered beneficial to neurons (7), but overactive astrocytes can be harmful (8). Astrocytosis in neurodegeneration has been intensively studied, but exactly how reactive astrocytes contribute to neurotoxicity remains to be determined.
Reactive astrocytes may lose neuroprotective functions or gain neurotoxic properties in neurodegenerative diseases. Astrocytes are responsible for the reuptake of the neurotransmitter glutamate, which is accomplished by excitatory amino acid transporter 2 (EAAT2 or GLT1) (9). In mice, GLT1 deficiency leads to synaptic glutamate accumulation and subsequent excitotoxicity (9). Astrocytes become reactive during neurodegeneration and gradually lose GLT1 function and expression (10–12). Stimulating GLT1 expression with antibiotics protects motor neurons in an ALS model (13). Because reactive astrocytes may lose their neuroprotective abilities, as observed during GLT1 deficiency (10–12), supplementing normal astrocytes to the areas of active neuropathology is expected to have a therapeutic effect. Indeed, transgenic rats with ALS are ameliorated by transplanting glia-restricted progenitor cells (14), which can differentiate into normal astrocytes. In addition to the loss of neuroprotective functions, increasing evidence strongly suggests that reactive astrocytes also gain neurotoxic properties. Cultured astrocytes expressing mutant superoxide dismutase 1 (SOD1) secrete unknown neurotoxic factors into the medium, and adding conditioned medium (CM) to cultured motor neurons can cause cell death (15, 16). Overexpression of mutant SOD1 in rats causes progressive loss of motor neurons and severe astrocyte activation (17). Reactive astrocytes isolated from mutant SOD1 transgenic rats damage cultured motor neurons (18), likely because of the secretion of unknown factors.
To identify neurotoxic molecules secreted by reactive astrocytes, we examined the organotypic cultures of brain slices from transgenic rats that express a mutant form of TAR DNA-binding protein 43 (TDP-43) in neurons. We reasoned that this model maintains the intrinsic environment of both neurons and glia from the CNS in vitro, thereby maximizing the likelihood of elucidating molecules that are relevant to neurodegeneration in vivo. Using 2D gel electrophoresis followed by mass spectrometry, we analyzed the composition of proteins that were secreted into culture medium from rat brain slices and identified lipocalin 2 (lcn2) as an inducible molecule secreted by reactive astrocytes that mediates neurotoxicity. We validated lcn2 as an astrocytic factor in transgenic rats expressing mutant TDP-43 or RNA-binding protein fused in sarcoma (FUS). Collectively, our data demonstrate that lcn2 is a potent neurotoxic factor secreted by reactive astrocytes.
Results
lcn2 Secretion Is Induced in Cultured Rat Brain Slice Expressing Mutant TDP-43.
To identify inducible factors secreted by activated glia in response to neurodegeneration, we used cultured organotypic rat brain slices. This model maintains the intrinsic interactions between neurons and glia, and secreted proteins can be enriched from culture medium and identified with mass spectrometry. To identify inducible factors that are true indicators of glial response to neurodegeneration rather than specific substrates of a given disease gene, we chose to express mutant human TDP-43 specifically in rat forebrain neurons. In our TDP-43 and calcium/calmodulin-dependent protein kinase II α (Camk2α)-tetracycline-responsive transactivator (tTA) double-transgenic rats (1, 2, 19), the Camk2α promoter drives tTA expression in forebrain neurons, and the tTA in turn drives expression of ALS-associated mutant TDP-43M337V, which is under the control of the tetracycline-responsive element (TRE) (Fig. 1 and Fig. S1). In contrast, human TDP-43 was not detected in Camk2α-tTA single-transgenic rats (Fig. 1A). Neuronal expression of mutant TDP-43 induced substantial microglia and astrocyte activation (Fig. 1 C–F), indicating that Camk2α-tTA/TRE-TDP43M337V transgenic rats are an appropriate model for isolating neurotoxic factors produced by reactive glia.
Fig. 1.

lcn2 is secreted from cultured brain slice expressing mutant human TDP-43. (A and B) Immunohistochemistry in rat cortex revealed that human TDP-43 with M337V substitution (hTDP43) was expressed in Camk2α-tTA/TRE-TDP43M337V double (B: M337V), but not Camk2α-tTA single (A: tTA), transgenic rats. (C–F) Immunofluorescent staining demonstrated that microglia (C and D) and astrocytes (E and F) were substantially activated in M337V rats (D and F) compared with tTA rats (C and E). Rats were analyzed at the age of 55 d. (Scale bar: A–F, 100 µm.) (G) Schematic diagrams of the protocols for culturing brain slices and detecting secreted proteins in culture medium. (H and I) Representative images show that the fluorescent intensity of a protein spot corresponding to lcn2 was increased in the culture medium of a M337V brain slice (I, red) compared with a tTA brain slice culture (H, green). The entire 2D gel images are shown in Fig. S2. The brain slices were cultured for 4 d in the presence of Dox to allow for recovery from slicing and were then cultured in Dox-free medium to allow hTDP43 expression. After 8 days in vitro (DIV), brain slices were further cultured in serum-free medium for 24 h and the culture medium was collected for analysis of secreted proteins by 2D gels and MS. (J) Immunoblotting detected lcn2 in the culture medium. Each lane was loaded with 10 µL culture medium. Each well of a six-well plate was cultured with three slices from the same regions of tTA or M337V rat brains.
