

Abstract
Glioblastoma (GB) is the most common and aggressive primary brain malignancy, with poor prognosis and a lack of effective therapeutic options. Accumulating evidence suggests that intratumor heterogeneity likely is the key to understanding treatment failure. However, the extent of intratumor heterogeneity as a result of tumor evolution is still poorly understood. To address this, we developed a unique surgical multisampling scheme to collect spatially distinct tumor fragments from 11 GB patients. We present an integrated genomic analysis that uncovers extensive intratumor heterogeneity, with most patients displaying different GB subtypes within the same tumor. Moreover, we reconstructed the phylogeny of the fragments for each patient, identifying copy number alterations in EGFR and CDKN2A/B/p14ARF as early events, and aberrations in PDGFRA and PTEN as later events during cancer progression. We also characterized the clonal organization of each tumor fragment at the single-molecule level, detecting multiple coexisting cell lineages. Our results reveal the genome-wide architecture of intratumor variability in GB across multiple spatial scales and patient-specific patterns of cancer evolution, with consequences for treatment design.
Keywords: tumor progression, high grade glioma
Glioblastoma (GB) is the most common primary brain malignancy in adults and one of the most aggressive cancers. The median survival in the general patient population is just 4.6 mo. Even in optimally treated patients, the median survival is 14 mo, with a 26% 2-y survival rate (1). Considering the average age at diagnosis, GB typically results in more than 20 y of life lost (2). Since the 1970s, primary treatment has involved surgery followed by radiotherapy (3). Recently, targeted chemotherapy approaches such as the alkylating agent temozolomide (1) also have been used, although with modest effects on survival. The impossibility of extensive tumor debulking and poor drug delivery in the brain contribute significantly to the lack of effective treatment options and poor prognosis.
Insights into the genetic regulatory landscape of GB have been achieved through The Cancer Genome Atlas (4) and other studies (5). Furthermore, patterns of gene expression have been collated to identify molecular subgroups with putative prognostic or predictive significance (5, 6). Nevertheless, the poor prognosis is compounded by the endemic problem of disease heterogeneity, which has been reported extensively for other cancer types (7–10). In glioblastoma, FISH has been used to identify receptor tyrosine kinase amplifications as markers for the generation of heterogeneity through clonal evolution (11, 12). These data are based on archival material from single tumor samples. Spatial heterogeneity within an individual tumor mass has not been investigated yet, and the impact of sampling bias has not been addressed. Furthermore, genome-wide studies of intratumor heterogeneity in GB have yet to be performed.
Real-time perioperative tumor sampling of GB may be confounded by tumor necrosis and the challenge of distinguishing disease from normal brain tissue. To address this, we have developed a fluorescence-guided multiple sampling (FGMS) approach (13) based on 5-aminolevulinic acid administration (14) to improve objective GB sampling in the operating theater. During surgery (Fig. 1A), viable tumor tissue can be identified by visible fluorescence (Fig. 1B). We adapted this technique to perform multiple sampling of objectively defined (visibly fluorescent) and spatially distinct GB tumor fragments from 11 patients (see Table S1 for sample details). During the operation, between four and six fragments (T1, T2, …) with a volume of 2–3 mm3 each were obtained from the neoplasm, with samples separated by at least 1 cm (Fig. 1C). The fragments were labeled in order of resection, with superficial fragments taken during the early stages of tumor debulking (T1, T2), followed by deeper fragments (T3, T4, …) taken later in the operation. In addition, we collected a further fragment from the bulk of the tumor mass (T) and a blood sample as a source of germline DNA to distinguish somatic copy number lesions. Histopathological analysis showed that all fragments had similar proliferative index, cellular atypia, and vascularization and were devoid of significant necrotic areas (as evident by fluorescence-aided resection). This sampling technique, performed directly in the operating theater, allowed us to collect a unique dataset to interrogate intratumor heterogeneity at the genomic level across the malignancy.
Fig. 1.
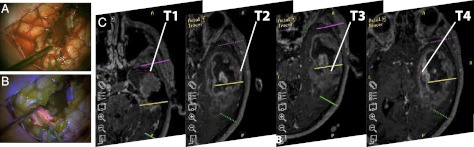
GB sample collection scheme. FGMS (Fluorescence-Guided Multiple Sampling) detects viable tumor tissue in bright pink color [(A): off; (B): on] while avoiding necrotic areas and normal brain tissue during surgery. (C) At the time of resection, multiple fluorescent tumor fragments (T1, T2, T3, …), ∼10 mm apart, were collected from 11 GB patients.
