

Abstract
Angiotensin-II (Ang-II) is associated with many conditions involving heart failure and pathologic hypertrophy. Ang-II induces the synthesis of monocyte chemoattractant protein-1 that mediates the uptake of CD34+CD45+ monocytic cells into the heart. These precursor cells differentiate into collagen-producing fibroblasts and are responsible for the Ang-II-induced development of non-adaptive cardiac fibrosis. In this study, we demonstrate that in vitro, using a human monocyte-to-fibroblast differentiation model, Ang-II required the presence of tumor necrosis factor-alpha (TNF) to induce fibroblast maturation from monocytes. In vivo, mice deficient in both TNF receptors did not develop cardiac fibrosis in response to 1 week Ang-II infusion. We then subjected mice deficient in either TNF receptor 1 (TNFR1-KO) or TNF receptor 2 (TNFR2-KO) to continuous Ang-II infusion. Compared to wild-type, in TNFR1-KO, but not in TNFR2-KO hearts, collagen deposition was greatly attenuated, and markedly fewer CD34+CD45+ cells were present. Quantitative RT-PCR demonstrated a striking reduction of key fibrosis-related, as well as inflammation-related mRNA expression in Ang-II-treated TNFR1-KO hearts. TNFR1-KO animals also developed less cardiac remodeling, cardiac hypertrophy, and hypertension compared to wild-type and TNFR2-KO in response to Ang-II. Our data suggest that TNF induced Ang-II-dependent cardiac fibrosis by signaling through TNFR1, which enhances the generation of monocytic fibroblast precursors in the heart.
Keywords: Tumor necrosis factor-alpha, Angiotensin-II, Fibrosis, Monocytes, Inflammation, Hypertrophy
1. INTRODUCTION
In the absence of cell death, fibrosis may develop on a reactive basis, filling the interstitial space between functional cardiomyocytes (non-adaptive fibrosis) [1,2]. Such deposition of collagen invariably accompanies ventricular remodeling and cardiac dysfunction and is therefore considered an important mechanism contributing to the symptoms and progression of heart failure [3,4]. The development of cardiac fibrosis is often associated with inflammation [5–7]. However, although several inflammatory cytokines and chemokines have been implicated in adverse remodeling, the cellular and molecular mechanisms by which inflammation contributes to fibrosis are still unclear.
Increased angiotensin-II (Ang-II) synthesis is associated with almost every condition leading to congestive heart failure, from hypertension to myocardial infarction, and stimulates the development of interstitial fibrosis intrinsically [8,9]. Ang-II also promotes the inflammatory response and alters metabolic regulation in the heart [10,11].
In our previous work, we established a murine model of continuous Ang-II infusion to study the mechanisms of non-adaptive fibrosis in the heart [12]. Our data demonstrated that Ang-II infusion resulted in interstitial deposition of collagen that was mediated by the monocyte chemoattractant protein-1 (MCP-1) -dependent influx of monocytic CD34+CD45+ fibroblast precursors into the heart. These cells were of myeloid origin and contributed to the development of cardiac fibrosis; in mice deficient in MCP-1 Ang-II infusion did not induce fibrosis despite the development of hypertension and cardiac hypertrophy. Among other inflammatory markers, Ang-II also increased the synthesis of tumor necrosis factor-alpha (TNF) in the heart, and genetic deletion of MCP-1 inhibited the increase of Ang-II-induced TNF transcription [12].
TNF is a major regulator of inflammation and immunity, but also of cardiac structure and function in health and disease [13–15]. Limited expression of TNF confers a short-term beneficial stress-response to the host, but like Ang-II, long-term expression or acutely high levels of TNF are deleterious [14,16]. The effects of TNF are initiated by engagement of two distinct TNF receptors, 55-kDa TNFR1 and 75-kDa TNFR2 [17,18]. Both receptors share homology in their extracellular domains, but differ considerably within their intracellular regions, suggesting that each receptor has distinct modes of signaling and cellular functions [17,18]. Engagement of TNFR1 is primarily thought to be responsible for the deleterious effects of TNF, whereas TNFR2 activation mediates protective mechanisms [19,20]. Thus, the net effects of TNF on heart failure may depend on the relative contribution of TNFR1 and TNFR2 signaling.
