

Abstract
Tumor stromal alternatively activated macrophages are important determinants of anti-tumor T lymphocyte responses, intratumoral neovascularization and metastatic dissemination. Our recent efforts to investigate the mechanism of macrophage migration inhibitory factor (MIF) in antagonizing anti-melanoma immune responses reveal that macrophage-derived MIF participates in macrophage alternative activation in melanoma-bearing mice. Both peripheral and tumor-associated macrophages (TAMs) isolated from melanoma bearing MIF-deficient mice display elevated pro-inflammatory cytokine expression and reduced anti-inflammatory, immunosuppressive and pro-angiogenic gene products compared to macrophages from tumor bearing MIF wildtype mice. Moreover, TAMs and myeloid-derived suppressor cells (MDSCs) from MIF-deficient mice exhibit reduced T lymphocyte immunosuppressive activities than do those from their wildtype littermates. Corresponding with reduced tumor immunosuppression and neoangiogenic potential by TAMs, MIF-deficiency confers protection against transplantable subcutaneous melanoma outgrowth and melanoma lung metastatic colonization. Finally, we report for the first time that our previously discovered MIF small molecule antagonist, 4-iodo-6-phenylpyrimidine (4-IPP), recapitulates MIF-deficiency in vitro and in vivo and attenuates tumor polarized macrophage alternative activation, immunosuppression, neoangiogenesis and melanoma tumor outgrowth. These studies describe an important functional contribution by MIF to tumor-associated macrophage alternative activation and provide justification for immunotherapeutic targeting of MIF in melanoma patients.
Introduction
Patients diagnosed with early stage (stage I) melanoma have a generally favorable prognosis while the five year survival rate for those diagnosed with stage IV metastatic disease is only 5 – 10% (1). Because of the highly immunogenic nature of melanocytic tumors, immunotherapeutic targeting strategies have largely focused on this malignancy (2). Despite this fact and encouraging results from clinical trials with anti-CTLA-4 in patients with late stage melanoma (3), overall survival percentages among advanced melanoma patients have not improved significantly (4). Contributing to the relative lack of clinical responses with current cancer immunotherapies is the exacerbation of both innate and adaptive immunosuppressive pathways by alternatively activated macrophages within the tumor microenvironment (5). Tumor-dependent polarization of peripheral and stromal macrophages promotes tumor progression by inducing regulatory T cell (Treg) generation (6), PD-1-dependent lymphocyte immunosuppression (7–9) and tumor-associated neoangiogenesis (10, 11). The identification of new immunotherapeutic targets that are readily “druggable” is critical to the elucidation of an individual or combinatorial immunotherapeutic strategy that will provide meaningful and durable clinical responses in late stage cancer patients.
Despite its well documented activities as a pro-inflammatory determinant of innate immune responses (12–14), macrophage migration inhibitory factor (MIF) has T lymphocyte immunosuppressive activities in malignant disease settings (15, 16). For example, systemic inhibition of MIF during murine tumor outgrowth significantly enhances anti-tumor cytotoxic T lymphocyte (CTL) Th1 responses (15). Moreover, MIF silencing in neuroblastoma cells induces robust anti-tumor CTL responses in transplanted mice compared to MIF-expressing neuroblastoma cells (16). Finally, MIF overexpression in ovarian and melanocytic cancers antagonizes natural killer (NK) cell-mediated cancer cell cytolysis (17, 18). Combined, these findings suggest that MIF actively suppresses anti-tumor lymphocyte responses and MIF inhibition breaks this immunosuppression resulting in enhanced cancer cytolytic responses. What is less clear is how MIF promotes immunosuppressive adaptive immune response phenotypes when MIF is so tightly linked to pro-inflammatory innate immune responses (19).
We now report that in malignant disease settings, MIF is an important mediator of macrophage alternative activation. MIF-deficiency or small molecule antagonism reduces B16 melanoma tumor outgrowth and B16F10 metastatic melanoma lung colonization in a manner that coincides with decreased anti-inflammatory and increased pro-inflammatory effector expression in bone marrow-derived, peripheral and tumor stromal macrophages. We also demonstrate that tumor-associated macrophage (TAM) pro-angiogenic effector expression and angiogenic potential are reduced in MIF-deficient and MIF small molecule inhibited macrophages. Combined, these findings provide important and novel evidence that MIF is a critical mediator of pro-tumorigenic macrophage alternative activation and ensuing melanomagenesis.
Materials and Methods
Mice
Wild-type male C57BL/6 mice (MIF+/+) were obtained from Harlan Laboratories (Dublin, VA). OT-1 transgenic mice were obtained from Jackson Laboratory (Bar Harbor, ME). Mice were used at 6–8 weeks of age. All mice were handled in accordance with the Association for Assessment and Accreditation of Laboratory Animals Care international guidelines, with the approval of the Institutional Animal Care and Use Committee at University of Louisville.
Tumor models
B16F0 melanoma cells were obtained from American Type Culture collection (Manassas, VA). To establish subcutaneous (s.c.) tumors, 1×105 B16 tumor cells were injected subcutaneously into the left flanks of MIF+/+ and MIF−/− C57BL/6 mice. The Lewis lung carcinoma (LLC) model used in some in vitro experiments was established by injecting 1×105 LLC tumor cells s.c. into mice. Tumor growth was monitored thrice a week by measuring with digital calipers. To establish the experimental metastasis model of melanoma, 1×105 B16-F10-luc2 cells (Caliper Life Sciences, Mountain View, CA) were injected into the tail vein of C57BL/6 MIF+/+ and MIF−/− mice. For in vivo imaging, mice were anesthetized with 75 mg/kg of ketamine and 7.5 mg/kg of xylazine and injected intraperitoneally (i.p.) with the reporter substrate, D-Luciferin, Potassium salt (150 mg/kg body weight; obtained from Gold Biotechnology, St. Louis, MO). Immediately after D-luciferin administration, mice were imaged using the PhotonIMAGER™ (Biospace Lab, Paris, France). Bioluminescence was quantified from the in vivo signals emitted from ventral views (day 18–25) using the Biospace software. The mean photon emission was calculated and expressed as photons/second/steredian (ph/s/sr). Lung tissues were also weighed and imaged ex vivo to assess gross lung tumor burden.
4-IPP treatment of tumor-bearing mice
MIF+/+ and MIF−/− C57BL/6 mice were inoculated s.c. with 1 × 105 B16 tumor cells, and 7 days later the mice received intraperitoneally (i.p.) either 4-IPP (80 mg/kg – a dose previously shown to inhibit steady state MIF when injected into naïve mice (20)) – dissolved in corn oil or vehicle control (corn oil) daily for the next 14 days. Tumor growth was monitored every 3–4 days in mice by measuring two opposing diameters with a set of calipers.
Isolation and treatment of peritoneal macrophages
Peritoneal exudate cells (PECs) were obtained by peritoneal lavage from naïve and B16 tumor-bearing, MIF+/+ and MIF−/− C57BL/6 mice, 18–21 days after tumor challenge. For isolation of CD11b+ macrophages, tumor bearing mice were injected i.p. 4 days before harvest with 2 ml of thioglycollate broth (BD Biosciences, San Jose, CA). The CD11b+ macrophages were enriched using the autoMACS ProSeparator (Miltenyi Biotec, Auburn, CA). Isolated cells were treated for 24 hours with 4-IPP (50 μM) or DMSO (vehicle control) in the presence of LPS (0.2 μg/ml from Escherichia coli 0111:B4, Sigma-Aldrich, St. Louis, MO). Please note that this dose of 4-IPP has previously been determined to be maximally inhibitory to cellular MIF (20) and that no monocyte/macrophage cytotoxicity is observed at this concentration (not shown). For anti-MIF antibody treatment, PECs were treated for 24 hours with either neutralizing anti-MIF antibody (100 μg; Clone 3D9 a generous gift of Dr. Richard Bucala, Yale University; IgG1 isotype) or IgG1 isotype control antibody (BD Biosciences) and LPS (0.2 μg/ml). To block Fc receptors from interacting with the antibodies, PECs from both treatment groups were initially pre-treated with CD16/CD32 antibody (2.4G2, Functional Grade; BD Biosciences) for 15 min at 4°C. At the end of the culture period, cells were harvested for RNA and supernatants were collected for protein measurements.