Here, we show that genome-wide GB intratumor genomic heterogeneity can be decomposed to reveal tumor evolution. Moreover, we report that based upon gene expression levels, tumor fragments from the same patient may be classified into different GB subtypes. Using single-molecule approaches, we also investigate the clonal composition of single fragments, revealing that a hierarchy of mitotic clones coexists within the same fragment. Our results show that tumor heterogeneity represents a specific signature that informs on GB evolutionary dynamics at the single-patient level.
Results
Subset of GB Putative Drivers Is Consistently Heterogeneous.
We profiled genome-wide DNA somatic copy number levels for 38 tumor fragments from nine patients from the cohort (see Table S1). To investigate the global patterns of copy number alteration (CNA) within each patient, we took the union of CNAs that occurred in at least one of the tumor fragments. We observed several frequent aberrations that have been reported in other GB cohorts, including partial loss of chromosome 10 (15) in eight patients and the focal (<10 Mb) deletion of the CDKN2A/B locus (4, 16) in seven patients, as well as the frequently co-occurring deletion of MTAP (17). Polysomy of chromosome 7 was found in all patients. Gain/amplification of EGFR, often presented as numerically unstable extrachromosomal double minutes (18), also was found in all patients (3/9 were focal high-level amplifications). Moreover, we identified aberrations in several other putative GB drivers linked to the Ras, p53, and Rb pathways, including amplification of MET (eight cases), CDK6 (eight cases), MDM4 (two cases), and AKT3 (two cases); focal amplification of PDGFRA (three cases), PIK3CA (two cases), CCND2 (one case), and MYCN (one case); deletion of PARK2 (one case), RB1 (one case), PTEN (nine cases, two of which were focal); and finally focal deletion of TP53 (two cases), NF1 (two cases), and CDKN2C (one case). CNAs in MDM2 or CDK4 were not detected in our cohort. We also noted copy-neutral loss of heterozygosity for TP53 (two cases), an important event for the inactivation of this gene (19). Similar to previous studies (4), we observed deregulation of the Ras, Rb, and p53 pathways in all patients, with aberrations in multiple components belonging to each pathway (Table S2).
Some of these putative driver aberrations were consistently heterogeneous within the same tumor (e.g., Fig. 2A), including copy number gain/amplification of the PDGFRA (3/3 heterogeneous tumors), MDM4 (2/2), and AKT3 (2/2) loci, and deletion of the genomic locus containing PTEN (6/9) (Fig. S1). Fig. 2B summarizes the CNAs in putative drivers for all patients, in which aberrations that displayed intratumor heterogeneity are emphasized. Moreover, the heterogeneity was not limited to regions of the genome containing genes previously associated with cancer. In fact, although the fragments from the same patient shared a common genomic profile, indicating the clonal origin of the tumor (Fig. S2), they displayed a striking variety of CNAs that were present in only a subset of the fragments (Fig. S3).
Fig. 2.
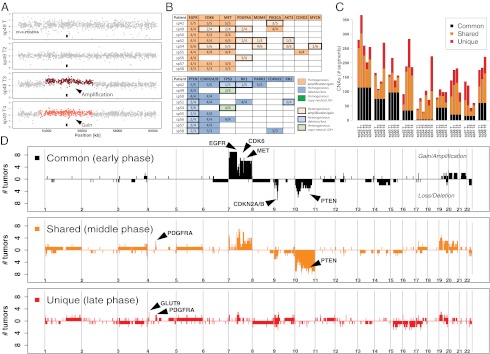
Landscape of intratumor heterogeneity at the copy number level. (A) Example of intratumor heterogeneity based on PDGFRA aberrations in patient sp49; whereas fragments T and T2 show no alterations, focal gain and amplification are evident in fragments T3 and T4, respectively (■, PDGFRA location). (B) For each patient, the number of fragments that exhibit CNAs in a putative driver gene is reported. In eight of nine patients, we found putative driver alterations that were not common to the whole tumor (enhanced borders), such as PDGFRA and PTEN. (C) CNAs were classified as common (found in all fragments), shared (found in more than one but not all fragments), and unique (found in only one fragment). All tumors displayed a large yet variable number of shared and unique aberrations. (D) The distribution of common/shared/unique altered probes delineates tumor evolution in the early/middle/late phases, respectively. Alterations focus on chr7 and chr10 as well as CDKN2A/B during an early phase, followed by a middle (shared) phase in which CNAs accumulate further on chr7 and 10, around the PDGFRA locus and on 19p13/12. Finally, during the late phase (corresponding to unique alterations), CNAs spread across the genome, with a peak on 4p16 (GLUT9).