In the current study, our data implicate a synergistic effect of Ang-II and TNF on the derivation of collagen-producing fibroblast precursors that mediate Ang-II-induced cardiac fibrosis, as well as on monocyte-to-fibroblast differentiation. Specifically, we provide evidence for the involvement of TNFR1 in the development of Ang-II-mediated development of cardiac fibrosis and adverse cardiac remodeling.
2. METHODS
2.1. In vitro transendothelial migration (TEM)
As detailed earlier [21], for a TEM assay, human peripheral blood mononuclear cells (PBMC) were allowed to migrate through human cardiac microvascular endothelial cells (HCMEC) in response to MCP-1. Some setups received stimulating agents (such as Ang-II, TNF, and/or specific inhibitors and agonists) in the top well. PBMC were allowed to migrate for 4 days, then the number of adherent fibroblast-shaped cells was counted in the bottom well (see Suppl. Fig. 1 for fibroblast identification). Each donor’s cells were measured in triplicate. Due to differences in absolute numbers between PBMC donors, the number of cells in each experimental well was divided by the mean of the control wells (no stimulating agents) to normalize the data.
2.2 Animals
B6;129S-Tnfrsf1atm1ImxTnfrsf1btm1Imx/J (“TNFR1R2-KO”) and their B6129SF2/J (“B6129”) wild-type control mice, as well as C57BL/6-Tnfrsf1atm1Imx/J (“TNFR1-KO”), B6.129S2-Tnfrsf1btm1Mwm/J C57BL/6J (“TNFR2-KO”) and their C57BL/6J (“C57”) wild-type control mice were purchased from Jackson Laboratory. Mice were infused with 1.5 μg/kg/min Ang-II via subcutaneously implanted osmotic pumps for 1 week [12]. Control animals were implanted with sterile saline-filled pumps. The investigation conformed with the Guide for the Care and Use of Laboratory Animals published by the US NIH. All animals were treated in accordance with the guidelines of the Baylor College of Medicine Animal Care and Research Advisory Committee.
2.3. Cardiac fibrosis
Hearts were arrested in diastole by infusion with cardioplegic solution, perfusion fixed, embedded in paraffin and sectioned as described earlier [12,22]. To measure collagen deposition, deparaffinized sections (4–6 per mouse) were stained with picrosirius red. Images within the left ventricle (4 per section) were scanned and collagen stained areas were calculated as percentages of the total myocardial area using ImagePro software. To evaluate macrophage influx, sections were stained with an antibody against Mac-2 and counterstained with eosin as described earlier [12].
2.4. Identification of fibroblast populations
Cardiac fibroblasts were isolated as described previously [12,22]. Freshly isolated cells were incubated with PE-conjugated anti-CD34, PE/Cy-5-conjugated anti-CD45, and either FITC-conjugated anti-collagen type I or calceinAM. Fluorescence intensities were measured on a Beckman Coulter Epics XL-MCL.
2.5. mRNA expression
Total RNA was isolated from the whole heart with TRIzol reagent, purified via columns, and cDNA was synthesized. Real-time RT-PCR amplifications were performed with SYBR Green on a C1000 Touch cycler (BioRad). Gene expression was measured by the ΔΔCT method and was normalized to 18S ribosomal RNA levels. The data are presented as the fold expression relative to the saline-treated wild-type group. All primer pairs were verified to adhere to the MIQE guidelines [23].
2.6. Cell size measurement
Perfusion-fixed heart sections (see 2.3.) were stained with tetramethylrhodamine-labeled wheat germ agglutinin; cell nuclei were counterstained with DAPI. Images were taken within the left ventricle (4 images per section, 2–4 sections per mouse). Cardiomyocyte size was measured in arbitrary units using ImagePro software.
2.7. Cardiovascular parameters
Cardiac function was obtained by 2D-directed M-mode echocardiography (Vevo770; Visual Sonics) and Doppler Ultrasound (Model DSPW, Indus Instruments) as previously described before and after 1 week of Ang-II infusion [12,22]. Functional data were stored and analyzed offline. Blood pressure measurements were obtained by the tail-cuff method (Visitech BP2000) before and after 1 week of Ang-II infusion.