Quantitative PCR analysis
For RNA extraction, cells were lysed in buffer RLT (Qiagen, Valencia, CA), homogenized and purified using RNeasy mini kit (Qiagen) following manufacturer’s instructions. Integrity of RNA was checked using a Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA). The RNA was reverse transcribed with MultiScribe reverse transcriptase and oligo(dT) primers (Applied Biosystems, Bedford, MA). Quantitative assessment of mRNA levels was done by real-time reverse transcription PCR on an ABI 7500 FAST instrument with either Taqman fast advanced master mix (Applied Biosystems) or SYBR green ROX qPCR master mix (Qiagen), 0.2 μM forward and reverse primers (Invitrogen, Grand Island, NY). Relative expression profiles of mRNAs were then calculated using the comparative CT method (ΔΔCT method). The ΔΔCT was calculated as the difference between the normalized CT values (ΔCT = CT of target gene – CT of endogenous control gene) of the treatment and the control samples: ΔΔCT = ΔCT treatment – ΔCT control. ΔΔCT was then converted to fold change by the following formula: fold change = 2−ΔΔCT. Primers and probes used in this study were: Stabilin 1, (PPM33734A; Qiagen); Taqman probes (Applied Biosystems) for genes 18S (Hs99999901.s1; VIC; endogenous control gene – works with human and murine mRNA), Interleukin 10 (Mm00439614_m1; FAM), TNF-α (Mm00443258_m1; FAM), Arginase 1 (Mm00475988_m1; FAM), VEGF-a (Mm01281449_m1; FAM), MMP-9 (Mm00442991_m1; FAM), NOS2 (Mm00440502_m1; FAM), Cox2 (Mm00478374_m1; FAM), Mrc1 (Mm00485148_m1; FAM), Chi313/Ym1 (Mm00657889_mH; FAM), Retnla/FIZZ1 (Mm 00445109_m1; FAM), IRF5 (Mm00496477_m1; FAM), IL-12 (Mm00434174_m1; FAM) were used according to manufacturer’s instructions.
Isolation and treatment of subcutaneous and lung tumor associated macrophages (TAMs)
Subcutaneous (s.c.) and lung TAMs were isolated from B16 tumor-bearing, MIF+/+ and MIF−/−C57BL/6 mice 18–21 days after tumor challenge. s.c and lung tumors were enzymatically digested and F4/80+ cells were positively selected using the autoMACS ProSeparator (Miltenyi Biotec). Purity of isolated cells was checked by flow cytometry. s.c. and lung F4/80+ TAMs were washed and treated for 16 hours with 4-IPP (50 μM) or DMSO (vehicle control) and LPS (0.2μg/ml, Sigma-Aldrich). For anti-MIF antibody treatment, TAMs were treated for 16 hours with either neutralizing anti-MIF antibody (100 μg; Clone 3D9 – a generous gift of Dr. Richard Bucala, Yale University; IgG1 isotype) or IgG1 isotype control antibody (BD Biosciences) and LPS (0.2 μg/ml). To block Fc receptors from interacting with the antibodies, TAMs were initially pre-treated with CD16/CD32 antibody (2.4G2, Functional Grade; BD Biosciences) for 15 min at 4°C.
Isolation of Splenic Myeloid Derived Suppressor cells (MDSCs)
Tumor-bearing MIF+/+ and MIF−/− C57BL/6 mice were sacrificed 18–21 days after tumor challenge. GR-1hi Ly-6G+ granulocytic and GR-1dim Ly-6G− monocytic MDSCs were isolated from the spleens using the MDSC isolation kit (Miltenyi Biotec). The purity of cell populations was confirmed by flow cytometry and was >98%.
Phenotypic and quantitative analysis of splenocytes, peritoneal cells and tumor infiltrating leukocytes (TILs)
Single cell suspensions from spleen, PECs and tumors were obtained from naïve or melanoma-bearing MIF+/+ and MIF−/− C57BL/6 mice and stained with relevant antibodies - F4/80, CD11b, CD206, CD23, MHC-II, CD11c, CD80, CD86, Ly6G, Ly6C (from BD Biosciences, San Jose, CA), for 30 min, after blocking with CD16/CD32 antibody (2.4G2; BD Biosciences, San Jose, CA) for 15 min at 4°C. Anti-CD45 antibody was used to selectively exclude CD45− tumor cells from analysis. Cell surface stained cells were analyzed on a FACSCalibur (Becton Dickinson and Company, Franklin Lakes, NJ) and results were analyzed with FlowJo software (TreeStar, Inc., Ashland, OR). To calculate the absolute number of a splenocyte subset in the total pool of splenocytes, the absolute number of total splenocytes (obtained via counting the cells on a haemocytometer) was multiplied by the relative prevalence of that subset (%) obtained via flow cytometry analysis. The results were expressed as the absolute number (106) of the population expressing the antigen of interest. Absolute number of macrophages in PECs and TILs were analysed as described above.
Functional Assays
Freshly isolated TAMs or MDSCs (Granulocytic or monocytic subpopulations) were added in triplicates to 96-well plates at the indicated cell number ratios and – in some cases - pre-treated with 4-IPP (50 μm) or DMSO (vehicle control) for 16 hours. Splenocytes from OT-1 mice were then added at the appropriate dilution in triplicate to wells containing TAMs or MDSCs in presence of the ovalbumin (200 μg/ml; Sigma-Aldrich) and cultured for an additional 72 hours. Eighteen hours before harvesting, co-cultures were pulsed with [3H]-thymidine (1 μCi/well; MP Bioscience). [3H]-Thymidine uptake was counted using a liquid scintillation counter and relative cpms were used to determine % inhibition of proliferation.
Migration and tube formation Assay (i) HUVEC migration assay
Human umbilical vein endothelial cells (HUVECs), (Cambrex, Walkersville, MD) were maintained in EGM media (Cambrex) supplemented with growth factors and Gentamicin/Amphotericin-B and passaged using TrypLE (Invitrogen, Carlsbad, CA). For functional assays, HUVECs were re-suspended in serum-free media, counted and plated on Matrigel transwell chambers (BD Biosciences, San Jose, CA) and co-cultured in supernatants from MIF+/+, MIF−/−, MIF+/+ control and MIF+/+ 4-IPP treated TAMs for 24 hours at 37°C with 5% CO2. The migrated cells were fixed with 4% formaldehyde and stained with Crystal violet (0.1% in 20% ethanol). HUVEC migration was quantitated by manually counting the number of cells on the inserts under low power at (40x) magnification. (ii) HUVEC tube formation assay - HUVECs were resuspended in conditioned media from MIF+/+, MIF−/−, MIF+/+ control and MIF+/+ 4-IPP treated and dispensed into wells pre-coated with matrigel and incubated for 24 hrs. Tubes were quantified by counting the number of connecting branches between discrete endothelial cells.
ELISAs
Cytokines were measured by ELISA in supernatants from PECs or TAMs cultures. ELISA kits used were the murine IL-10, TNF-α, VEGF, MMP-9 and IL-12 kits obtained from R&D Systems (Minneapolis, MN).
Western blotting
B16 and B16F10 cells were cultured overnight in serum free media and 500 μl of media was concentrated ~ 25x using 10kD MW Microcon concentrators (Millipore). Concentrated supernatants and lysates of adherent B16 and B16F10 cells were probed with an antibody that recognizes murine MIF (Torrey Pines Biolabs).
Arginase activity and nitric oxide assays
Arginase activity was quantified in cell lysates by measuring the production of urea using the QuantiChrom™ arginase Assay Kit (DARG-200, BioAssays Systems). Nitrite concentrations in culture supernatants were determined using Greiss reagent (Sigma-Aldrich).
Generation and treatment of bone marrow-derived macrophages (BMDMφ)
Bone marrow cells were harvested from femur and tibias of 6–10-wk-old C57BL/6 mice. The cells were cultured in RPMI 1640 supplemented with 10% FBS (GIBCO), recombinant mouse M-CSF (10 ng/ml; R&D Systems) and L929 conditioned medium (15%) in Nunclon surface cell culture plates. Non-adherent cells were collected after 24 hours and were cultured for 7 days in the supplemented medium in Corning/Costar ultralow attachment polystyrene culture plates, changing the medium once on day 4. On day 7, live cells were purified by centrifugation over Fico/Lite-LM (Atlanta Biologicals). The resulting cell population was >98% CD11b+. The cells were resuspended in RPMI 1640 with 10% FBS, dispensed into 24-well cell culture plates and treated for 24 hours with 4-IPP (50 μM) or DMSO (vehicle control) in the presence of LPS (0.2 μg/ml, Sigma-Aldrich).
Statistical analysis
GraphPad Prism 5.0 software (GraphPad Prism Software, Inc., La Jolla, CA) was used for all statistical analyses. Comparisons between groups were done by two tailed Student’s t tests. For all tests, statistical significance was assumed where p<0.05.