As outlined in Fig. 1G, postnatal day 3 (P3) rat forebrain was cut into coronal slices at 300-µm intervals and every three-brain-slice was cultured on a nest in a 2.5-cm dish in doxycycline (Dox)-containing medium for 4 d and then switched to Dox-free medium to allow neuronal expression of mutant TDP-43. After 8 d in vitro (20), brain slices were switched to serum-free medium. After culturing overnight, the medium was harvested for protein composition analysis. Proteins that were precipitated from the culture medium of Camk2α-tTA single or Camk2α-tTA/TRE-TDP43M337V double-transgenic slices were labeled with different fluorescent dyes and mixed in a 1:1 ratio. Fluorescent-labeled proteins were resolved on 2D gels, and selected protein spots were cut out for analysis by MS when they met the following three criteria. First, the spots should be present in both WT and mutant brain slice cultures. This criterion eliminated neuronal factors that were specifically induced by mutant TDP-43 expression in the same cells. Second, the spots needed to have a stronger signal in mutant slices than in WT. When cultured in vitro, brain slices undergo damage and restructuring, which trigger glial reactions even in WT slices; however, this reaction should be greater in slices that express mutant TDP-43. Third, the spots should be moderately abundant in culture medium. This criterion was designed to eliminate the most abundant housekeeping proteins that may be released due to tissue damage in culture. Additionally, we sought signaling molecules that are induced in response to neuronal damage caused by short-term (5 d) expression of mutant TDP-43 that are expected to be secreted at moderate levels. Using these three criteria, we selected four spots and identified them as mitofusin-1 [protein molecular weight (MW) 83896; protein pI 6.1; accession no. gi|149048649], rCG60414 isoform CRA_b (MW 28373; pI 6.5; accession no. gi|149033819), neutrophil gelatinase-associated lipocalin (NGAL or lcn2; MW 22461; pI 7.7; accession no. gi|306526224), and lcn2 (MW 20482; pI 7.9; accession no. gi|225733895). We examined the responsiveness of these four proteins to mutant TDP-43 expression in rat brain slice cultures and determined lcn2 as the protein whose secretion was significantly increased in mutant brain slices compared with WT (Fig. 1 H–J and Fig. S2). lcn2 is often glycosylated, which alters its MW and pI. Accordingly, two lcn2 spots were identified in CM.
lcn2 Is Induced in Reactive Astrocytes in Transgenic Rats Expressing Mutant TDP-43, FUS, or SOD1.
To validate the inducibility of lcn2 gene expression in vivo, we first examined Camk2α-tTA/TRE-TDP43M337V double-transgenic rats that selectively expressed mutant human TDP-43 in forebrain neurons. Immunostaining revealed that lcn2 was induced in the brain of the double-transgenic rats compared with the tTA single-transgenic rat (Fig. 2 A and B). Immunofluorescence staining revealed that lcn2 colocalized with the astrocyte marker glial acid fibrillary protein (GFAP, Fig. 2 C–E) and did not colocalize with the microglia marker ionized calcium binding adaptor molecule 1 (Iba1, Fig. 2 F–H), thus indicating that lcn2 was induced in reactive astrocytes in the brain of TDP-43 transgenic rats. To determine whether lcn2 is also induced during motor neuron degeneration, we examined lcn2 expression in the spinal cord of double-transgenic rats [choline acetyltransferase (Chat)-tTA/TRE-TDP-43M337V] that express mutant TDP-43M337V and develop motor neuron degeneration and ALS phenotypes (19). Lcn2 was markedly up-regulated in the spinal cord of mutant TDP-43 transgenic rats compared with control rats (Fig. 2 I and J). Similar to its localization in the forebrain, lcn2 colocalized with the astrocyte marker GFAP (Fig. 2 K–M).
Fig. 2.

lcn2 is induced in reactive astrocytes in transgenic rats expressing mutant TDP-43, FUS, or SOD1. (A and B) Immunohistochemistry in rat cortex revealed significant lcn2 induction in Camk2α-tTA/TRE-TDP43M337V double (B, CamK2α/M337V), but not Camk2α-tTA single (A, CamK2α-tTA), transgenic rats. Rats were analyzed at 55 d of age. (C–H) Double-labeling fluorescent staining revealed that lcn2 (C and F) colocalized with GFAP (D and E) but not with Iba1 (G and H). (I and J) Immunohistochemistry revealed that lcn2 was substantially up-regulated in the spinal cord of ChAT-tTA/TRE-TDP43M337V double-transgenic rats (ChAT/M337V) compared with ChAT-tTA single-transgenic rats. (K–M) Immunofluorescence staining revealed that lcn2 colocalized with GFAP in ChAT/M337V rat spinal cord. ChAT-tTA and ChAT/M337V rats were analyzed at 58 d of age. (Scale bar: A and B, 100 µm; C–H, 30 µm; I and J, 200 µm; and K–M, 50 µm.) (N and O) Immunofluorescence staining revealed that lcn2 was diffusely expressed in mutant rat cortex at disease end-stages. (Scale bar: 30 µm.) (P) Immunoblotting revealed that lcn2 was up-regulated in rats expressing mutant TDP-43 or FUS. Rat frontal cortex was analyzed for lcn2 expression. (Q) Quantitative PCR revealed that lcn2 was up-regulated in rats expressing mutant TDP-43, FUS, or SOD1. Lcn2 mRNA was normalized to L17 mRNA and was calculated as a ratio of mutant rats to age-matched normal littermates. Data are mean ± SEM (n = 3–5). (R) ELISA lcn2 levels in the CSF of tTA and M337V transgenic rats. Data are mean ± SEM (n = 7), *P < 0.05.