2.8. Statistical analysis
All data are expressed as mean ± SEM. One-way ANOVA was used to evaluate differences between all groups (control and treated) and post-hoc testing (Tukey-Kramer Method) was performed when appropriate (Fig. 1–6, % change in Table 1). Additionally, in Table 1, the difference between baseline and after Ang-II treatment means in mice of the same genetic background was analyzed by paired sample two-tailed Student’s t-test. A P-value <0.05 was considered statistically significant.
Fig. 1. In vitro differentiation of human monocytes to fibroblasts required the presence of both Ang-II and TNF.

Human PBMC were placed on top of HCMEC and allowed to migrate through the confluent cell layer in response to MCP-1. After culturing the successfully migrated cells for 4 days, adherent fibroblast-shaped cells were counted (see also Suppl. Fig. 1) [21]. For each donor, the amount of fibroblasts in the absence of any stimuli was designated as 100% (= control value, bold line). In separate setups, cells were exposed to Ang-II, TNF, or both in the top well during migration, in the absence or presence of valsartan, an AT1 receptor inhibitor. Final data are expressed as % increase over control values. n=5/group without valsartan, n=3/group with valsartan. *P<0.05 compared to the Ang-II only group. #P<0.05 compared to the Ang-II/TNF group.
Fig. 6. In vitro differentiation of human monocytes to fibroblasts in response to Ang-II and TNF was mediated via TNFR1.

As described for Fig. 1, mononuclear cells in the top well were exposed to the indicated combinations of Ang-II, TNF, or an antibody agonist for TNFR1 and its control IgG, before they were allowed to migrate through the HCMEC layer in response to MCP-1 into the bottom well. Final data are expressed as % of control values (no stimuli = 100%; bold line). n=4–5/group; *P<0.05 compared to the Ang-II only group. #P<0.05 compared to the Ang-II/TNF group. NS = no significant difference.
Table 1. Cardiovascular Parameters.
The hemodynamic and anatomic responses to 1 week Ang-II infusion (discussion in text). Baseline values (before treatment) and experimental values (after 1 week) are expressed as mean ± SEM. The % change was calculated within the same mouse with parameters measured before and after Ang-II infusion. BW: body weight, HR: heart rate, LVEDD and LVESD: left ventricular end diastolic/systolic diameter, LVEDV and LVESV: left ventricular end diastolic/systolic volume, LVPW and LVAW: left ventricular posterior/anterior wall thickness, -d/-s: during diastole/systole, E-lin Dec Time: E-linear deceleration time, Peak Vel: peak velocity, EjT: ejection time, Tei Index (= myocardial performance index) was calculated as [(Isovolumetric Relaxation Time + Isovolumetric Contraction Time)/Ejection Time] and is heart rate independent. SBP: systolic blood pressure.
| C57 | C57 | C57 | TNFR1-KO | TNFR1-KO | TNFR1- KO | TNFR2- KO | TNFR2-KO | TNFR2- KO | |
|---|---|---|---|---|---|---|---|---|---|
| baseline | 1 week | % change | baseline | 1 week | % change | baseline | 1 week | % change | |
| Global Parameters | |||||||||
| n | 6 | 6 | 6 | 8 | 8 | 8 | 8 | 8 | 8 |
| BW, g | 26.6±0.6 | 24.8±0.5 # | 7±1 | 24.0±0.9 | 23.0±0.8 | 4±1 | 22.5±0.7 | 21.1±0.9 | 7±2 |
| HR, bpm | 446±21 | 483±21 | 9±6 | 474±25 | 516±17 | 10±6 | 506±17 | 495±20 | 1±5 |