Results
MIF-deficiency or small molecule antagonism reduces murine melanoma outgrowth and enhances peripheral macrophage pro-inflammatory responses
Because MIF promotes evasion from anti-tumor cytotoxic T lymphocyte (CTL) responses (15, 16), we set out to determine how or whether MIF regulates pro-tumorigenic phenotypes and macrophage responses in tumor bearing mice. B16 murine melanoma cells were injected subcutaneously into syngeneic C57Bl/6 MIF+/+ and MIF−/− mice and tumor outgrowth was followed by caliper measurements. As shown in Fig. 1A, melanomas from MIF-deficient mice grew out at a significantly slower rate than those developing in mice with functional MIF (Fig. 1A). Consequently, MIF-deficient mice showed increased survival when compared to MIF wildtype mice (Fig. 1B). We next determined the relative pro-inflammatory status of peritoneal macrophages from MIF+/+ and MIF−/− tumor bearing mice using surrogate pro-inflammatory marker expression in peritoneal exudate cells (PECs). As shown in Fig. 1C, TNF-α mRNA and protein levels, IL-12 mRNA/protein, COX-2 mRNA and inducible NOS (iNOS) mRNA and corresponding nitric oxide levels were substantially higher in PECs derived from tumor bearing MIF-deficient mice than those from MIF wildtype mice suggesting that MIF functionally promotes a reduced pro-inflammatory macrophage phenotype in tumor bearing mice. In support of a functional role for host effector cell-derived MIF – as opposed to implanted tumor cell-derived MIF – in suppressing the inflammatory phenotype observed in tumor bearing mice (Fig. 1C), PECs from B16 tumor bearing MIF+/+ mice treated ex vivo with the MIF small molecule suicide antagonist, 4-iodo-6-phenylpyrimidine (4-IPP) (20), expressed significantly higher levels of TNF-α mRNA and protein than control PECs (Fig. 1D).
Figure 1. Host cell-derived MIF promotes B16 melanoma outgrowth and reduces pro-inflammatory macrophage phenotypes in mice.

(A) MIF-deficiency impairs in vivo tumor growth in B16 transplanted mice. C57BL/6 MIF+/+ and MIF−/− mice were challenged with B16 cells s.c. and tumor volumes were plotted. The data represent the average tumor volumes of 8 mice/group ± SEM representative of three independent experiments. (B) Tumor growth was monitored daily in all animals until sacrifice due to tumors exceeding 5% of body weight. (C, D) Resident PECs from MIF+/+ and MIF−/− C57BL/6 mice bearing a s.c. melanoma tumor (n = 10) were pooled and activated in vitro with (C) LPS alone or (D) either LPS and DMSO (vehicle control) or LPS and 4-IPP (50 μM) for 24 hours. mRNA and protein expression was analyzed by qPCR and ELISA. Nitric oxide levels were measured using Greiss reagent. Data represents the average ± SEM of duplicate samples and are representative of three independent experiments. P values = *, p≤0.05; **, p≤0.005; ***, p≤0.0005.
We next tested whether systemic in vivo neutralization of MIF with 4-IPP recapitulates MIF-deficiency in reducing established B16 melanoma progression. 4-IPP treatment resulted in a significant impairment of B16 outgrowth and progression but with only a modest increase in survival rates compared to vehicle alone control mice (Fig 2A and 2B). Importantly, PECs from 4-IPP treated mice expressed pro-inflammatory effectors at significantly higher levels than those from control mice (Fig. 2C) – effectively phenocopying MIF-deficiency (Fig. 1C). To ascertain that 4-IPP treatment had no off-target effects, MIF−/− mice were treated with 4-IPP or vehicle control and B16 melanoma tumor growth was monitored. No difference in tumor progression or percent survival was observed between vehicle control and 4-IPP treated MIF-deficient mice (Supplemental Fig. 1A, 1B). Furthermore, the pro-inflammatory cytokine responses of peripheral macrophages were similar in vehicle and 4-IPP treated MIF-deficient mice confirming the lack of any residual in vivo 4-IPP activity in the absence of its target – MIF (Supplemental Fig. 1C).
Figure 2. Pharmacologic inhibition of MIF delays in vivo melanoma tumor outgrowth and enhances peripheral macrophage pro-inflammatory profiles.

(A) C57BL/6 MIF+/+ mice were injected with B16 cells (s.c.). 7 days post tumor inoculation, mice were treated i.p. with 4-IPP (80 mg/kg in corn oil) or vehicle control (corn oil) for 14 days and tumor volumes were plotted. Data represents the average tumor volumes of 10 mice/group ± SEM and are representative two independent experiments. (B) Tumor growth was monitored daily in all animals until sacrifice due to tumors exceeding 5% of body weight. (C) Resident PECs from C57BL/6 mice bearing a s.c. melanoma tumor (n = 10) were pooled and activated in vitro with LPS and either DMSO (vehicle control) or 4-IPP (50 μM) for 24 hours. mRNA, protein and nitric oxide levels were analyzed from indicated cells. Data represents the average of ± SEM of duplicate samples representative of three independent experiments. P values = *, p≤0.05; **, p≤0.005; ***, p≤0.0005.
MIF promotes alternative activation markers and reduces classical activation markers of peripheral macrophage polarization in tumor bearing mice
Prior studies have shown that tumor-bearing mice develop alternatively activated macrophages in distal sites such as the spleen and peritoneal cavity that ultimately serve to suppress peripheral inflammatory and immune responses (21, 22). Consistent with these reports, resident peritoneal cells from B16 melanoma-bearing mice display increased pro-tumoral, M2-type marker expression – ARG-1, IL-10, VEGF-A – and decreased M1-type marker expression – TNF-α and IL-12 – compared to peritoneal cells from naïve mice (data not shown), indicating that, in tumor-bearing hosts, peripheral macrophages are skewed toward an M2-like, tumor-promoting phenotype. Hypothesizing that the pro-inflammatory polarization profile observed in MIF-deficient/inhibited PECs was indicative of reduced peripheral alternative macrophage activation (23, 24), we determined the expression of several M2 markers, arginase-1 (ARG-1) (25), IL-10 (25) and stabilin-1 (STAB-1) (26) in relation to the classical (M1) macrophage activation marker, TNF-α in 4-IPP treated MIF+/+ peritoneal macrophages. As shown in Fig. 3, ARG-1 mRNA (Fig. 3A) and activity (Fig. 3B), IL-10 mRNA/protein (Figs. 3A, 3B), and STAB-1 mRNA (Fig. 3A) expression was significantly reduced – and TNF-α mRNA/protein was increased (Fig. 3A, 3B) – in 4-IPP treated CD11b purified peritoneal macrophages isolated from melanoma bearing MIF+/+ mice. Importantly, this same trend was observed in 4-IPP-treated bone marrow-derived macrophages isolated from MIF+/+ tumor bearing mice (data not shown).
Figure 3. Alternative activation of peritoneal macrophages from tumor-bearing mice is altered by in vitro 4-IPP treatment.
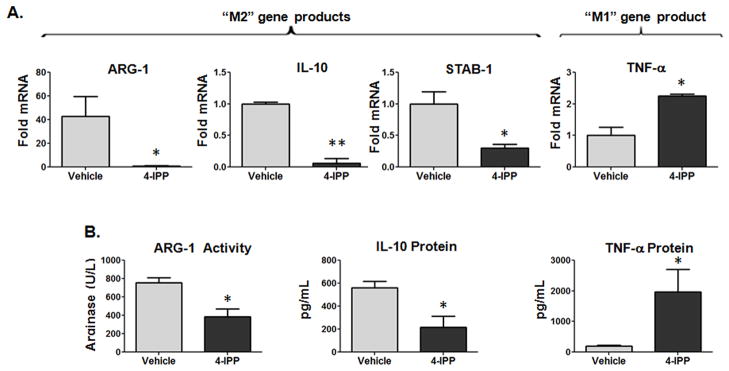
(A, B) CD11b+ peritoneal macrophages from melanoma bearing C57BL/6 MIF+/+ mice (n = 10) and treated in vitro with LPS and either DMSO (vehicle control) or 4-IPP (50 μM) for 24 hours. (A) mRNA; (B) arginase activity and protein expression was analyzed from indicated cells. Data represents the average ± SEM of duplicate samples representative of two independent experiments. P values = *, p≤0.05; **, p≤0.005.
Because alternatively activated stromal macrophages within malignant lesions are important determinants of local tumor immunosuppression (5), we next investigated the polarization profiles and functional activities of B16 melanoma tumor-associated macrophages (TAMs) from MIF wildtype and MIF-deficient mice. As shown in Fig. 4A, F4/80+ cells isolated from B16 lesions from MIF−/− mice had significantly reduced expression of the alternative activation marker, ARG-1 and increased classical activation marker, TNF-α mRNA (Fig. 4A, top panel) and activity/protein (Fig. 4A, bottom panel). In contrast to peritoneal monocyte/macrophage populations (Fig. 1C), IL-12 expression was undetectable in F4/80+ TAMs from MIF+/+ wildtype mice consistent with prior studies (27–29) and MIF-deficiency/inhibition was unable to reverse this effect in TAMs (not shown). Consistent with a reduced alternative activation immunosuppressive phenotype, MIF-deficient TAMs were less active in suppressing antigen (ovalbumin)-induced splenocyte activation/proliferation (30) than TAMs from MIF wildtype mice (Fig. 4B).