Copy Number Heterogeneity Allows Tumor Evolution to Be Inferred.
For each tumor, we classified CNAs as “common” (all tumor fragments had the CNA), “shared” (more than one but not all fragments had the CNA), and “unique” (only one fragment had the CNA). The total number of aberrations (Fig. 2C) was highly variable among tumors, with sample sp42 having the largest number of CNAs (average 273) and sp54 the smallest (average 91). We observed that for every tumor, only a proportion of CNAs were common to all fragments (ranging from 1.3% in sp54 T6 to 70% in sp50 T3; mean = 31%; Fig. S4), with shared plus unique alterations representing most of the CNAs in 31/38 fragments. We confirmed that the observed heterogeneity was not the result of differences in normal contamination by demonstrating the absence of significant correlation between the estimated cellularity [using ASCAT (Allele Specific Copy number Analysis of Tumors) (20)] and the fraction of shared (Spearman’s ρ = 0.025, P value = 0.88) and unique (Spearman’s ρ = 0.043, P value = 0.8) alterations.
Because all fragments taken from the same tumor mass almost certainly evolved from the same tumor-initiating clone, we may infer that common aberrations arose earlier in the development of the malignancy with respect to alterations that are found only in a subset of tumor cells (21). Hence, shared or unique events likely represent changes that occurred later in tumor development. Given this reasoning, we can deduce that multiple sampling of spatially separated tumor regions enables interrogation of the mutational spectrum of the malignancy both spatially and temporally. Indeed, when we examine the distribution of CNAs across the genome for all patients (Fig. 2D), we find that in the inferred early tumor growth phase (common), CNAs largely target chromosomes 7 and 10, on which the most frequent putative GB drivers, such as EGFR, CDK6, MET, and PTEN, are located. We also report common copy number deletions of CDKN2A/B on chr9 (seven patients) and of the 10p12 locus during the inferred early phase. During the middle phase (shared), amplifications accumulate on chr7 (7p22, 7p11, 7q22, 7q36) and on chr19p12/13, whereas deletions arise almost exclusively on chr10, further targeting PTEN. Additionally, we observe the focal amplification of PDGFRA during this middle phase. During what we inferred to be the late (unique) phase, alterations are more broadly scattered across the genome with respect to the middle phase. We also found a focal peak of amplification containing GLUT9 (4p16), a gene previously shown to be involved in cancer glucose metabolism (22).
To investigate tumor evolution within each patient in more detail, we used TuMult (23) to reconstruct phylogenies based on the underlying copy number data (Fig. S5). The results are consistent with the analysis above: During the first appearance of a malignant clone, loss of CDKN2A/B and amplification of EGFR, CDK6, and MET occur. Later malignant events most often are copy number aberrations in genomic regions containing PDGFRA, PTEN, and TP53. For example, the expansion of tumor sp52 was initiated by alterations in EGFR, CDK6, MET, CDKN2A, and NF1. Subsequently, the initial tumor clone separated into one subclone with an amplification of PDGFRA and another subclone with loss of PTEN and RB1. The latter diversified further into three subclones, one of which showed amplification of AKT3 (Ras pathway) and MDM4 (p53 pathway).
Distinct GB Subtypes Are Present Within the Same Tumor.
To characterize intratumor variation at the level of transcription, we used microarrays to profile gene expression levels in 51 tumor fragments from 10 patients, 8 of whom were common to the copy number set (Table S1). Hierarchical clustering confirmed patient-specific cancer profiles, with most samples from the same tumor falling into the same cluster, with only three exceptions (sp42T4, sp54T3, and sp56T3). We assigned each sample to one of four subtypes: “proneural,” “neural,” “classical,” and “mesenchymal” using the Verhaak classifier (5), which is based on an 840-gene signature. Each subgroup has a characteristic copy number profile, different survival, and variable response to treatment; hence, this classification is believed to be relevant for patient stratification and thus therapy design (24). We found that in 6 of 10 cases (sp42, sp49, sp52, sp54, sp55, and sp56), the fragments from the same tumor mass were classified into at least two different GB subgroups (Fig. 3 A and B). This indicates that tumor clones with different phenotypic profiles coexist within the same malignancy.