|
| |||||||||
| Cardiac Echocardiographic Parameters | |||||||||
| LVEDD, mm | 3.76±0.12 | 3.81±0.18 | 2±7 | 3.64±0.13 | 3.47±0.17 | −5±3 | 3.66±0.08 | 3.29±0.08 # | −10±3 |
| LVEDV, mm3 | 68.4±4.6 | 63.4±7.2 | −3±15 | 63.2±5.1 | 51.1±5.9 # | −20±6 | 61.3±2.9 | 44.1±2.5 # | −27±6 |
| LVESD, mm | 2.76±0.13 | 2.75±0.24 | 2±10 | 2.66±0.11 | 2.71±0.17 | 2±5 | 2.65±0.09 | 2.54±0.13 | −2±8 |
| LVESV, mm3 | 27.6±2.6 | 30.1±6.8 | 18±31 | 24.3±3.1 | 28.6±4.4 | 19±13 | 24.7±2.3 | 24.1±2.9 | 10±21 |
| Fractional Shortening | 33.1±1.6 | 29.7±1.3 | −9±6 | 33.3±1.5 | 31.2±1.7 | −4±8 | 31.9±1.5 | 31.6±1.4 | 1±6 |
| Ejection Fraction | 62.7±1.9 | 55.7±2.3 | −10±5 | 62.4±2.1 | 59.6±2.6 | −3±6 | 60.4±2.2 | 60.5±2.1 | 1±5 |
| LVPW-d, mm | 0.78±0.08 | 1.21±0.09 # | 58±6 | 0.78±0.06 | 0.79±0.04 | 4±7 *† | 0.73±0.04 | 1.00±0.07 # | 37±5 |
| LVPW-s, mm | 1.00±0.09 | 1.38±0.08 # | 44±12 | 0.92±0.07 | 0.95±0.04 | 8±9 * | 0.96±0.04 | 1.19±0.08 # | 24±9 |
| LVAW-d, mm | 0.59±0.03 | 0.86±0.05 # | 48±12 | 0.69±0.06 | 0.67±0.05 | 0±9 *† | 0.57±0.03 | 0.78±0.04 # | 40±11 |
| LVAW-s, mm | 0.79±0.07 | 1.15±0.09 | 59±29 | 0.76±0.07 | 0.99±0.06 # | 36±11 | 0.72±0.05 | 1.07±0.06 # | 55±14 |
| LV Mass (calc), mg | 88.0±7.9 | 138.9±7.2 # | 63±12 | 88.4±7.5 | 85.3±8.1 | −1±10 *† | 76.8±4.9 | 99.6±6.8 # | 33±10 |
|
| |||||||||
| Cardiac Doppler Parameters | |||||||||
| E-lin Dec Time, ms | 49.9±7.3 | 20.2±1.5 # | 56±5 | 39.2±5.9 | 36.5±5.7 | 2±18 * | 56.0±4.5 | 40.9±5.3 # | 25±9 |
| E-Peak Vel, cm/s | 75.2±2.7 | 81.9±5.0 | 10±9 | 70.9±1.3 | 76.4±3.6 | 8±5 | 78.8±2.1 | 71.6±4.7 | 9±5 |
| Peak Vel, cm/s | 109±7 | 107±11 | 2±8 | 105±6 | 110±8 | 5±5 | 125±8 | 104±6 | 16±6 |
| Pre-EjT, ms | 15.7±1.1 | 17.7±0.4 | 15±9 | 17.9±0.5 | 16.1±0.5 | 10±3 *† | 15.4±0.3 | 18.0±0.5 # | 18±5 |
| Pre-EjT/EjT | 0.32±0.03 | 0.38±0.02 | 22±9 | 0.36±0.01 | 0.35±0.02 | 3±4 *† | 0.33±0.01 | 0.41±0.01 # | 26±4 |
| Tei Index | 0.61±0.07 | 0.70±0.04 | 17±7 | 0.68±0.02 | 0.65±0.03 | 4±6 *† | 0.67±0.03 | 0.78±0.03 # | 18±4 |
| Blood Pressure Parameters | |||||||||
| SBP, mm Hg | 117±5 | 155±4 # | 34±8 | 115±2 | 126±4 | 9±4 *† | 120±3 | 151±4 # | 25±5 |
P< 0.05 between baseline and 1 week of corresponding group
P< 0.05 between TNFR1-KO and C57
P< 0.05 between TNFR1-KO and TNFR2-KO
3. RESULTS
3.1. In vitro differentiation of human monocytes to fibroblasts required the presence of both Ang-II and TNF
Using our previously developed TEM assay [21], we found that the presence of Ang-II alone, TNF alone, or valsartan (an Ang-II type 1 (AT1) receptor blocker) alone during cell migration, did not increase the differentiation of human adherent PBMC into fibroblasts after their migration through HCMEC in response to MCP-1 (Fig. 1; see also Suppl. Fig. 1). However, when Ang-II and TNF were both present, the number of fibroblasts was significantly higher, increased by 88% compared to the control group (no stimuli). This suggested that both agents were necessary for increased fibroblast maturation. The presence of valsartan in addition to both agents reduced this number below control levels, indicating that Ang-II signaling though the AT1 receptor was actively involved in Ang-II/TNF-mediated fibroblast maturation. Exposure of monocytes to TNF did not increase AT1 expression as measured by flow cytometry (data not shown).