Figure 4. TAM alternative activation profile and phenotype is attenuated by MIF-deficiency and 4-IPP treatment.

(A, C) F4/80+ TAMs from MIF+/+ and MIF−/− C57BL/6 mice (n = 10) bearing a s.c. melanoma tumor were pooled and activated in vitro with (A) LPS alone or (C) LPS in the presence of either DMSO (vehicle control) or 4-IPP (50 μM) for 24 hours. Cell lysates were analyzed for mRNA, protein expression and arginase activity. (B, D) F4/80+ TAMs from tumor-bearing MIF+/+ and MIF−/− C57BL/6 mice (n = 10) were untreated or pre-treated for 16 hours with 4-IPP (50 μm) or DMSO (vehicle control). Splenocytes from OT-1 mice were added in triplicate to wells containing TAMs in the presence of ovalbumin (200 μg/ml) and cultured for 72 hours. Eighteen hours before harvesting, co-cultures were pulsed with [3H]-thymidine. Data represents the average ± SEM of duplicate samples (A, C) or average ± SD of triplicate samples (B, D) representative of three independent experiments. P values = *, p≤0.05; **, p≤0.005; ***, p≤0.0005.
To further evaluate the relative importance of macrophage-derived MIF in promoting the polarization profile and phenotype of alternatively activated tumor stromal macrophages, TAMs from MIF+/+ mice were isolated, treated ex vivo with 4-IPP followed by expression and phenotypic profiling. As shown in Fig. 4C, 4-IPP effectively reduced ARG-1 expression and increased TNF-α similar to the trend observed with peritoneal macrophages (Fig. 3) and MIF-deficient TAMs (Fig. 4A). Importantly, 4-IPP pre-treatment of TAMs reduced the immunosuppressive phenotype of alternatively activated TAMs (Fig. 4D) similar to that observed with MIF-deficiency (Fig. 4B) consistent with the reduced anti-inflammatory – and enhanced pro-inflammatory – expression profile (Fig. 4C). For further validation of a functional role for MIF in promoting macrophage alternative activation in tumor bearing mice, qPCR analyses of PECs and TAMs revealed that the mRNA expression of several additional well characterized M2 alternative activation markers (Retnla/FIZZ1, Mrc-1 and Chi313/Ym1) were reduced in MIF-deficient PECs (Supplemental Fig. 2A) and TAMS (Supplemental Fig. 2B). Importantly, ex vivo treatment of MIF+/+ PECs and TAMs similarly resulted in significantly decreased FIZZ-1, Mrc-1 and YM1 expression (not shown). PECs from MIF-deficient mice also displayed significantly increased expression of the M1 classical activation marker IRF5 (Supplemental Fig. 2A), while IRF5 expression was unchanged in TAMs from MIF−/− mice (Supplemental Fig 2B). Flow cytometric analyses of CD45+ F4/80+ MIF+/+ and MIF−/− TAMs also revealed that the expression of cell surface-associated M2 markers, CD206 and CD23, were reduced, while M1 cell surface-associated markers, MHC-II, CD11c, CD80, and CD86, were increased in MIF−/− TAMs compared to MIF+/+ TAMs (Supplemental Fig. 2C). Finally, it is important to note that the total numbers of CD11b+F4/80+ macrophages within splenocytes, peritoneal cells and CD45+ tumor infiltrating leukocytes were not significantly different between MIF+/+ and MIF−/− B16 tumor-bearing mice (not shown). Combined, these studies strongly indicate that macrophage-derived MIF is an important determinant of peripheral and intratumoral macrophage alternative activation in B16 melanoma-bearing mice.
MIF promotes melanoma metastases and TAM polarization in colonized organs
Alternatively activated macrophages promote metastatic dissemination and colonization (31, 32). To investigate whether stromal MIF contributes to melanoma pulmonary metastases and colonization, luciferase-expressing B16F10 metastatic melanoma cells were injected intravenously into syngeneic MIF+/+ or MIF−/− mice and progression followed by whole body luminescence. Imaging on day 21 post intravenous injection of B16F10 cells (Fig. 5A, 5B) or days 18, 21 and 23 (Fig. 5B) revealed significantly reduced lung tumor burden in MIF−/− mice compared to MIF+/+ mice. Lungs from MIF-deficient mice exhibited reduced metastatic tumor burden both visually (Fig. 5C) and as a function of total lung mass (Fig. 5D). To determine whether macrophage polarization was altered in the lungs of MIF-deficient vs. wildtype mice, F4/80+ cells were purified from metastatic tumor bearing lungs from MIF+/+ and MIF−/− mice and evaluated for M2 and M1 marker expression by qPCR. As shown in Fig. 6A, the immunosuppressive effectors, ARG-1 and IL-10 mRNA and activity/protein, were significantly reduced while TNF-α mRNA and protein were elevated in lung TAMs from MIF-deficient mice. It is important to note that MIF is expressed and secreted at very high levels in both B16 and B16F10 murine melanoma cells (Fig. 6B), suggesting that tumor cell-derived MIF does not independently dictate intratumoral macrophage polarization – at least in initial melanoma outgrowth – and that macrophage-derived MIF dominates the macrophage phenotype. This is consistent with our observations that 4-IPP – added ex vivo to polarized MIF+/+ TAMs similarly reverts TAMs from an M2 expression profile towards an M1 profile (Fig. 4C and Supplemental Fig. 3A).
Figure 5. MIF-deficiency decreases lung metastasis in an experimental metastasis model of melanoma.
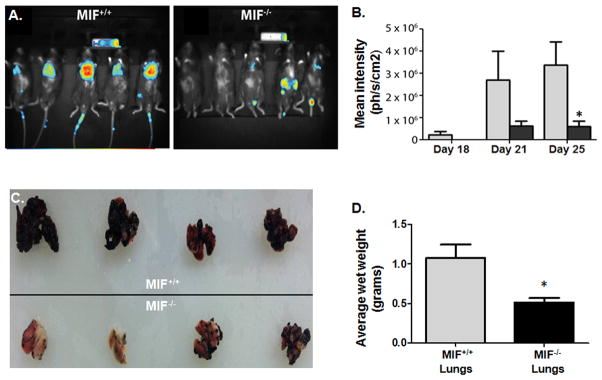
B16-F10-luc2 cells were injected into the tail vein of MIF+/+ and MIF−/− mice (n=10). (A) Ventral images taken on day 21 are shown for representative mice (n = 5). (B) Bioluminescence was quantified from the in vivo signals emitted from ventral views. Data shown are representative of three independent experiments (n = 5). (C) Mice were sacrificed on day 25 and lungs were perfused, harvested and photographed. (D) Lung tumor burden in tumor-bearing MIF+/+ and MIF−/− mice. Data represents the average weights of 4 sets of lungs/group ± SEM and are representative of two independent experiments. P value = *, p≤0.05.
Figure 6. Host effector cell-derived MIF promotes macrophage alternative activation in metastatic melanoma bearing lungs.

(A) Lung TAM polarization. F4/80+ TAMs were isolated from the lungs of B16-F10-bearing MIF+/+ and MIF−/− C57BL/6 mice (n = 10). Cells were pooled and activated in vitro with LPS for 24 hours. mRNA, protein expression and arginase activity was analyzed from indicated cells. (B) Concentrated supernatants and cell lysates from B16 and B1610 cells were immunoblotted for MIF content determination. (C) F4/80+ TAMs from lungs of B16-F10 tumor-bearing MIF+/+ and MIF−/− C57BL/6 mice (n = 10) were pooled and cultured in triplicates at the indicated cell number ratios for 16 hours. Splenocytes from OT-1 mice were added to wells containing lung TAMs in presence of ovalbumin (200 μg/ml) and cultured for 72 hours. Eighteen hours before harvesting, co-cultures were pulsed with [3H]-thymidine. Data represents the average ± SEM of duplicate (A) or average ± SD of triplicate (C) samples representative of two independent experiments. P values = *, p≤0.05; **, p≤0.005.
Because MIF-deficient TAMs from subcutaneous melanoma lesions were found to be less immunosuppressive than those from MIF wildtype mice (Fig. 4B), we next tested whether MIF-deficient lung TAM immunosuppression was similarly dysfunctional. As shown in Fig. 6C, TAM-mediated inhibition of antigen-induced splenocyte proliferation was reduced in MIF-deficient lung TAMs when compared with lung TAMs from MIF wildtype mice. Additionally, and consistent with the defective macrophage alternative activation profile observed with ex vivo inhibition of macrophage-derived MIF (Supplemental Fig. 3A), pre-treatment of MIF+/+ lung TAMs with 4-IPP reduced the immunosuppressive potential of these cells (Supplementary Fig. 3B).