Using immunoblotting, we further examined the effects of disease progression on lcn2 induction in FUS and TDP-43 transgenic rats. Lcn2 was substantially induced at disease onset and gradually increased as neurodegeneration progressed in the brain of transgenic rats that specifically expressed either mutant FUS or TDP-43 in forebrain neurons (Fig. 2P). Immunofluorescence staining revealed that lcn2 was diffusely expressed in rats at advanced disease stages (Fig. 2 N and O). Quantitative PCR confirmed that lcn2 was significantly up-regulated in the brain of rats developing FTLD phenotypes and in the spinal cord of rats developing ALS phenotypes (Fig. 2Q). lcn2 was also induced in transgenic rats expressing mutant SOD1 (Fig. 2Q). As disease progressed in mutant TDP-43 transgenic rats, lcn2 was progressively elevated in the cerebrospinal fluid (CSF, Fig. 2R), suggesting that lcn2 in the CSF is a useful marker of disease progression, at least in this rat model.
Megalin and brain-type organic cation transporter [also called solute carrier family 22 member 17 (SLC22A17)] are two known receptors for lcn2 (21, 22). We examined the expression of megalin and SLC22A17 in mutant TDP-43 transgenic rats. Lcn2 and its receptor megalin were barely detectable in normal rats (Fig. 3). In contrast, both lcn2 and megalin were markedly increased in rats expressing mutant TDP-43 (Fig. S3). Like lcn2, megalin colocalized with GFAP but not Iba1 (Fig. S3), indicating that the lcn2 receptor megalin was induced in reactive astrocytes. Unlike megalin, SLC22A17 was robustly expressed in neurons but not in quiescent astrocytes in normal rats (Fig. S3). Upon neuronal expression of mutant TDP-43, SLC22A17 appeared to be induced in reactive astrocytes and continued to be expressed in diseased neurons (Fig. S3). Thus, expression of the lcn2 receptors megalin and SLC22A17 were both induced in reactive astrocytes, although SLC22A17 was also expressed in normal neurons.
Fig. 3.

lcn2 is selectively toxic to neurons. (A–F) Phase-contrast micrographs show that synthetic lcn2 induced progressive neuronal death. Cortical neurons were prepared from WT SD rat embryos and were cultured at low density for 10 d before lcn2 or sham treatment. (Scale bar: A–F, 50 µm.) (G) Cell counting revealed that lcn2 induced significant neuronal death. Cortical neurons were cultured in gridded chambers and examined daily under a microscope. The fates of chosen neurons were monitored throughout lcn2 treatment. Data are mean ± SEM (n = 6), *P < 0.05. (H–L) Micrographs show a dose-dependent response of cortical neurotoxicity to lcn2. Cortical neurons were grown at a high density and treated with varying doses of lcn2 for 4 d. Cell viability was examined with a cell Live/Dead assay. (Scale bar: H–L, 50 µm.) (M) Cell counting was performed to count the number of cortical neurons after lcn2 treatment. Data are mean ± SEM (n = 6), *P < 0.05. (N) Cell Live/Dead assay revealed the susceptibility of motor neurons to lcn2 toxicity. Mixed neurons were prepared from WT SD rat embryos and cultured for 10 d before they were treated with lcn2 for 4 d. Data are mean ± SEM (n = 7), *P < 0.05. (O–Q) Cell Live/Dead assay revealed the susceptibility of glial cells to lcn2 toxicity. Glial cells were prepared from WT SD rats (P3) and separated to enrich astrocytes, microglia, or oligodendrocytes. Enriched glial cells were treated with lcn2 for 4 d and analyzed for viability. Data are mean ± SEM (n = 5), *P < 0.05.
lcn2 Is Selectively Toxic to Neurons.
lcn2 has been reported to stimulate apoptosis signaling pathways in blood cells (22–24). To determine whether lcn2 is also toxic to CNS cells, we added synthetic lcn2 to neuron and glia cultures. Synthetic lcn2 induced cortical neuron death in a time- (Fig. 3 A–G) and dose-dependent manner (Fig. 3 H–M), with a lethal concentration 50 of about 100 ng/mL (4.5 nM). A similar neurotoxic potency was observed for spinal cord neurons (Fig. 3N), cerebellar granule neurons, and dopaminergic neurons (Fig. S4). lcn2 toxicity is correlated with cell density (25). Cells cultured at low density were highly sensitive to lcn2 (Fig. 3 A–G), whereas cells of high density were less sensitive to lcn2 (Fig. 3 H–M). In contrast, lcn2 did not induce a detectable loss of primary astrocytes, microglia, and oligodendrocytes when it was applied at 1 µg/mL, the highest dose tested (Fig. 3 O–Q). These results indicate that lcn2 is selectively toxic to neurons.
lcn2 is reported to induce apoptosis in nonneuronal cells through iron depletion (22). We examined the effects of iron and iron-loaded transferrin on lcn2 cytotoxicity in primary neurons. Although iron ion induced a cell death in primary neurons, both iron and transferrin did not produce a detectable effect on lcn2 toxicity to neurons (Fig. S5). lcn2 may produce distinct effects in different cell types.
To determine whether disease gene-expressing neurons had enhanced sensitivity to lcn2 cytotoxicity, we added lcn2 to cultured neurons that express mutant FUSR521C (Fig. 4). Expression of mutant FUS further sensitized neurons to lcn2 toxicity (Fig. 4). Under our culture conditions, expression of mutant FUS did not cause significant neuronal death compared with control neurons that express tTA alone (Fig. 4G). Although applying lcn2 to control neurons caused some neurons to die, applying lcn2 to mutant FUS-expressing neurons caused more neurons to die. Thus, expression of mutant FUS sensitizes neurons to lcn2 toxicity (Fig. 4). We further examined whether disease gene expression sensitized motor neurons to lcn2 toxicity. Similar to cortical neurons, spinal motor neurons were sensitized to lcn2 toxicity when they expressed human TDP-43 with a pathogenic mutation (Fig. S6).