3.2. Infusion of Ang-II to mice deficient in TNF signaling did not induce interstitial fibrosis in the heart
Compared to saline-treated wild-type mice, Ang-II-treated wild-type mice developed interstitial cardiac fibrosis in response to Ang-II infusion (Fig. 2A) as measured by picrosirious red staining (5.1±0.8 % vs 1.0±0.1% collagen area). These data were consistent with our previous work [12]. However, when mice deficient in both TNF receptors, TNFR1 and TNFR2, were infused with Ang-II, the induction of reactive fibrosis was obviated (1.9±0.2 % collagen area vs 1.0±0.2% in saline-treated hearts; also Fig. 2A). Interstitial collagen deposition in the left ventricle of Ang-II-infused TNFR1R2-KO mouse hearts was comparable to values from untreated control mice. These data indicated that Ang-II-induced deposition of collagen in the heart depended on TNF signaling.
Fig. 2. Infusion of Ang-II to mice deficient in TNFR1 did not induce interstitial fibrosis in the heart.
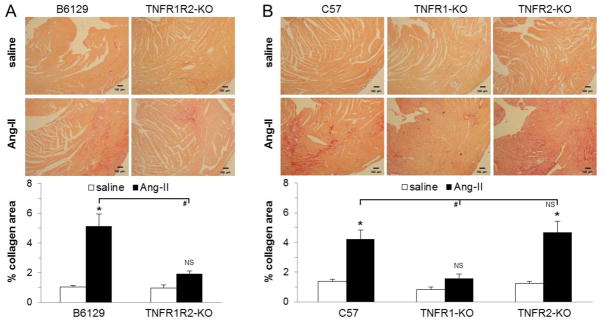
Tissue sections were stained with picrosirius red (image magnification: x100) after 1 week of continuous Ang-II treatment (n=5–6/group) and collagen deposition was evaluated in the left ventricle. Control mice received saline (n=3/group). A) In contrast to their corresponding wild-type mice (B6129), mice deficient in both TNF receptors (TNFR1R2-KO) were protected from Ang-II exposure, as interstitial collagen deposition was lower than in wild-type mice and comparable to saline values. B) Mice deficient in TNFR1 (TNFR1-KO) did not develop interstitial collagen deposition in response to Ang-II when compared to their corresponding wild-type group (C57) or to mice deficient in TNFR2 (TNFR2-KO), the latter two not being different from each other (see also Suppl. Fig. 2). *P<0.05 between Ang-II- and saline-treated groups (same genetic background). #P<0.05 compared to Ang-II-treated wild-type group. NS = no significant difference.
3.3. Infusion of Ang-II to mice deficient in TNFR1 did not induce interstitial fibrosis in the heart
Because both our in vitro and in vivo experiments required TNF signaling for Ang-II-induced fibroblast formation and development of cardiac fibrosis, we next investigated if TNFR1 or TNFR2 (or both) were necessary for signal transmission. As seen in Fig. 2B, we found that in TNFR1 deficient mice, infusion of Ang-II did not lead to increased collagen deposition in the heart compared to saline-treated mice (1.6±0.3% vs 0.8±0.2% collagen area). By contrast, in TNFR2 deficient mice Ang-II infusion resulted in cardiac fibrosis, as TNFR2-KO mice developed significant collagen deposition in the heart, which was not different from the corresponding Ang-II-treated wild-type mice (4.7±0.7% vs 4.2±0.6% collagen area; Fig. 2B) (see also Suppl. Fig. 2). These data indicated that the Ang-II/TNF-mediated reactive cardiac fibrosis involved signaling through TNFR1, but not via TNFR2.