Fig. 3.

Intratumor heterogeneity at the transcriptional level. (A) Clustering based on the gene expression profiles of the full set of 16,811 transcripts results in the grouping of most tumor specimens by patient, with three exceptions (sp42-T4, sp54-T3, and sp56-T3). (B) Despite the overall patient-specific signature, samples taken from the same tumor were classified into different GB subtypes in 6 of 10 patients. (C) Gene ontology analysis of the genes that exhibit intratumor heterogeneity in each of the 10 patients revealed that they were involved in biological processes related to neuron generation/development, cell morphogenesis, and tumor angiogenesis.
To further investigate heterogeneity at the gene expression level, we selected the 100 most heterogeneous genes within each tumor (see SI Materials and Methods for details) and found that 5 of them were common to more than six patients (Table S3). Gene ontology analysis (25) revealed that the most heterogeneous genes from each patient were involved in the same biological processes (Fig. 3C), such as brain cell proliferation/differentiation (neurogenesis, generation of neurons, CNS development, neuron projection development), neuronal activity (regulation of membrane potential, neurotransmitter secretion), morphogenesis (cell morphogenesis, cell morphogenesis involved in differentiation) as well as tumor angiogenesis (blood vessel development, vasculature development), and cell migration and invasion (extracellular matrix organization). Moreover, we found that many of these genes are putatively regulated by three transcription factors (E12, LEF1, and NFAT; Table S4), all of which are known to regulate neural progenitor cell proliferation/differentiation either through the Wnt/β-catenin signaling pathway (LEF1, NFAT) or through basic-loop-helix factors (E12) (26–29).
Each Tumor Fragment Contains a Complex Hierarchy of Clone Lineages.
Given the heterogeneity at both the DNA copy number and transcriptional levels within each malignancy, an important question is the depth and complexity of this subclonal structure at the resolution of individual cells. A clone is a group of cells with a common genomic profile, yet because of the large spectrum of possible alterations, identifying distinct subclones and their common mutations within a mixed population of cells is challenging. The definition of a clone implies that cells within the same clone are related in terms of proliferation. Hence, an optimal approach to identifying subclones is to exploit a measure of mitotic distance (number of cell divisions) between the cells within a fragment. This also allows the reconstruction of the phylogeny of the cells and the relationship between the subclones. Here, we use neutral methylation loci as molecular clocks that report on the number of cell divisions. Previously, we used this approach in normal (30–32) and cancerous tissues (33) to investigate mitotic heterogeneity at the single-molecule level and to infer patient-specific tumor parameters (34). In brief, a molecular clock is a genomic locus that records proliferative events within a cell, thereby allowing mitotic relationships to be traced at single-molecule resolution within a tumor fragment.
In this study, we used the IRX2 locus as a molecular clock for eight patients, using 454 sequencing to generate single-molecule reads from each tumor fragment. Fig. 4A illustrates the fraction of the different mitotic clones in each sample (one mitotic clone corresponds to a unique methylation tag). Interestingly, in none of the 38 fragments examined from eight GB cases was a single dominant clone (defined as >60% of cells) observed. Fig. 4B shows the phylogenetic reconstruction of the mitotic clones for three representative tumor fragments, in which leaf thickness indicates the abundance of each clone. This indicates that not only are there different subclones in each fragment, but they belong to clearly distinct cell lineages, representative of a complex hierarchy.
Fig. 4.

Multiple mitotic clones coexist within each GB fragment. (A) Fraction of the five most common mitotic clones in each tumor fragment based on molecular clock analysis of eight patients (sp57 was excluded because of PCR failure). (B) Phylogenetic reconstruction within each tumor fragment based on the molecular clock analyses. In these three representative cases, the existing mitotic clones are represented by the leaves of the tree, where a leaf´s thickness is proportional to the abundance of the clone within the fragment. The results indicate clonal heterogeneity within each fragment and the presence of multiple cell lineages that correspond to distinct mitotic clones.