MIF-dependent TAM polarization promotes macrophage angiogenic potential
Alternative activation of tumor-associated macrophages represents a critically important component of neo-angiogenic potential in developing neoplasms (33, 34). Because MIF has previously been shown to promote the expression of pro-angiogenic growth factors from macrophages in in vitro settings (35), we hypothesized that macrophage-derived MIF contributes to not only the anti-inflammatory and immunosuppressive phenotype of TAMs, but may also promote pro-angiogenic TAM potential within malignant lesions. F4/80+ TAMs from the lungs of MIF+/+ and MIF−/− mice were examined for relative expression of pro-angiogenic effectors. As shown in Fig. 7A, MIF-deficient lung TAMs expressed reduced mRNA and protein levels of both VEGF and MMP-9 – two critically important mediators of TAM angiogenic potential (11, 36) compared to MIF wildtype lung TAMs. To determine whether MIF-deficient lung TAMs were functionally defective in promoting tumor vascularization, supernatants from MIF+/+ and MIF−/− lung TAMs were co-cultured with human umbilical vein endothelial cells (HUVECs) and endothelial cell migration and tube formation was assessed. Supernatants from MIF+/+ lung TAMs were significantly more active in inducing HUVEC migration (Fig. 7B) and tube formation (Fig. 7C) in vitro than lung TAMs from MIF-deficient mice. Similarly, decreased VEGF and MMP-9 expression as well as HUVEC tube forming potential was reduced by ex vivo treatment of MIF wildtype lung TAMs with 4-IPP (Supplemental Fig. 4A, 4B). Finally, 4-IPP ex vivo treatment of MIF+/+ TAMs from B16 subcutaneous lesions (Supplemental Fig. 4C), PECs from B16 tumor bearing MIF+/+ mice (Supplemental Fig. 4D) and bone marrow-derived macrophages from B16 MIF+/+ mice (Supplemental Fig. 4E) resulted in a similar reduction of VEGF and MMP-9 expression. Combined, these studies indicate that TAM-derived MIF is a critical determinant of melanoma TAM polarization, immunosuppression and angiogenic potential.
Figure 7. MIF contributes to pro-angiogenic potential of TAMs.

(A) F4/80+ TAMs were isolated from the lungs of B16-F10-bearing MIF+/+ and MIF−/− C57BL/6 mice (n = 10). Cells were pooled and activated in vitro with LPS for 24 hours. mRNA and protein expression was analyzed in cell lysates. (B) 1 × 105 HUVECs were plated in the upper chamber on collagen coated Transwell filters and supernatants from F4/80+ TAMs (A) were added to the bottom chambers. 24 hours later, migrated cells on the bottom of the filter were fixed, stained and manually counted under low power (40x). (C) Supernatants from F4/80+ TAMs (A) were used to re-suspend 2 × 105 HUVECs and then plated on GF-depleted Matrigel plugs for 16 hours. Tubes were quantified by counting the number of connecting branches between discrete endothelial cells. Data represents the average ± SEM of duplicate (A) or triplicate (B, C) samples and are representative of two independent experiments. P values = *, p≤0.05; **, p≤0.005; ***, p≤0.0005.
MIF-deficiency or small molecule inhibition reduces splenic MDSC immune suppression in tumor bearing mice
Myeloid-derived suppressor cells (MDSCs) promote tumor progression through both immune-dependent (37) and immune-independent (38) mechanisms. When MDSCs populate the tumor microenvironment they become F4/80+ (39) and, therefore, represent an important subpopulation of the immunosuppressive TAM population in malignant lesions (Figs. 4, 6 and 7). To determine whether MIF participates in measureable MDSC-specific immunosuppression, we determined relative MDSC immunosuppressive activities in the spleens of tumor bearing MIF+/+ and MIF−/− mice. Initial attempts to isolate sufficient numbers of splenic MDSCs from B16 tumor bearing mice were unsuccessful. To circumvent this problem, MIF+/+ and MIF−/− mice were inoculated with the well characterized peripheral MDSC-eliciting cell line, Lewis lung carcinoma (LLC) (40, 41). Pooled spleens were used to purify both GR-1hi Ly-6G+ granulocytic, and GR-1dim Ly-6G− monocytic MDSCs from either MIF+/+ or MIF−/− LLC tumor bearing mice and evaluated for their relative suppression of ovalbumin-induced lymphocyte proliferation (40). GR-1hi Ly-6G+ and GR-1dim Ly-6G MDSCs isolated from LLC-bearing MIF-deficient mice were significantly less immunosuppressive than MIF wildtype MDSCs (Fig. 8A, 8B). Importantly, MIF-deficiency caused no significant changes in the raw numbers of spleen-derived CD11b+GR1hi or CD11b+GR1dim MDSCs or in the total MDSCs when compared to MDSCs numbers obtained in MIF+/+ mice (not shown). Finally, this MIF-deficient MDSC phenotype was fully recapitulated by 4-IPP pre-treatment of MIF+/+ granulocytic and monocytic MDSCs isolated from LLC tumor bearing mice (Fig. 8C, 8D) again suggesting that effector cell-derived MIF is necessary for the observed alleviation of MDSC immunosuppressive activity.
Figure 8. MIF-deficiency or small molecule inhibition reduces splenic MDSC immune suppression in tumor bearing mice.
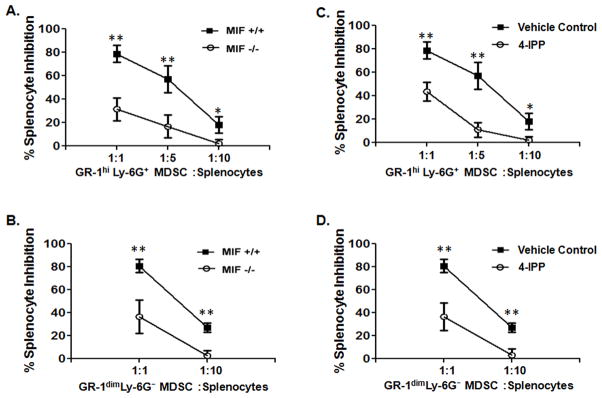
GR-1hi Ly-6G+ granulocytic and GR-1dim Ly-6G−monocytic MDSCs were isolated from spleens of MIF+/+ and MIF−/− C57BL/6 mice bearing a s.c. melanoma tumor (n = 10). (A, B) Cells were either untreated or (C, D) pre-treated with 4-IPP (50 μm) or DMSO (vehicle control). Cells were cultured in triplicates at the indicated cell number ratios for 16 hours. Splenocytes from OT-1 mice were then added to wells containing MDSCs in presence of the ovalbumin (200 μg/ml) and cultured for additional 72 hours. Eighteen hours before harvesting, co-cultures were pulsed with [3H]-thymidine. Data represents the average ± SD of triplicate samples representative of two independent experiments. P values = *, p≤0.05; **, p≤0.005.
Discussion
Our findings suggest that monocyte/macrophage-derived MIF contributes to TAM polarization and functionally promotes an immune-suppressive, pro-angiogenic and alternatively activated (M2-like) phenotype in tumor bearing mice. MIF induces pro-inflammatory cytokine responses in several disease pathologies including arthritis (42), acute respiratory distress syndrome (43), atherogenesis (44), glomerulonephritis (45), and Type I diabetes (46). However, our results clearly indicate that, in malignant disease settings, MIF is a central participant in macrophage M2-like, anti-inflammatory cytokine responses. As an autocrine-acting cytokine, MIF exerts signal initiating events via interaction with its cognate cell surface receptor, CD74 (47). Pro-inflammatory signaling in macrophages by MIF requires CD74 and its first identified co-receptor, CD44 (48, 49). However, recent studies show that CD74 also forms MIF-sensitive co-receptor complexes with several chemokine receptors including CXCR2, CXCR4 and CXCR7 (50–52). Given this differential co-receptor usage by soluble MIF, it is tempting to speculate that MIF signals to pro-inflammatory, classically activated pathways or anti-inflammatory, alternatively activated pathways in a manner that is dictated by which CD74 co-receptors are preferentially expressed on a given cell type. That being said, in vitro neutralization of MIF with a monoclonal antibody was unable to recapitulate MIF-deficiency or 4-IPP antagonism in decreasing M2 and increasing M1 marker expression profiles in PECs from tumor bearing mice (data not shown). Since 4-IPP interferes with MIF secretion (53), it is conceivable that there is a distinct and stringent requirement for local MIF secretion and/or receptor dynamics in macrophage populations that is absent in other cell types where autocrine acting MIF is readily inhibited by neutralizing antibodies (54, 55).