3.4. Infusion of Ang-II to mice deficient in TNFR1 did not induce the presence of myeloid fibroblast precursors in the heart
We have previously demonstrated that Ang-II-induced cardiac fibrosis was mediated by myeloid precursor cells that matured into fibroblasts [12]. Thus, we infused TNF receptor deficient mice with Ang-II, isolated cardiac non-myocyte cells and subjected them to flow cytometry. In mice deficient in both TNF receptors, we found no increase in CD34+CD45+ fibroblast precursors in the heart in response to Ang-II exposure (0.4±0.1 % [Ang-II-treated; n=7] vs 0.1±0.1 % [saline-treated; n=3]; p>0.05). Next, as shown in Fig. 3A, we found that in the absence of TNFR1, mice did not accumulate monocytic CD34+CD45+ fibroblast precursors in the heart. By contrast, deletion of TNFR2 did not inhibit the presence of this cell population, as the number of viable CD34+CD45+ cells found in TNFR2-KO mouse hearts was similar to levels of the Ang-II-treated wild-type hearts. Further, the amount of CD34+CD45+ cells that also produced collagen (collagen type I+ CD34+CD45+) was reduced in the Ang-II-treated TNFR1-KO heart (Fig. 3B). These data indicated that Ang-II and TNF induced the presence of myeloid fibroblast precursor cells in the heart via signaling through TNFR1, but not through TNFR2. In a separate set of experiments, we stained cardiac tissue for the presence of macrophages. Fig. 3C illustrates that the amount of Mac-2+ cells did not differ between Ang-II-treated wild-type, TNFR1-KO, and TNFR2-KO hearts (see also Suppl. Fig. 3). These data indicate that the overall influx of macrophages in response to Ang-II was independent of TNF signaling.
Fig. 3. Infusion of Ang-II to mice deficient in TNFR1 did not induce the presence of myeloid fibroblast precursors in the heart.
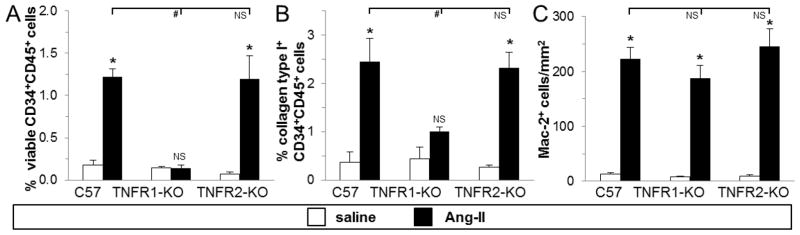
Hearts were removed and non-myocytes isolated. A) Dispersed cells were analyzed for both CD34 and CD45 expression in the presence of calcein (viability marker) by flow cytometry. B) Cells were analyzed for collagen type I, CD34 and CD45 expression. Note that these cells were fixed and so their numbers are not directly comparable to those in graph 3A (which were live cells). C) Cardiac tissue sections were stained for Mac-2+ cells (see also Suppl. Fig. 3). Control mice received saline (n=3/group), and treated mice received Ang-II for 1 week (n=5/group). *P<0.05 between Ang-II- and saline-treated groups (same genetic background). #P<0.05 compared to Ang-II-treated wild-type group. NS = no significant difference.
3.5. Infusion of Ang-II to mice deficient in TNFR1 did not upregulate mRNA levels of fibrosis- and inflammation-related genes in the heart
We examined the transcriptional activation of critical factors associated with fibrotic and inflammatory cardiomyopathies. As shown in Fig. 4A–E, in mouse hearts lacking TNFR1 signaling we found significantly smaller increases in collagen types I and III, and osteopontin mRNA expression, and no increase in alpha-smooth muscle actin (α-SMA) and transforming growth factor beta-1 (TGF-β1) mRNA expression after 1 week of Ang-II exposure. However, in mice deficient in TNFR2 signaling, transcriptional activation of these genes was similarly upregulated and comparable to wild-type levels. Type I collagen and α-SMA levels were even higher in TNFR2-KO than in wild-type, while osteopontin levels were slightly lower, but still significantly higher than in TNFR1-KO mouse hearts. Further, as shown in Fig. 4F–I, in mouse hearts lacking TNFR1 signaling we found no increase in MCP-1 mRNA, a small but not statistically significant increase in TNF mRNA, and little increases in CCR2 (ligand for MCP-1), and IL-6 mRNA expression after 1 week of Ang-II exposure. In mice deficient in TNFR2 signaling, transcriptional activation of these genes were either comparable (CCR2, IL-6) or slightly lower (MCP-1, TNF) than wild-type levels, but still higher than in TNFR1-KO hearts. There were no Ang-II-induced compensatory increases in TNFR1 mRNA expression in TNFR2-KO mice (2.8±0.3 [wild-type] vs 2.7±0.8 [TNFR2-KO] fold increase over saline-treated; n=7/group, p>0.05) or in TNFR2 mRNA expression in TNFR1-KO mice (2.6±0.4 [wild-type] vs 1.7±0.2 [TNFR1-KO] fold increase over saline-treated; n=7/group, p>0.05). These data indicated that signaling through TNFR1 was involved in Ang-II-induced fibrotic and inflammatory gene transcription.