Discussion
From an evolutionary perspective (7, 35), the divergent development of subpopulations of cancer cells within the same tumor likely is at the root of therapy failure, the development of treatment resistance, and ultimately recurrence of the malignancy (36–40). A detailed understanding of the evolutionary dynamics of tumor progression will provide insight into the associated molecular genetic mechanisms and will allow us to construct order from apparent chaos.
In GB, intratumor heterogeneity at the level of the tyrosine kinases EGFR, PDGFRA, and MET was demonstrated previously using FISH (11). Moreover, cell lines with different EGFR/PDGFRA profiles derived from the same GB showed differential response to growth factors (12). However, a comprehensive genomic analysis of intratumor heterogeneity and tumor evolution in GB has not been previously described. More importantly, the mechanisms behind intratumor heterogeneity and its consequences remain largely unknown. To date, analyses in GB as well as in many other cancer types are based on single tissue samples from individual patients. Here, we show that an objective multiple sampling scheme is the key to interrogating intratumor heterogeneity and deconvoluting the underlying cancer dynamics. A single biopsy is unlikely to represent the full set of mutations present in a particular cancer because it may underestimate the landscape of alterations that are present, as recently reported in renal carcinoma (10). For GB, in which the tumor is not resected as a solid mass but in a piecemeal fashion, multiple sampling from the same neoplasm is challenging and is done most easily at the time of surgery. Using FGMS (Fluorescence-Guided Multiple Sampling), we have applied a real-time perioperative multiple sampling approach during GB resection, which partially conserves information on the spatial organization of the samples. All patients underwent primary GB resection with no preoperative treatment that would have introduced biases in the analysis of genetic alterations.
Here, we demonstrate that GB exhibits a landscape of heterogeneous mutations across the whole genome at the copy number level; this represents a signature of the history of the malignancy from the first tumor-founding cell(s). By deconvoluting such a signature, we may infer the temporal sequence of alterations that have occurred in the malignancy. Further, using gene expression data we observed that different samples from the same tumor were classified into different GB subtypes (Fig. 3A). This shows that the impact of sampling bias must be considered when establishing molecular criteria for patient stratification. Supporting this assertion, the morphological data available from individual tissue biopsies may provide misleading information, resulting in diagnostic errors (39). Hence, these results support the development of personalized treatments based upon a multimodal approach that uses information from multiple samples from the same GB.
As an example of how our data might be integrated with the current standard of clinical practice, Fig. 5 shows how the anatomical positions of the five tumor fragments collected for patient sp42 (A) may be coupled with the reconstructed evolution of the tumor mass (B). During the operation, fragments are numbered in order of resection, approximately corresponding to the depth of the sample within the brain. This coupling reveals a complex evolutionary pattern with the accumulation of malignant traits in different parts of the neoplasm (C). Our analysis suggests that the founder clone displayed amplification/gain of EGFR, CDK6, and MET, and loss/deletion of CDKN2A/B, PTEN, and PARK2 (hence targeting the Ras and Rb pathways), before splitting into two populations (subclones 1 and 2), the first of which generated T2 and T3, with T3 gaining a copy of chromosome 3 (which contains PIK3CA). The second subclone subsequently acquired further mutations manifested in T4, which also displayed an altered gene expression profile belonging to a mesenchymal subtype, despite the rest of the tumor fragments being proneural. Subclone 2 gave rise to another independent +PIK3CA subclone (again with chromosome 3 gain). The latter then expanded to form T1 and, with the partial loss of chromosome 17 (containing NF1 and TP53) and therefore alteration of the p53 pathway, generated T. Of note, the occurrence of new lesions, such as loss of NF1 and TP53 in fragment T, may not necessarily represent a fast clonal expansion event, but also a slower process of selection of a preexisting rare clone that becomes dominant within a fragment and, therefore, detectable by copy number profiling.
Fig. 5.

Reconstruction of GB progression in time and space. The combination of sampling information (A), reconstructed tumor phylogeny (B), gene expression profiles, and molecular clock data enables temporal and spatial reconstruction of tumor ontogeny (C), as shown here for sp42. The evolution of the malignancy is illustrated by the accumulation of CNAs in different parts of the tumor and the corresponding variation in the gene expression profile, which reveal that T4 is classified as a different subtype (mesenchymal) with respect to the rest of the neoplasm (proneural). Moreover, we report the presence of variable numbers of subclones in each tumor fragment by molecular clock analysis.