In vivo reprogramming of macrophage polarization by loss or inhibition of host MIF coincides with a significant reduction in subcutaneous and metastatic melanoma tumor growth and progression (Figs. 1, 2 and 5). Our observed defects in MIF-deficient TAM and MDSC immunosuppressive activities (Figs. 4, 6 and 8), coupled with prior studies showing that MIF neutralization in tumor bearing hosts induces cytotoxic T lymphocyte activities (15), is suggestive of an important role for MIF in tumor-induced innate and adaptive immune tolerance and immunosuppression. One caveat to these studies is that tumor size differences could, in theory, influence relative TAM polarization states and phenotypes in an MIF-independent manner. However, our results indicate that when equal sized tumors across different comparison groups are analyzed, we still find significant re-orientation of TAM polarization/immunosuppressive function in MIF-deficient and 4-IPP-treated mice suggesting that TAM re-polarization associated with MIF-deficiency/inhibition is not attributable to differences in tumor size.
It is interesting to note that, like other M2-elicited cytokines/growth factors, MIF promotes functional tumor-promoting phenotypes in malignant cells (56–59). These autocrine tumor promoting activities of secreted MIF on malignant disease processes coincide with paracrine effects on stromal macrophages. For example, tumor cell-derived MIF regulates CXCL8 (IL-8) and VEGF expression in human monocytes (35, 60), consistent with our findings (Fig. 7). As such, soluble MIF elicited from TAMs within a tumor’s stromal compartment likely promotes not only M2 TAM polarization in an autocrine manner but also amplifies paracrine-signaling on tumor cells. In this scenario, tumor-derived MIF would also be expected to promote M2 TAM polarization. Interestingly, our studies in MIF-deficient mice suggest that the bulk of MIF’s M2 promoting activity in early melanoma outgrowth/progression (Figs. 1 and 5) derives primarily from stromal, host-derived MIF as B16 and B16F10 melanoma cell lines express copious amounts of MIF (Fig. 6B). In further support of this scenario, 4-IPP has negligible anti-tumor efficacy in immuno-deficient SCID mouse models of established tumorigenesis (data not shown) suggesting that 4-IPP inhibition of established melanoma outgrowth (Fig. 2A) is independent of melanoma-derived MIF inhibition and dependent on inhibition of host immune effector cell-derived MIF (Fig. 1C, 2B).
During the preparation of this manuscript a study was published by Wang and colleagues describing a contributing role for MIF in teratoma progression through its ability to promote macrophage-dependent angiogenesis (61). Our study validates and extends these findings by demonstrating an important role for MIF in murine melanoma TAM alternative activation. We further demonstrate that, in addition to regulating TAM angiogenic potential, MIF contributes to TAM and MDSC immunosuppressive phenotypes in tumor bearing mice. An important component of the Wang study was the use of MIF−/− embryonic stem cells to demonstrate the importance of stromal cell-derived MIF to teratoma outgrowth (61). In contrast, our efforts to utilize lentiviral shRNA to deplete melanoma MIF in B16 and B16F10 cell lines were unsuccessful (not shown) but the findings by Wang support our conclusion that – at least during initial melanoma outgrowth – stromal macrophage MIF contributes dominantly to macrophage polarization. In further support for this conclusion, a study by the Dranoff group provides clinically relevant evidence that MIF promotes tumor promoting TAM angiogenic properties in late stage malignant melanoma patients (62). MIF autoantibodies were identified in chemotherapy responding melanoma patients and these neutralizing MIF antibodies functionally attenuated Tie-2 and matrix metalloproteinase-9 (MMP-9) expression in human TAMs leading to disrupted tumoral vasculature and lymphocyte/granulocyte infiltrates (62).
Simpson et al very recently reported that in an aggressive 4T-1 model of metastatic breast cancer, tumor-derived MIF promotes tumor growth and pulmonary metastasis by influencing the differentiation of inflammatory cells within the tumor microenvironment (63). Our studies additionally indicate that the MIF-dependent effects on alternative activation observed with PECs and TAMs are not restricted to melanoma tumors. Analysis of PECs and TAMs from Lewis Lung Carcinoma (LLC)-bearing mice reveals that MIF-deficiency and 4-IPP inhibition similarly skews TAM from an M2-like expression pattern/phenotypic profile towards an M1-like expression pattern/phenotype (data not shown). Clearly more studies are needed to fully demonstrate the precise contribution of aberrant macrophage polarization to defective melanoma outgrowth and lung metastases in MIF-deficient mice but, combined with other recent studies (61–63), there is now considerable evidence to suggest that MIF is an important determinant of TAM pro-tumorigenic activities.
In conclusion, these studies describe a functional role for MIF in contributing to the alternative activation of macrophages in tumor bearing hosts. Perhaps more importantly, our discovery that the loss or inhibition of MIF effectively re-programs TAM polarization coinciding with attenuated melanoma progression and pulmonary metastases suggests that immunotherapeutic targeting of MIF may one day provide significant clinical benefits for melanoma patients.
Supplementary Material
Acknowledgments
Grant Support: This work was supported in part by NIH CA129967 (R.A.M), and NIH/NCRR P20RR018733 (K.Y.). J.W.E is supported by the Commonwealth of Kentucky Research Trust Fund.
R.A.M is an inventor on patents pertaining to 4-IPP as a novel anti-cancer therapeutic agent targeting MIF. The authors wish to acknowledge Drs. Jill Suttles, Robert Stout and Jun Yan for helpful discussion and insight on TAM alternative activation. We also thank Mr. Huaiyu Zheng for his assistance with mouse tumor imaging and imaging analyses.
Reference List
- 1.Wolchok JD, Saenger Y. The mechanism of anti-CTLA-4 activity and the negative regulation of T-cell activation. Oncologist. 2008;13(Suppl 4):2–9. doi: 10.1634/theoncologist.13-S4-2. [DOI] [PubMed] [Google Scholar]
- 2.Dranoff G. Targets of protective tumor immunity. Ann N Y Acad Sci. 2009;1174:74–80. doi: 10.1111/j.1749-6632.2009.04938.x. [DOI] [PubMed] [Google Scholar]
- 3.Hoos A, Ibrahim R, Korman A, Abdallah K, Berman D, Shahabi V, Chin K, Canetta R, Humphrey R. Development of ipilimumab: contribution to a new paradigm for cancer immunotherapy. Semin Oncol. 2010;37:533–46. doi: 10.1053/j.seminoncol.2010.09.015. [DOI] [PubMed] [Google Scholar]
- 4.Korn EL, Liu PY, Lee SJ, Chapman JA, Niedzwiecki D, Suman VJ, Moon J, Sondak VK, Atkins MB, Eisenhauer EA, Parulekar W, Markovic SN, Saxman S, Kirkwood JM. Meta-analysis of phase II cooperative group trials in metastatic stage IV melanoma to determine progression-free and overall survival benchmarks for future phase II trials. J Clin Oncol. 2008;26:527–34. doi: 10.1200/JCO.2007.12.7837. [DOI] [PubMed] [Google Scholar]
- 5.Mantovani A, Sica A. Macrophages, innate immunity and cancer: balance, tolerance, and diversity. Curr Opin Immunol. 2010;22:231–7. doi: 10.1016/j.coi.2010.01.009. [DOI] [PubMed] [Google Scholar]
- 6.Melief CJ. Cancer immunotherapy by dendritic cells. Immunity. 2008;29:372–83. doi: 10.1016/j.immuni.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 7.Loke P, Allison JP. PD-L1 and PD-L2 are differentially regulated by Th1 and Th2 cells. Proc Natl Acad Sci U S A. 2003;100:5336–41. doi: 10.1073/pnas.0931259100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huber S, Hoffmann R, Muskens F, Voehringer D. Alternatively activated macrophages inhibit T-cell proliferation by Stat6-dependent expression of PD-L2. Blood. 2010;116:3311–20. doi: 10.1182/blood-2010-02-271981. [DOI] [PubMed] [Google Scholar]