Fig. 4. Infusion of Ang-II to mice deficient in TNFR1 did not upregulate mRNA levels of fibrosis-and inflammation-related genes in the heart.

Total myocardial RNA was isolated and subjected to real-time RT-PCR using specific primers for the indicated genes and SYBR Green. mRNA expression for each gene was calculated as fold induction compared to the saline-treated wild-type (C57) group. Control mice received saline (n=3–5/group), treated mice received Ang-II for 1 week (n=8/group). *P<0.05 between Ang-II- and saline-treated groups (same genetic background). #P<0.05 compared to Ang-II-treated wild-type group. NS = no significant difference.
3.6. Infusion of Ang-II to mice deficient in TNFR1 resulted in reduction of adverse left ventricular (LV) remodeling and hypertension
In Table 1, we examined indices of LV remodeling, cardiac function, and blood pressure after 1 week of Ang-II infusion. Using echocardiography, we found no significant effects in fractional shortening or ejection fraction resulting from the Ang-II infusion protocol used in any of the mouse groups over the period studied. However, we found evidence of significant thickening of the anterior and posterior LV walls in the wild-type and TNFR2-KO mice in response to Ang-II. The change in the TNFR1-KO was not statistically significant. In the hemodynamic studies with Doppler, we found no differences in peak Early filling velocity, but the E-linear deceleration time was significantly decreased after Ang-II infusion in the wild-type and TNFR2-KO mice (but not in the TNFR1-KO) suggesting that there was less stiffness associated with the reduced fibrosis in the TNFR1-KO group. The sensitive Tei index (a time interval index that combines both systolic and diastolic cardiac performance [24]) was also increased in the wild-type and TNFR2-KO mice after Ang-II infusion, suggesting subtle ventricular dysfunction present in these mice. The Tei Index was unchanged in the TNFR1-KO mice after the Ang-II infusion. Using tail-cuff measurements, Table 1 also shows that Ang-II infusion in both wild-type and TNFR2-KO mice resulted in increased systolic blood pressure within 1 week; however, in TNFR1-KO mice, systolic blood pressure was only minimally increased. Taken together, these data indicated that signaling through TNFR1 was involved in the Ang-II-mediated development of adverse LV remodeling and hypertension.
3.7. Infusion of Ang-II to mice deficient in TNFR1 resulted in less development of cardiac hypertrophy than in wild-type mice
One-week Ang-II infusion to all mice resulted in an immediate loss in body weight, which was not different between groups (Fig. 5A). However, whole heart weights in wild-type and TNFR2-KO, but not in TNFR1-KO mice, increased in response to Ang-II (Fig. 5B). This resulted in a significantly higher heart weight-to-body weight ratio in wild-type and TNFR2-KO mice than in TNFR1-KO mice (6.1±0.1 vs 5.9±0.1 vs 5.2±0.04; Fig. 5C). These data indicated that TNFR1 was involved in Ang-II mediated cardiac hypertrophy (see also Suppl. Fig. 2). To investigate this aspect further, we measured cell size in wheat germ agglutinin-stained heart sections, as well as quantified mRNA transcription of selected hypertrophy-related genes. Fig. 5D (see also Suppl. Fig. 4) demonstrates that the average wild-type cardiomyocyte size increased by 43% after Ang-II treatment; similarly, TNFR2-deficient cells increased in size by 40%. TNFR1-deficient cells also increased in size by 20% after Ang-II treatment; however, this increase was significantly smaller compared to wild-type and TNFR2-KO hearts (4075±80 vs 5055±275 vs 4863±250 arbitrary units; Fig. 5D). As further seen in Fig. 5E and 5F, we found that the mRNA level of atrial natriuretic peptide (ANP) and beta-myosin heavy chain (β-MHC), two hypertrophy-related genes, increased in all three mouse groups, but this increase was significantly greater in wild-type and TNFR2-KO hearts than in TNFR1-KO hearts after Ang-II infusion. These data indicated that TNFR1 was involved in the development of cardiac hypertrophy due to Ang-II exposure.