Fig. 4.
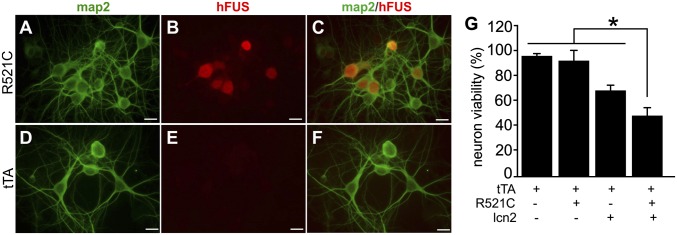
Expression of mutant FUS sensitizes cortical neurons to lcn2 toxicity. (A–F) Double immunofluorescent labeling revealed that human FUS with R521C substitution (hFUS) colocalized with the neuronal marker MAP2 in cortical neurons. hFUS expression was detected in the neurons of CamK2α-tTA/TRE-FUS-R521C double-transgenic rats (R521C) but not in the neurons of CamK2α-tTA single-transgenic rats (tTA). Enriched cortical neurons were prepared from rat embryos (E19) and cultured in the absence of Dox to allow for mutant hFUS expression. Neurons were examined for transgene expression after 10 d in culture. (Scale bar: A–F, 20 µm.) (G) Cell viability assay revealed the susceptibility of neurons to lcn2 toxicity. Enriched cortical neurons were cultured for 10 d and then treated with synthetic lcn2 (10 ng/mL) for 4 d before the viability assay. Data are mean ± SEM (n = 7), *P < 0.05.
lcn2 Is a Potent Mediator of Cytotoxicity in the CNS.
To validate lcn2 as a potent mediator of neurotoxicity in CM, we depleted lcn2 from CM by immunoprecipitation and tested the neurotoxic properties of CM with or without lcn2. We cultured brain slices from Camk2α-tTA/TRE-GFP double-transgenic rats to use in cell viability assays (Fig. 5 A–G). lcn2 was selectively induced in reactive astrocytes in response to neurodegeneration caused by mutant TDP-43 expression and was abundant in CM from brain slice cultures (Figs. 1J, 2P, and 5H). After immunodepletion, lcn2 was largely, although not completely, depleted from CM (Fig. 5H, lane 2). As expected, CM-mediated cytotoxicity to neurons was significantly mitigated by partial lcn2 depletion from CM (Fig. 5 I–L). The concentration of lcn2 in CM was ∼8 ng/mL, which is far above its toxic dosage (Fig. 5M). These results demonstrate that lcn2 is a significant mediator of neurotoxicity in CM.
Fig. 5.

lcn2 in CM from rat brain slice cultures is a potent mediator of neurotoxicity. (A–F) Representative images show that cortical neurons are sensitive to CM from brain slice cultures expressing mutant human TDP-43 (M337V-CM). Hippocampal slices (300 µm) were prepared from CamK2α-tTA/TRE-GFP double-transgenic rats (P1) that expressed GFP in neurons. Hippocampal slices were cultured in normal medium for 4 d before they were cultured in CM collected from brain slice cultures carrying CamK2α-tTA single (tTA) or Camk2α-tTA/TRE-TDP43M337V double (M337V) transgenes. Arrows indicate two GFP-labeled neurons that were lost by 10 d in culture. (Scale bar: A–F, 40 µm.) (G) Cell viability assay was used to quantify CM-induced cytotoxicity in rat brain slices expressing mutant human TDP-43. GFP-labeled neuron viability in hippocampal slices was determined after culture in CM for 10 d. Data are mean ± SEM (n = 5), *P < 0.05. (H) Immunoblotting revealed that lcn2 was partially depleted from the CM by immunoprecipitation (IP). CM from M337V slice cultures was immunoprecipitated with an lcn2 antibody, and the efficiency of lcn2 IP was examined by immunoblotting. Each lane was loaded with 10 µL CM. (I–K) Representative images illustrate the sensitivity of cortical neurons to CM with or without lcn2 depletion. Neurons were examined for viability after culture in CM for 10 d. (Scale bars: I–K, 40 µm.) (L) Cell Live/Dead assay revealed that CM-induced cytotoxicity was reduced after lcn2 immunoprecipitation. Data are mean ± SEM (n = 7), *P < 0.05. (M) ELISA revealed the levels of rat lcn2 in CM from tTA or M337V rat brain slice cultures. Data are mean ± SEM (n = 5), *P < 0.01.
lcn2 Is Up-regulated in the Brain of FTLD Patients.
To further validate lcn2 as a mediator of neurotoxicity, we examined its expression in brain tissue from three patients with FTLD and two control subjects without neurological disease (Fig. S7). As a secretory protein, lcn2 can be clearly localized at early disease stages in TDP-43 transgenic rats (Fig. 2 C–M), but lcn2 was diffusely expressed at advanced disease stages in the transgenic rats (Figs. 2 N and O and 6K). We anticipated difficulty in pinpointing lcn2 localization in the brain of FTLD patients. Compared with the control subjects (Fig. S7 A–C), FTLD patients exhibited up-regulated lcn2 expression and punctate lcn2 immunostaining partially colocalized with GFAP immunostaining (Fig. S7 D–I).
Fig. 6.

Schematic diagram illustrating the hypothesized astrocytic secretion of lcn2 in response to neurodegeneration. Degenerating neurons activate quiescent astrocytes, and reactive astrocytes secrete lcn2 to promote neuronal death. Reactive astrocytes may also secrete lcn2 to promote astrocyte and microglia activation.