Our study presents an integrated analysis of intratumor heterogeneity at the genotype level (copy number), cellular phenotype (gene expression), and single-molecule mitotic level (molecular clocks). To our knowledge, the multiple sampling scheme and genomic data have never before been integrated in this way to describe GB evolution at the individual patient level.
Taken together, our results shed light upon intratumor heterogeneity and the clonal evolution of GB. Based on these results, we propose that patient-specific dynamics of tumor heterogeneity underlie variation in response to treatment. Specifically, after therapy, the surviving population may not be a single resistant cancer clone, but rather a heterogeneous population of malignant cells with genetic aberrations that allow them to survive the initial treatment. This view extends the concept of clonal evolution by allowing selection to play a role limited only by the spatial structure of the neoplasm, in which multiple clones with different fitness coexist within the same cancer (41). Instead of benefitting single clones, this would favor the whole tumor population by converting heterogeneity into an asset to resist treatment (7, 42). This hypothesis implies that patterns of heterogeneity might be used to stratify individual patients and to select an appropriate multimodal therapeutic strategy. To test this hypothesis, multiple sampling data (paired primary tumor and recurrence) from a larger population are required. Furthermore, future studies should characterize the interaction of cell populations within tumor fragments, as this would improve our understanding of complex microenvironmental forces that contribute to tumor development.
Materials and Methods
Using our established FGMS approach (13), we obtained between four and six tumor fragments from different areas of the visibly fluorescent tumor mass that were at least 10 mm apart. Tissue collection protocols were compliant with the UK Human Tissue Act 2004 (HTA Licence ref 12315) and approved by the Local Regional Ethics Committee (LREC ref 04/Q0108/60). Informed consent was obtained from each patient before surgery. Samples were subject to genome-wide copy number analysis on the Affymetrix SNP6 platform (Fig. S6), gene expression profiling on the Illumina HT12 platform, and methylation molecular clock analysis as described elsewhere (34). See SI Materials and Methods for further details.
Supplementary Material
Acknowledgments
We thank Dr. Nicholas Marko for helpful discussions and Dr. Frank Desmoulin for sharing the R package used for tumor classification. We acknowledge the Human Research Tissue Bank and Biomedical Research Centre for the tissue being accessed through the Human Research Tissue Bank. The Human Research Tissue Bank is supported by the NIHR Cambridge Biomedical Research Centre. This study was supported by Cancer Research UK Programme Grant C14303/A10825 (to A.S., I.S., A.T., and S.T.), the Addenbrooke’s Charitable Trust, the National Institute for Health Research (NIHR) Cambridge Biomedical Research Centre, the Higher Education Funding Council for England, the Royal College of Surgeons of Edinburgh, the European Commission Seventh Framework Programme [Marie Curie Intra-European Fellowship (to S.G.M.P.)], European Bioinformatics Institute funding (A.T. and J.C.M.), and Keck School of Medicine of the University of Southern California (C.C. and A.S.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: Copy number arrays and gene number arrays reported in this paper have been deposited in the ArrayExpress Archive, www.ebi.ac.uk/arrayexpress/ (accession nos. E-MTAB-1215 and E-MTAB-1129).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1219747110/-/DCSupplemental.