- 9.Terrazas LI, Montero D, Terrazas CA, Reyes JL, Rodriguez-Sosa M. Role of the programmed Death-1 pathway in the suppressive activity of alternatively activated macrophages in experimental cysticercosis. Int J Parasitol. 2005;35:1349–58. doi: 10.1016/j.ijpara.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 10.Crowther M, Brown NJ, Bishop ET, Lewis CE. Microenvironmental influence on macrophage regulation of angiogenesis in wounds and malignant tumors. J Leukoc Biol. 2001;70:478–90. [PubMed] [Google Scholar]
- 11.Luo Y, Zhou H, Krueger J, Kaplan C, Lee SH, Dolman C, Markowitz D, Wu W, Liu C, Reisfeld RA, Xiang R. Targeting tumor-associated macrophages as a novel strategy against breast cancer. J Clin Invest. 2006;116:2132–41. doi: 10.1172/JCI27648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bernhagen J, Calandra T, Mitchell RA, Martin SB, Tracey KJ, Voelter W, Manogue KR, Cerami A, Bucala R. MIF is a pituitary-derived cytokine that potentiates lethal endotoxaemia. Nature. 1993;365:756–9. doi: 10.1038/365756a0. [DOI] [PubMed] [Google Scholar]
- 13.Calandra T, Spiegel LA, Metz CN, Bucala R. Macrophage migration inhibitory factor is a critical mediator of the activation of immune cells by exotoxins of Gram-positive bacteria. Proc Natl Acad Sci U S A. 1998;95:11383–8. doi: 10.1073/pnas.95.19.11383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McDevitt MA, Xie J, Shanmugasundaram G, Griffith J, Liu A, McDonald C, Thuma P, Gordeuk VR, Metz CN, Mitchell R, Keefer J, David J, Leng L, Bucala R. A critical role for the host mediator macrophage migration inhibitory factor in the pathogenesis of malarial anemia. J Exp Med. 2006;203:1185–96. doi: 10.1084/jem.20052398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abe R, Peng T, Sailors J, Bucala R, Metz CN. Regulation of the CTL response by macrophage migration inhibitory factor. J Immunol. 2001;166:747–53. doi: 10.4049/jimmunol.166.2.747. [DOI] [PubMed] [Google Scholar]
- 16.Zhou Q, Yan X, Gershan J, Orentas RJ, Johnson BD. Expression of macrophage migration inhibitory factor by neuroblastoma leads to the inhibition of antitumor T cell reactivity in vivo. J Immunol. 2008;181:1877–86. doi: 10.4049/jimmunol.181.3.1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krockenberger M, Dombrowski Y, Weidler C, Ossadnik M, Honig A, Hausler S, Voigt H, Becker JC, Leng L, Steinle A, Weller M, Bucala R, Dietl J, Wischhusen J. Macrophage migration inhibitory factor contributes to the immune escape of ovarian cancer by down-regulating NKG2D. J Immunol. 2008;180:7338–48. doi: 10.4049/jimmunol.180.11.7338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Repp AC, Mayhew ES, Apte S, Niederkorn JY. Human uveal melanoma cells produce macrophage migration-inhibitory factor to prevent lysis by NK cells. J Immunol. 2000;165:710–5. doi: 10.4049/jimmunol.165.2.710. [DOI] [PubMed] [Google Scholar]
- 19.Greven D, Leng L, Bucala R. Autoimmune diseases: MIF as a therapeutic target. Expert Opin Ther Targets. 2010;14:253–64. doi: 10.1517/14728220903551304. [DOI] [PubMed] [Google Scholar]
- 20.Winner M, Meier J, Zierow S, Rendon BE, Crichlow GV, Riggs R, Bucala R, Leng L, Smith N, Lolis E, Trent JO, Mitchell RA. A novel, macrophage migration inhibitory factor suicide substrate inhibits motility and growth of lung cancer cells. Cancer Res. 2008;68:7253–7. doi: 10.1158/0008-5472.CAN-07-6227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23:549–55. doi: 10.1016/s1471-4906(02)02302-5. [DOI] [PubMed] [Google Scholar]
- 22.Stout RD, Jiang C, Matta B, Tietzel I, Watkins SK, Suttles J. Macrophages sequentially change their functional phenotype in response to changes in microenvironmental influences. J Immunol. 2005;175:342–9. doi: 10.4049/jimmunol.175.1.342. [DOI] [PubMed] [Google Scholar]
- 23.Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity. 2010;32:593–604. doi: 10.1016/j.immuni.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 24.Prieto-Lafuente L, Gregory WF, Allen JE, Maizels RM. MIF homologues from a filarial nematode parasite synergize with IL-4 to induce alternative activation of host macrophages. J Leukoc Biol. 2009;85:844–54. doi: 10.1189/jlb.0808459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest. 2007;117:175–84. doi: 10.1172/JCI29881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kzhyshkowska J, Workman G, Cardo-Vila M, Arap W, Pasqualini R, Gratchev A, Krusell L, Goerdt S, Sage EH. Novel function of alternatively activated macrophages: stabilin-1-mediated clearance of SPARC. J Immunol. 2006;176:5825–32. doi: 10.4049/jimmunol.176.10.5825. [DOI] [PubMed] [Google Scholar]
- 27.Sica A, Saccani A, Bottazzi B, Polentarutti N, Vecchi A, van Damme J, Mantovani A. Autocrine production of IL-10 mediates defective IL-12 production and NF-kappa B activation in tumor-associated macrophages. J Immunol. 2000;164:762–7. doi: 10.4049/jimmunol.164.2.762. [DOI] [PubMed] [Google Scholar]
- 28.Biswas SK, Gangi L, Paul S, Schioppa T, Saccani A, Sironi M, Bottazzi B, Doni A, Vincenzo B, Pasqualini F, Vago L, Nebuloni M, Mantovani A, Sica A. A distinct and unique transcriptional program expressed by tumor-associated macrophages (defective NF-kappaB and enhanced IRF-3/STAT1 activation) Blood. 2006;107:2112–22. doi: 10.1182/blood-2005-01-0428. [DOI] [PubMed] [Google Scholar]
- 29.Torroella-Kouri M, Ma X, Perry G, Ivanova M, Cejas PJ, Owen JL, Iragavarapu-Charyulu V, Lopez DM. Diminished expression of transcription factors nuclear factor kappaB and CCAAT/enhancer binding protein underlies a novel tumor evasion mechanism affecting macrophages of mammary tumor-bearing mice. Cancer Res. 2005;65:10578–84. doi: 10.1158/0008-5472.CAN-05-0365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Curtsinger JM, Lins DC, Mescher MF. CD8+ memory T cells (CD44high, Ly-6C+) are more sensitive than naive cells to (CD44low, Ly-6C-) to TCR/CD8 signaling in response to antigen. J Immunol. 1998;160:3236–43. [PubMed] [Google Scholar]
- 31.Solinas G, Schiarea S, Liguori M, Fabbri M, Pesce S, Zammataro L, Pasqualini F, Nebuloni M, Chiabrando C, Mantovani A, Allavena P. Tumor-conditioned macrophages secrete migration-stimulating factor: a new marker for M2-polarization, influencing tumor cell motility. J Immunol. 2010;185:642–52. doi: 10.4049/jimmunol.1000413. [DOI] [PubMed] [Google Scholar]
- 32.Redente EF, Dwyer-Nield LD, Merrick DT, Raina K, Agarwal R, Pao W, Rice PL, Shroyer KR, Malkinson AM. Tumor progression stage and anatomical site regulate tumor-associated macrophage and bone marrow-derived monocyte polarization. Am J Pathol. 2010;176:2972–85. doi: 10.2353/ajpath.2010.090879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen W, Ma T, Shen XN, Xia XF, Xu GD, Bai XL, Liang TB. Macrophage-induced tumor angiogenesis is regulated by the TSC2-mTOR pathway. Cancer Res. 2012;72:1363–72. doi: 10.1158/0008-5472.CAN-11-2684. [DOI] [PubMed] [Google Scholar]
- 34.Chen P, Huang Y, Bong R, Ding Y, Song N, Wang X, Song X, Luo Y. Tumor-associated macrophages promote angiogenesis and melanoma growth via adrenomedullin in a paracrine and autocrine manner. Clin Cancer Res. 2011;17:7230–9. doi: 10.1158/1078-0432.CCR-11-1354. [DOI] [PubMed] [Google Scholar]
- 35.White ES, Strom SR, Wys NL, Arenberg DA. Non-small cell lung cancer cells induce monocytes to increase expression of angiogenic activity. J Immunol. 2001;166:7549–55. doi: 10.4049/jimmunol.166.12.7549. [DOI] [PubMed] [Google Scholar]
- 36.Varney ML, Johansson SL, Singh RK. Tumour-associated macrophage infiltration, neovascularization and aggressiveness in malignant melanoma: role of monocyte chemotactic protein-1 and vascular endothelial growth factor-A. Melanoma Res. 2005;15:417–25. doi: 10.1097/00008390-200510000-00010. [DOI] [PubMed] [Google Scholar]
- 37.Talmadge JE. Pathways mediating the expansion and immunosuppressive activity of myeloid-derived suppressor cells and their relevance to cancer therapy. Clin Cancer Res. 2007;13:5243–8. doi: 10.1158/1078-0432.CCR-07-0182. [DOI] [PubMed] [Google Scholar]