Discussion
Increasing evidence has suggested that reactive astrocytes gain neurotoxic properties. Reactive astrocytes isolated from ALS rats induce motor neuron death in vitro, which is likely mediated by astrocytic secretion of unidentified factors into culture medium (15, 16, 18). In this study, we identified lcn2 as a potent mediator of astrocytic neurotoxicity. lcn2 is secreted from cultured rat brain slices that specifically express mutant TDP-43 in forebrain neurons and develop rapid and severe astrocyte and microglia activation (2). Second, lcn2 is likely secreted from reactive astrocytes. lcn2 is selectively induced in reactive astrocytes in the brain and spinal cord of transgenic rats that express mutant TDP-43 restrictedly in neurons, indicating that its induction is a response of astrocytes to the degenerative changes initiated in neurons and does not require the expression of disease genes in astrocytes themselves. Third, synthetic lcn2 is highly toxic to neurons but is not toxic to astrocytes, microglia, and oligodendrocytes. Fourth, lcn2 is particularly toxic to unhealthy neurons expressing a disease gene such as mutant FUS or TDP-43 that is associated with ALS. Fifth, CM containing secreted lcn2 induces neuronal death in primary neuron culture and in organotypic slice culture, and the neurotoxicity is significantly reduced after lcn2 is depleted from CM. Taken together, these data demonstrate that lcn2 is a potent molecule secreted by reactive astrocytes and suggest that lcn2 may be a significant contributor to neuron death in several disease states.
lcn2 belongs to the lipocalin family, which is characterized by the ability to bind and transport lipids and other hydrophobic molecules (26–29). In culture, lcn2 induces blood cell death by stimulating an apoptotic signaling pathway (22–24). Interestingly, lcn2 is secreted from cultured astrocytes upon lipopolysaccharide stimulation and can serve as an autocrine mediator and stimulate surrounding astrocytes and microglia to become reactive (4, 30). Although disruption of lcn2 expression in mice does not result in detectable phenotypes (31), it is essential for the progression of renal lesions in mouse models (32). Recent studies have demonstrated that lcn2 is increased in the CSF of patients with Alzheimer’s disease (33), and its concentration is also indicative of disease progression in rat models of neurodegeneration (Fig. 2R). lcn2 is also up-regulated in the frontal cortex of patients with FTLD (Fig. 6). Further extending these findings, our in vitro data demonstrate that lcn2 is induced and secreted by reactive astrocytes under neurodegenerative conditions and is a potent mediator of neuronal toxicity. Our findings suggest that this function of lcn2 likely plays a significant role in neurodegenerative diseases.
The inducibility of lcn2 during neuronal death raises the possibility that lcn2 could be developed as a marker for the diagnosis and prognosis of neurological diseases. Lcn2 is a small secretory protein that is resistant to degradation. It is readily detected in urine and is used as an indicator of renal lesions (32). In the CNS, lcn2 secreted by reactive astrocytes may quickly diffuse throughout tissue. Indeed, lcn2 expression was immunohistochemically detected only in certain disease stages in transgenic rats, although immunoblotting showed steady increases in expression (Fig. 2). The lipocalin family comprises 20 small, soluble, often secreted proteins (23). Because lcn2 shares homologous sequences with other family members, antibodies against lcn2 often recognize other lipocalin members as well. We tested several lcn2 antibodies and found only one that was specific to rat lcn2. To further study the roles of lcn2 and to determine whether lcn2 can be used as a biomarker for neurological diseases, better antibodies against lcn2 are needed in the future.
lcn2 is known to act through the target cell receptors megalin and SLC22A17 (21, 22). In the brain of normal Sprague–Dawley (SD) rats, we detected substantial SLC22A16 neuronal expression and minimal megalin expression on glia (Fig. S3). In diseased rat brain, megalin and SLC22A17 were both up-regulated in reactive astrocytes (Fig. S3). Because there is no specific antagonist for lcn2, it remains to be determined whether lcn2 mediates astrocytic neurotoxicity by binding to specific receptors. However, lcn2 is involved in the disease of several peripheral organs, including kidney and liver, and tremendous efforts are being devoted to developing effective antagonists for lcn2. These efforts may also lead to developing a treatment for related neurological diseases.
During neuronal cell death, unhealthy neurons interact with quiescent astrocytes to promote astrocyte activation. Reactive astrocytes secrete lcn2, which facilitates neuronal cell death and may also promote the activation of quiescent astrocytes and microglia (4, 5) (Fig. 6). In summary, our findings suggest that reactive astrocytes secrete lcn2, which accelerates or propagates neuronal cell death.
Methods
Generation of Transgenic Rats.
Camk2α-tTA, ChAT-tTA, TRE-TDP43M337V, and TRE-FUSR521C rats were created as described previously (1, 19, 34). These transgenic rats have been deposited to the Rat Resource and Research Center (RRRC) (www.rrrc.us) for free distribution to academic investigators. Similar to the TRE-FUSR521C transgenic construct, the TRE-GFP construct was built by replacing the FUSR521C ORF with the GFP ORF. TRE-GFP transgenic founder rats were created by injecting the transgenic construct into the pronuclei of single-cell, fertilized rat embryos. TRE-GFP transgenic rats were identified by PCR analysis of tail DNA with the following primers: 5′-CACTACCTGAGCACCCAGTC-3′ (forward) and 5′-TGGTGATACAAGGGACATCT-3′ (reverse). A single expression line was established and used in this study. SOD1G93A transgenic rats were obtained from Taconic and were maintained on an SD genomic background. In our colony, SOD1G93A rats developed paralysis by 195 d of age and reached disease end-stage at 200 d of age.