References
- 1.Stupp R, et al. European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups National Cancer Institute of Canada Clinical Trials Group Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 2.Burnet NG, Jefferies SJ, Benson RJ, Hunt DP, Treasure FP. Years of life lost (YLL) from cancer is an important measure of population burden—and should be considered when allocating research funds. Br J Cancer. 2005;92(2):241–245. doi: 10.1038/sj.bjc.6602321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Walker MD, et al. Randomized comparisons of radiotherapy and nitrosoureas for the treatment of malignant glioma after surgery. N Engl J Med. 1980;303(23):1323–1329. doi: 10.1056/NEJM198012043032303. [DOI] [PubMed] [Google Scholar]
- 4.Cancer Genome Atlas Research Network Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455(7216):1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Verhaak RG, et al. Cancer Genome Atlas Research Network Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17(1):98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Phillips HS, et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell. 2006;9(3):157–173. doi: 10.1016/j.ccr.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 7.Marusyk A, Polyak K. Tumor heterogeneity: Causes and consequences. Biochim Biophys Acta. 2010;1805(1):105–117. doi: 10.1016/j.bbcan.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Navin N, et al. Inferring tumor progression from genomic heterogeneity. Genome Res. 2010;20(1):68–80. doi: 10.1101/gr.099622.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yachida S, et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature. 2010;467(7319):1114–1117. doi: 10.1038/nature09515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gerlinger M, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366(10):883–892. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Snuderl M, et al. Mosaic amplification of multiple receptor tyrosine kinase genes in glioblastoma. Cancer Cell. 2011;20(6):810–817. doi: 10.1016/j.ccr.2011.11.005. [DOI] [PubMed] [Google Scholar]
- 12.Szerlip NJ, et al. Intratumoral heterogeneity of receptor tyrosine kinases EGFR and PDGFRA amplification in glioblastoma defines subpopulations with distinct growth factor response. Proc Natl Acad Sci USA. 2012;109(8):3041–3046. doi: 10.1073/pnas.1114033109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Piccirillo SGM, et al. Fluorescence-guided surgical sampling of glioblastoma identifies phenotypically distinct tumour-initiating cell populations in the tumour mass and margin. Br J Cancer. 2012;107(3):462–468. doi: 10.1038/bjc.2012.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stummer W, et al. ALA-Glioma Study Group Fluorescence-guided surgery with 5-aminolevulinic acid for resection of malignant glioma: a randomised controlled multicentre phase III trial. Lancet Oncol. 2006;7(5):392–401. doi: 10.1016/S1470-2045(06)70665-9. [DOI] [PubMed] [Google Scholar]
- 15.Horiguchi H, Hirose T, Sano T, Nagahiro S. Loss of chromosome 10 in glioblastoma: Relation to proliferation and angiogenesis. Pathol Int. 1999;49(8):681–686. doi: 10.1046/j.1440-1827.1999.00934.x. [DOI] [PubMed] [Google Scholar]
- 16.Ichimura K, Schmidt EE, Goike HM, Collins VP. Human glioblastomas with no alterations of the CDKN2A (p16INK4A, MTS1) and CDK4 genes have frequent mutations of the retinoblastoma gene. Oncogene. 1996;13(5):1065–1072. [PubMed] [Google Scholar]
- 17.Christopher SA, Diegelman P, Porter CW, Kruger WD. Methylthioadenosine phosphorylase, a gene frequently codeleted with p16(cdkN2a/ARF), acts as a tumor suppressor in a breast cancer cell line. Cancer Res. 2002;62(22):6639–6644. [PubMed] [Google Scholar]
- 18.Lopez-Gines C, et al. New pattern of EGFR amplification in glioblastoma and the relationship of gene copy number with gene expression profile. Mod Pathol. 2010;23(6):856–865. doi: 10.1038/modpathol.2010.62. [DOI] [PubMed] [Google Scholar]
- 19.Beroukhim R, et al. Assessing the significance of chromosomal aberrations in cancer: Methodology and application to glioma. Proc Natl Acad Sci USA. 2007;104(50):20007–20012. doi: 10.1073/pnas.0710052104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Van Loo P, et al. Allele-specific copy number analysis of tumors. Proc Natl Acad Sci USA. 2010;107(39):16910–16915. doi: 10.1073/pnas.1009843107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nik-Zainal S, et al. Breast Cancer Working Group of the International Cancer Genome Consortium The life history of 21 breast cancers. Cell. 2012;149(5):994–1007. doi: 10.1016/j.cell.2012.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Levine AJ, Puzio-Kuter AM. The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science. 2010;330(6009):1340–1344. doi: 10.1126/science.1193494. [DOI] [PubMed] [Google Scholar]