- 38.Yang L, DeBusk LM, Fukuda K, Fingleton B, Green-Jarvis B, Shyr Y, Matrisian LM, Carbone DP, Lin PC. Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell. 2004;6:409–21. doi: 10.1016/j.ccr.2004.08.031. [DOI] [PubMed] [Google Scholar]
- 39.Sica A, Bronte V. Altered macrophage differentiation and immune dysfunction in tumor development. J Clin Invest. 2007;117:1155–66. doi: 10.1172/JCI31422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Corzo CA, Cotter MJ, Cheng P, Cheng F, Kusmartsev S, Sotomayor E, Padhya T, McCaffrey TV, McCaffrey JC, Gabrilovich DI. Mechanism regulating reactive oxygen species in tumor-induced myeloid-derived suppressor cells. J Immunol. 2009;182:5693–701. doi: 10.4049/jimmunol.0900092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ozao-Choy J, Ma G, Kao J, Wang GX, Meseck M, Sung M, Schwartz M, Divino CM, Pan PY, Chen SH. The novel role of tyrosine kinase inhibitor in the reversal of immune suppression and modulation of tumor microenvironment for immune-based cancer therapies. Cancer Res. 2009;69:2514–22. doi: 10.1158/0008-5472.CAN-08-4709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mikulowska A, Metz CN, Bucala R, Holmdahl R. Macrophage migration inhibitory factor is involved in the pathogenesis of collagen type II-induced arthritis in mice. J Immunol. 1997;158:5514–7. [PubMed] [Google Scholar]
- 43.Donnelly SC, Haslett C, Reid PT, Grant IS, Wallace WA, Metz CN, Bruce LJ, Bucala R. Regulatory role for macrophage migration inhibitory factor in acute respiratory distress syndrome. Nat Med. 1997;3:320–3. doi: 10.1038/nm0397-320. [DOI] [PubMed] [Google Scholar]
- 44.Pan JH, Sukhova GK, Yang JT, Wang B, Xie T, Fu H, Zhang Y, Satoskar AR, David JR, Metz CN, Bucala R, Fang K, Simon DI, Chapman HA, Libby P, Shi GP. Macrophage migration inhibitory factor deficiency impairs atherosclerosis in low-density lipoprotein receptor-deficient mice. Circulation. 2004;109:3149–53. doi: 10.1161/01.CIR.0000134704.84454.D2. [DOI] [PubMed] [Google Scholar]
- 45.Hoi AY, Hickey MJ, Hall P, Yamana J, O’Sullivan KM, Santos LL, James WG, Kitching AR, Morand EF. Macrophage migration inhibitory factor deficiency attenuates macrophage recruitment, glomerulonephritis, and lethality in MRL/lpr mice. J Immunol. 2006;177:5687–96. doi: 10.4049/jimmunol.177.8.5687. [DOI] [PubMed] [Google Scholar]
- 46.Stosic-Grujicic S, Stojanovic I, Maksimovic-Ivanic D, Momcilovic M, Popadic D, Harhaji L, Miljkovic D, Metz C, Mangano K, Papaccio G, Al-Abed Y, Nicoletti F. Macrophage migration inhibitory factor (MIF) is necessary for progression of autoimmune diabetes mellitus. J Cell Physiol. 2008;215:665–75. doi: 10.1002/jcp.21346. [DOI] [PubMed] [Google Scholar]
- 47.Leng L, Metz CN, Fang Y, Xu J, Donnelly S, Baugh J, Delohery T, Chen Y, Mitchell RA, Bucala R. MIF signal transduction initiated by binding to CD74. J Exp Med. 2003;197:1467–76. doi: 10.1084/jem.20030286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shi X, Leng L, Wang T, Wang W, Du X, Li J, McDonald C, Chen Z, Murphy JW, Lolis E, Noble P, Knudson W, Bucala R. CD44 is the signaling component of the macrophage migration inhibitory factor-CD74 receptor complex. Immunity. 2006;25:595–606. doi: 10.1016/j.immuni.2006.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Naujokas MF, Morin M, Anderson MS, Peterson M, Miller J. The chondroitin sulfate form of invariant chain can enhance stimulation of T cell responses through interaction with CD44. Cell. 1993;74:257–68. doi: 10.1016/0092-8674(93)90417-o. [DOI] [PubMed] [Google Scholar]
- 50.Bernhagen J, Krohn R, Lue H, Gregory JL, Zernecke A, Koenen RR, Dewor M, Georgiev I, Schober A, Leng L, Kooistra T, Fingerle-Rowson G, Ghezzi P, Kleemann R, McColl SR, Bucala R, Hickey MJ, Weber C. MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nat Med. 2007;13:587–96. doi: 10.1038/nm1567. [DOI] [PubMed] [Google Scholar]
- 51.Weber C, Kraemer S, Drechsler M, Lue H, Koenen RR, Kapurniotu A, Zernecke A, Bernhagen J. Structural determinants of MIF functions in CXCR2-mediated inflammatory and atherogenic leukocyte recruitment. Proc Natl Acad Sci U S A. 2008;105:16278–83. doi: 10.1073/pnas.0804017105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tarnowski M, Grymula K, Liu R, Tarnowska J, Drukala J, Ratajczak J, Mitchell RA, Ratajczak MZ, Kucia M. Macrophage migration inhibitory factor is secreted by rhabdomyosarcoma cells, modulates tumor metastasis by binding to CXCR4 and CXCR7 receptors and inhibits recruitment of cancer-associated fibroblasts. Mol Cancer Res. 2010;8:1328–43. doi: 10.1158/1541-7786.MCR-10-0288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Merk M, Baugh J, Zierow S, Leng L, Pal U, Lee SJ, Ebert AD, Mizue Y, Trent JO, Mitchell R, Nickel W, Kavathas PB, Bernhagen J, Bucala R. The Golgi-associated protein p115 mediates the secretion of macrophage migration inhibitory factor. J Immunol. 2009;182:6896–906. doi: 10.4049/jimmunol.0803710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mitchell RA, Metz CN, Peng T, Bucala R. Sustained mitogen-activated protein kinase (MAPK) and cytoplasmic phospholipase A2 activation by macrophage migration inhibitory factor (MIF). Regulatory role in cell proliferation and glucocorticoid action. J Biol Chem. 1999;274:18100–6. doi: 10.1074/jbc.274.25.18100. [DOI] [PubMed] [Google Scholar]
- 55.Liao H, Bucala R, Mitchell RA. Adhesion-dependent signaling by macrophage migration inhibitory factor (MIF) J Biol Chem. 2003;278:76–81. doi: 10.1074/jbc.M208820200. [DOI] [PubMed] [Google Scholar]
- 56.Rendon BE, Roger T, Teneng I, Zhao M, Al-Abed Y, Calandra T, Mitchell RA. Regulation of human lung adenocarcinoma cell migration and invasion by macrophage migration inhibitory factor. J Biol Chem. 2007;282:29910–8. doi: 10.1074/jbc.M704898200. [DOI] [PubMed] [Google Scholar]
- 57.Winner M, Koong AC, Rendon BE, Zundel W, Mitchell RA. Amplification of tumor hypoxic responses by macrophage migration inhibitory factor-dependent hypoxia-inducible factor stabilization. Cancer Res. 2007;67:186–93. doi: 10.1158/0008-5472.CAN-06-3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liao B, Zhong BL, Li Z, Tian XY, Li Y, Li B. Macrophage migration inhibitory factor contributes angiogenesis by up-regulating IL-8 and correlates with poor prognosis of patients with primary nasopharyngeal carcinoma. J Surg Oncol. 2010;102:844–51. doi: 10.1002/jso.21728. [DOI] [PubMed] [Google Scholar]
- 59.Ren Y, Chan HM, Li Z, Lin C, Nicholls J, Chen CF, Lee PY, Lui V, Bacher M, Tam PK. Upregulation of macrophage migration inhibitory factor contributes to induced N-Myc expression by the activation of ERK signaling pathway and increased expression of interleukin-8 and VEGF in neuroblastoma. Oncogene. 2004;23:4146–54. doi: 10.1038/sj.onc.1207490. [DOI] [PubMed] [Google Scholar]
- 60.White ES, Flaherty KR, Carskadon S, Brant A, Iannettoni MD, Yee J, Orringer MB, Arenberg DA. Macrophage migration inhibitory factor and CXC chemokine expression in non-small cell lung cancer: role in angiogenesis and prognosis. Clin Cancer Res. 2003;9:853–60. [PubMed] [Google Scholar]
- 61.Wang X, Chen T, Leng L, Fan J, Cao K, Duan Z, Zhang X, Shao C, Wu M, Tadmori I, Li T, Liang L, Sun D, Zheng S, Meinhardt A, Young W, Bucala R, Ren Y. MIF produced by bone marrow-derived macrophages contributes to teratoma progression after embryonic stem cell transplantation. Cancer Res. 2012;72:2867–78. doi: 10.1158/0008-5472.CAN-11-3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schoenfeld J, Jinushi M, Nakazaki Y, Wiener D, Park J, Soiffer R, Neuberg D, Mihm M, Hodi FS, Dranoff G. Active immunotherapy induces antibody responses that target tumor angiogenesis. Cancer Res. 2010;70:10150–60. doi: 10.1158/0008-5472.CAN-10-1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Simpson KD, Templeton DJ, Cross JV. Macrophage migration inhibitory factor promotes tumor growth and metastasis by inducing myeloid-derived suppressor cells in the tumor microenvironment. J Immunol. 2012;189:5533–40. doi: 10.4049/jimmunol.1201161. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.