Because the TRE promoter lacked intrinsic transcription activity and needed to be activated by tTA, all TRE-driven transgenes were bred onto a Camk2α-tTA or ChAT-tTA transgenic background. The expression of TRE-driven transgenes depended on tTA activity and was regulated by Dox. Breeding and postnatal rats received Dox in drinking water (50 µg/mL) to suppress TRE-driven transgene expression. To induce disease phenotypes in adult rats, Camk2α-tTA/TRE-TDP43M337V rats were deprived of Dox at the age of 35 d, and Camk2α-tTA/TRE-FUSR521C rats were deprived of Dox at the age of 30 d (1). To induce rapid lcn2 induction in reactive astrocytes, ChAT-tTA/TRE-TDP43M337V rats were deprived of Dox at 40 d of age so that ALS phenotypes were induced by the age of 60 d.
Quantitative PCR and ELISA Analyses.
lcn2 mRNA in transgenic rats was measured and calculated as the ratio to that in age-matched nontransgenic littermates. Total RNA was extracted from rat tissues with an Oligotex mRNA kit (Qiagen) and reverse-transcribed into cDNA pools for quantitative PCR. A fragment of rat cDNA spanning a large intron was amplified with a pair of primers: 5′-CTGGCAGCGAATGCGGTCCA-3′ (forward) and 5′-TGTTCTGATCCAGTAGCGAC-3′ (reverse). A fragment of rat L17 cDNA was simultaneously amplified as an internal control with the primers 5′-TGGTTCGCTACTCCCTTGAC-3′ (forward) and 5′-CTTGATGGCCTGGGCAGTT-3′ (reverse). lcn2 mRNA was normalized to L17 mRNA and was calculated as a ratio of mutant rats to normal control rats. Rat lcn2 protein in the CM of rat brain slice cultures and the CSF of transgenic rats was quantified by ELISA following the manufacturer’s instruction (KA0560; Abnova).
Immunohistochemistry and Immunofluorescent Staining.
Immunohistochemistry and immunofluorescence staining were done as described in our publications (1, 19, 34). Briefly, rat forebrain and spinal cord were cut into coronal or transverse sections with a cryostat, respectively. Immunohistochemistry was performed for human TDP-43 (2E2-D3; Abnova) and rat lcn2 (MAB2560; Abnova ). To detect lcn2 induction, four antibodies against lcn2 purchased from the following companies were tested: Abnova (MAB2560, diluted at 1:200), Millipore (AB2267, diluted at 1:100), Santa Cruz Biotechnology (SC50351, diluted at 1:200), and R&D Systems (MAB1757, diluted at 1:50). Only the lcn2 antibody from Abnova was specific, and lcn2 immunoprecipitated from rat brain with this antibody was validated by MS. Immunostained sections were visualized using an ABC kit in combination with diaminobenzidine (Vector). Some immunostained sections were counterstained lightly with hematoxylin to show nuclei. Double-labeling immunofluorescent staining was performed to assess colocalization. Immunofluorescence staining was done for human TDP-43 (2E2-D3; Abnova), human FUS (made in-house), GFAP (DAKO), Iba-1 (Wako Chemicals), lcn2 (MAB2560; Abnova), SLC22A17 (4651; ProSci Inc.), and megalin (SC16478; Santa Cruz Biotechnology). Fluorescent staining was documented with a digital camera attached to a Nikon fluorescence microscope.
Rat Brain Slice Culture and CM Preparation.
The rat brain slice culture was adapted from published protocols (35, 36). Postnatal rats (P3) were killed, and their forebrains were removed, washed in PBS solution, and coronal slices (300 µm) were made with a Stoelting tissue chopper. To produce CM, every third whole forebrain slice was cultured on a net in dishes (2.5-cm diameter). Brain slices were cultured in Dox-containing medium for 4 d and then switched to culture medium without Dox. Culture medium was changed every other day. CM was collected and filtered through 0.25-µm porous membranes to eliminate large cellular debris. Freshly harvested CM was used to assess cytotoxicity. Rat hippocampal slices were cultured using published protocols (35, 36).
Immunoprecipitation was performed to deplete lcn2 from CM. To precipitate lcn2 from CM, a monoclonal antibody recognizing rat lcn2 (MAB2560; Abnova) was used with a direct immunoprecipitation kit (26148; Pierce) that was used according to the manufacturer’s instruction. After antibody conjugation, the beads and the column were thoroughly washed with fresh culture medium to eliminate chemical residues that could induce cytotoxicity.
CM Protein Composition Analysis and Immunoblotting.
For protein composition analysis, anatomically similar brain slices were taken from Camk2α-tTA single- and Camk2α-tTA/TRE-TDP43M337V double-transgenic rats, such that the concentrations of secreted proteins from the normal and the diseased tissues were comparable. Conditioned culture medium was collected and submitted to Applied Biomics Inc. for protein composition analysis. Proteins were precipitated from equal volumes of CM and were mixed after labeling with cyanine dye (Cy) 2 or Cy3. Fluorescent-labeled proteins were resolved on 2D gels, and protein spots with increased intensity in diseased cultures were selected and analyzed with MS. MS and 2D gel analyses were performed by Applied Biomics Inc. lcn2 in CM and in rat tissues was estimated by immunoblotting with a monoclonal antibody (MAB2560; Abnova). Immunoblotting was done as described previously (1, 19, 34).
Primary Culture and Cell Viability Assay.