- 23.Letouzé E, Allory Y, Bollet MA, Radvanyi F, Guyon F. Analysis of the copy number profiles of several tumor samples from the same patient reveals the successive steps in tumorigenesis. Genome Biol. 2010;11(7):R76. doi: 10.1186/gb-2010-11-7-r76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sulman EP, Aldape K. The use of global profiling in biomarker development for gliomas. Brain Pathol. 2011;21(1):88–95. doi: 10.1111/j.1750-3639.2010.00456.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nogales-Cadenas R, et al. GeneCodis: Interpreting gene lists through enrichment analysis and integration of diverse biological information. Nucleic Acids Res. 2009;37(Web Server issue):W317-22. doi: 10.1093/nar/gkp416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang T, et al. Nuclear factor of activated T cells (NFAT) proteins repress canonical Wnt signaling via its interaction with Dishevelled (Dvl) protein and participate in regulating neural progenitor cell proliferation and differentiation. J Biol Chem. 2011;286(43):37399–37405. doi: 10.1074/jbc.M111.251165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Seki T, Sawamoto K, Alvarez-Buylla A, Parent JM. Neurogenesis in the Adult Brain I: Neurobiology. Tokyo: Springer; 2011. [Google Scholar]
- 28.Ota S, et al. NLK positively regulates Wnt/β-catenin signalling by phosphorylating LEF1 in neural progenitor cells. EMBO J. 2012;31(8):1904–1915. doi: 10.1038/emboj.2012.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sun J, Gong X, Purow B, Zhao Z. Uncovering microRNA and transcription factor mediated regulatory networks in glioblastoma. PLOS Comput Biol. 2012;8(7):e1002488. doi: 10.1371/journal.pcbi.1002488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yatabe Y, Tavaré S, Shibata D. Investigating stem cells in human colon by using methylation patterns. Proc Natl Acad Sci USA. 2001;98(19):10839–10844. doi: 10.1073/pnas.191225998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nicolas P, Kim KM, Shibata D, Tavaré S. The stem cell population of the human colon crypt: Analysis via methylation patterns. PLOS Comput Biol. 2007;3(3):e28. doi: 10.1371/journal.pcbi.0030028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sottoriva A, Tavaré S. Integrating approximate Bayesian computation with complex agent-based models for cancer research. In: Saporta G, Lechevallier Y, editors. Proceedings of COMPSTAT 2010. Heidelberg: Springer; 2010. pp. 57–66. [Google Scholar]
- 33.Siegmund KD, Marjoram P, Woo YJ, Tavaré S, Shibata D. Inferring clonal expansion and cancer stem cell dynamics from DNA methylation patterns in colorectal cancers. Proc Natl Acad Sci USA. 2009;106(12):4828–4833. doi: 10.1073/pnas.0810276106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sottoriva A, Spiteri I, Shibata D, Curtis C, Tavaré S. Single-molecule genomic data delineate patient-specific tumor profiles and cancer stem cell organization. Cancer Res. 2013;73(1):41–49. doi: 10.1158/0008-5472.CAN-12-2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Merlo LM, Pepper JW, Reid BJ, Maley CC. Cancer as an evolutionary and ecological process. Nat Rev Cancer. 2006;6(12):924–935. doi: 10.1038/nrc2013. [DOI] [PubMed] [Google Scholar]
- 36.Greaves M, Maley CC. Clonal evolution in cancer. Nature. 2012;481(7381):306–313. doi: 10.1038/nature10762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gillies RJ, Verduzco D, Gatenby RA. Evolutionary dynamics of carcinogenesis and why targeted therapy does not work. Nat Rev Cancer. 2012;12(7):487–493. doi: 10.1038/nrc3298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Marusyk A, Almendro V, Polyak K. Intra-tumour heterogeneity: A looking glass for cancer? Nat Rev Cancer. 2012;12(5):323–334. doi: 10.1038/nrc3261. [DOI] [PubMed] [Google Scholar]
- 39.Yap TA, Gerlinger M, Futreal PA, Pusztai L, Swanton C. Intratumor heterogeneity: Seeing the wood for the trees. Sci Transl Med. 2012;4(127):127ps10. doi: 10.1126/scitranslmed.3003854. [DOI] [PubMed] [Google Scholar]
- 40.Yates LR, Campbell PJ. Evolution of the cancer genome. Nat Rev Genet. 2012;13(11):795–806. doi: 10.1038/nrg3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sottoriva A, et al. Cancer stem cell tumor model reveals invasive morphology and increased phenotypical heterogeneity. Cancer Res. 2010;70(1):46–56. doi: 10.1158/0008-5472.CAN-09-3663. [DOI] [PubMed] [Google Scholar]
- 42.Sottoriva A, Vermeulen L, Tavaré S. Modeling evolutionary dynamics of epigenetic mutations in hierarchically organized tumors. PLOS Comput Biol. 2011;7(5):e1001132. doi: 10.1371/journal.pcbi.1001132. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.