Cortical and spinal neurons were cultured using published protocols (37). Rat embryos at embryonic day 19 (E19) were used for culturing cortical neurons, and rat embryos at E16 were used for culturing mixed spinal neurons. Primary neurons were cultured for 10 d before use in cytotoxic experiments. Dopaminergic neurons were obtained from rat embryos at E15 using a published protocol (38). Primary glial culture was obtained from P3 rats. Astrocytes, microglia, and oligodendrocytes were separated and enriched by shaking in a flask at 180 rpm for a defined time using published protocols (39, 40). Enriched glial cells were then tested for lcn2 cytotoxicity. Cell viability was quantified with a Live/Dead assay kit for mammalian cells (L3224; Invitrogen). Synthetic human lcn2 was purchased from Sino Biological Inc. and was purified from human cells. lcn2 was diluted in culture medium and was administered to cultures at defined concentrations.
Statistics Analysis.
Difference between mean values was analyzed with unpaired t tests. P values less than 0.05 were considered statistically significant.
Study Approval.
Animal use was in accord with National Institutes of Health (NIH) guidelines and was approved by the Institutional Animal Care and Use Committees at Thomas Jefferson University. The Committee for Oversight of Research Involving the Dead at the University of Pittsburgh approved the use of human tissue from the ALS Tissue Bank. Age-matched tissue sections from FTLD patients and nonneurological disease controls were used for this study.
Supplementary Material
Acknowledgments
We thank Ms. Xiaotao Wei for technical assistance and Dr. Qilin Cao for assistance with primary culture. This work is supported by the National Institutes of Health/National Institute of Neurological Disorders and Stroke Grants NS072696 and NS072113 (to X.-G.X.) and Grant NS073829 (to H.Z.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1218497110/-/DCSupplemental.
References
- 1.Huang C, et al. Entorhinal cortical neurons are the primary targets of FUS mislocalization and ubiquitin aggregation in FUS transgenic rats. Hum Mol Genet. 2012;21(21):4602–4614. doi: 10.1093/hmg/dds299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tong J, et al. XBP1 depletion precedes ubiquitin aggregation and Golgi fragmentation in TDP-43 transgenic rats. J Neurochem. 2012;123(3):406–416. doi: 10.1111/jnc.12014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Escartin C, Bonvento G. Targeted activation of astrocytes: A potential neuroprotective strategy. Mol Neurobiol. 2008;38(3):231–241. doi: 10.1007/s12035-008-8043-y. [DOI] [PubMed] [Google Scholar]
- 4.Lee S, et al. Lipocalin-2 is an autocrine mediator of reactive astrocytosis. J Neurosci. 2009;29(1):234–249. doi: 10.1523/JNEUROSCI.5273-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee S, et al. A dual role of lipocalin 2 in the apoptosis and deramification of activated microglia. J Immunol. 2007;179(5):3231–3241. doi: 10.4049/jimmunol.179.5.3231. [DOI] [PubMed] [Google Scholar]
- 6.Landreth G, Jiang Q, Mandrekar S, Heneka M. PPARgamma agonists as therapeutics for the treatment of Alzheimer’s disease. Neurotherapeutics. 2008;5(3):481–489. doi: 10.1016/j.nurt.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Okada S, et al. Conditional ablation of Stat3 or Socs3 discloses a dual role for reactive astrocytes after spinal cord injury. Nat Med. 2006;12(7):829–834. doi: 10.1038/nm1425. [DOI] [PubMed] [Google Scholar]
- 8.Custer SK, et al. Bergmann glia expression of polyglutamine-expanded ataxin-7 produces neurodegeneration by impairing glutamate transport. Nat Neurosci. 2006;9(10):1302–1311. doi: 10.1038/nn1750. [DOI] [PubMed] [Google Scholar]
- 9.Tanaka K, et al. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science. 1997;276(5319):1699–1702. doi: 10.1126/science.276.5319.1699. [DOI] [PubMed] [Google Scholar]
- 10.Behrens PF, Franz P, Woodman B, Lindenberg KS, Landwehrmeyer GB. Impaired glutamate transport and glutamate-glutamine cycling: downstream effects of the Huntington mutation. Brain. 2002;125(Pt 8):1908–1922. doi: 10.1093/brain/awf180. [DOI] [PubMed] [Google Scholar]
- 11.Howland DS, et al. Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS) Proc Natl Acad Sci USA. 2002;99(3):1604–1609. doi: 10.1073/pnas.032539299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Trotti D, Rolfs A, Danbolt NC, Brown RH, Jr, Hediger MA. SOD1 mutants linked to amyotrophic lateral sclerosis selectively inactivate a glial glutamate transporter. Nat Neurosci. 1999;2(5):427–433. doi: 10.1038/8091. [DOI] [PubMed] [Google Scholar]
- 13.Rothstein JD, et al. Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature. 2005;433(7021):73–77. doi: 10.1038/nature03180. [DOI] [PubMed] [Google Scholar]
- 14.Lepore AC, et al. Focal transplantation-based astrocyte replacement is neuroprotective in a model of motor neuron disease. Nat Neurosci. 2008;11(11):1294–1301. doi: 10.1038/nn.2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nagai M, et al. Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat Neurosci. 2007;10(5):615–622. doi: 10.1038/nn1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Di Giorgio FP, Carrasco MA, Siao MC, Maniatis T, Eggan K. Non-cell autonomous effect of glia on motor neurons in an embryonic stem cell-based ALS model. Nat Neurosci. 2007;10(5):608–614. doi: 10.1038/nn1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nagai M, et al. Rats expressing human cytosolic copper-zinc superoxide dismutase transgenes with amyotrophic lateral sclerosis: Associated mutations develop motor neuron disease. J Neurosci. 2001;21(23):9246–9254. doi: 10.1523/JNEUROSCI.21-23-09246.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Díaz-Amarilla P, et al. Phenotypically aberrant astrocytes that promote motoneuron damage in a model of inherited amyotrophic lateral sclerosis. Proc Natl Acad Sci USA. 2011;108(44):18126–18131. doi: 10.1073/pnas.1110689108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang C, Tong J, Bi F, Zhou H, Xia XG. Mutant TDP-43 in motor neurons promotes the onset and progression of ALS in rats. J Clin Invest. 2012;122(1):107–118. doi: 10.1172/JCI59130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ju S, et al. A yeast model of FUS/TLS-dependent cytotoxicity. PLoS Biol. 2011;9(4):e1001052. doi: 10.1371/journal.pbio.1001052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hvidberg V, et al. The endocytic receptor megalin binds the iron transporting neutrophil-gelatinase-associated lipocalin with high affinity and mediates its cellular uptake. FEBS Lett. 2005;579(3):773–777. doi: 10.1016/j.febslet.2004.12.031. [DOI] [PubMed] [Google Scholar]
- 22.Devireddy LR, Gazin C, Zhu X, Green MR. A cell-surface receptor for lipocalin 24p3 selectively mediates apoptosis and iron uptake. Cell. 2005;123(7):1293–1305. doi: 10.1016/j.cell.2005.10.027. [DOI] [PubMed] [Google Scholar]
- 23.Kehrer JP. Lipocalin-2: Pro- or anti-apoptotic? Cell Biol Toxicol. 2010;26(2):83–89. doi: 10.1007/s10565-009-9119-9. [DOI] [PubMed] [Google Scholar]
- 24.Devireddy LR, Teodoro JG, Richard FA, Green MR. Induction of apoptosis by a secreted lipocalin that is transcriptionally regulated by IL-3 deprivation. Science. 2001;293(5531):829–834. doi: 10.1126/science.1061075. [DOI] [PubMed] [Google Scholar]
- 25.Lee S, Lee WH, Lee MS, Mori K, Suk K. Regulation by lipocalin-2 of neuronal cell death, migration, and morphology. J Neurosci Res. 2012;90(3):540–550. doi: 10.1002/jnr.22779. [DOI] [PubMed] [Google Scholar]
- 26.Bolignano D, Coppolino G, Lacquaniti A, Buemi M. From kidney to cardiovascular diseases: NGAL as a biomarker beyond the confines of nephrology. Eur J Clin Invest. 2010;40(3):273–276. doi: 10.1111/j.1365-2362.2010.02258.x. [DOI] [PubMed] [Google Scholar]
- 27.Devarajan P. Neutrophil gelatinase-associated lipocalin: new paths for an old shuttle. Cancer Ther. 2007;5(B):463–470. [PMC free article] [PubMed] [Google Scholar]
- 28.Devarajan P. Neutrophil gelatinase-associated lipocalin: A promising biomarker for human acute kidney injury. Biomarkers Med. 2010;4(2):265–280. doi: 10.2217/bmm.10.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Srisawat N, et al. Recovery from acute kidney injury: Determinants and predictors. Contrib Nephrol. 2010;165:284–291. doi: 10.1159/000313768. [DOI] [PubMed] [Google Scholar]
- 30.Zamanian JL, et al. Genomic analysis of reactive astrogliosis. J Neurosci. 2012;32(18):6391–6410. doi: 10.1523/JNEUROSCI.6221-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Berger T, Cheung CC, Elia AJ, Mak TW. Disruption of the Lcn2 gene in mice suppresses primary mammary tumor formation but does not decrease lung metastasis. Proc Natl Acad Sci USA. 2010;107(7):2995–3000. doi: 10.1073/pnas.1000101107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Viau A, et al. Lipocalin 2 is essential for chronic kidney disease progression in mice and humans. J Clin Invest. 2010;120(11):4065–4076. doi: 10.1172/JCI42004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Naudé PJ, et al. Lipocalin 2: Novel component of proinflammatory signaling in Alzheimer’s disease. FASEB J. 2012;26(7):2811–2823. doi: 10.1096/fj.11-202457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang C, et al. FUS transgenic rats develop the phenotypes of amyotrophic lateral sclerosis and frontotemporal lobar degeneration. PLoS Genet. 2011;7(3):e1002011. doi: 10.1371/journal.pgen.1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hulse RE, Swenson WG, Kunkler PE, White DM, Kraig RP. Monomeric IgG is neuroprotective via enhancing microglial recycling endocytosis and TNF-alpha. J Neurosci. 2008;28(47):12199–12211. doi: 10.1523/JNEUROSCI.3856-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lein PJ, Barnhart CD, Pessah IN. Acute hippocampal slice preparation and hippocampal slice cultures. Methods Mol Biol. 2011;758:115–134. doi: 10.1007/978-1-61779-170-3_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu A, Zhuang Z, Hoffman PW, Bai G. Functional analysis of the rat N-methyl-D-aspartate receptor 2A promoter: Multiple transcription starts points, positive regulation by Sp factors, and translational regulation. J Biol Chem. 2003;278(29):26423–26434. doi: 10.1074/jbc.M211165200. [DOI] [PubMed] [Google Scholar]
- 38.Schneider JS, Huang FN, Vemuri MC. Effects of low-level lead exposure on cell survival and neurite length in primary mesencephalic cultures. Neurotoxicol Teratol. 2003;25(5):555–559. doi: 10.1016/s0892-0362(03)00018-7. [DOI] [PubMed] [Google Scholar]
- 39.Albuquerque C, Joseph DJ, Choudhury P, MacDermott AB. 2009. Dissection, plating, and maintenance of cortical astrocyte cultures. Cold Spring Harb Protoc pdb.prot5273.
- 40.Cao Q, et al. Functional recovery in traumatic spinal cord injury after transplantation of multineurotrophin-expressing glial-restricted precursor cells. J Neurosci. 2005;25(30):6947–6957. doi: 10.1523/JNEUROSCI.1065-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.