Fig. 5. Infusion of Ang-II to mice deficient in TNFR1 resulted in less development of cardiac hypertrophy than in wild-type mice.
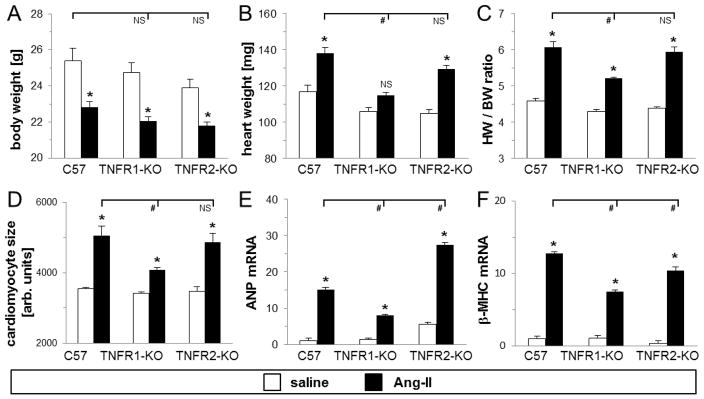
A–C) Body weights and whole heart weights were recorded at time of heart isolation after 1 week Ang-II treatment (n=8/saline, n=16/Ang-II groups). D) The average cardiomyocte size (in arbitrary units) within the left ventricle was measured by staining heart sections with wheat germ agglutinin (n=3/saline, n=5–6/Ang-II groups) (see also Suppl. Fig. 4). E–F) Real-time transcriptional regulation of ANP and β-MHC was determined using specific primers and SYBR Green (n=5/saline, n=6–8/Ang-II groups). *P<0.05 between Ang-II- and saline-treated groups (same genetic background). #P<0.05 compared to Ang-II-treated wild-type group. NS = no significant difference.
3.8. In vitro differentiation of human monocytes to fibroblasts in response to Ang-II and TNF was mediated via TNFR1
To corroborate the role of TNFR1 signaling in our in vitro transendothelial migration assay, we utilized a goat antibody that specifically bound and activated mouse TNFR1 (TNFR1-agonist). As seen in Fig. 6, when we added the TNFR1-agonist together with Ang-II we found equal numbers of fibroblasts in the bottom well compared to the numbers obtained with TNF and Ang-II. A control goat IgG antibody together with Ang-II did not increase fibroblast formation. These data indicated that signaling through TNFR1 was sufficient for the in vitro Ang-II-induced monocyte-to-fibroblast differentiation, validating our murine in vivo results by using human cells.
4. DISCUSSION
We have recently demonstrated that continuous Ang-II infusion to mice induced the synthesis of MCP-1 and concurrent uptake of myeloid CD34+CD45+ fibroblast precursor cells into the heart that differentiated into collagen-producing fibroblasts and mediated the development of cardiac interstitial fibrosis [12]. Infusion of Ang-II to mice deficient in MCP-1 did not induce the presence of this fibroblast precursor population and cardiac fibrosis was not observed in these mice. Genetic deletion of MCP-1 also inhibited the Ang-II-driven synthesis of TNF in the myocardium [12]. Because of TNF’s strong pro-inflammatory properties as well as its reported pro-fibrotic actions [13,15,25], we were intrigued by the possibility that TNF may be involved in the Ang-II-mediated initial inflammatory response that results in cardiac fibrosis.
To begin answering this question, we utilized an in vitro monocyte-to-fibroblast assay that we have developed previously [21]. In this assay, human PBMC were allowed to migrate in response to MCP-1 through an endothelial cell layer in the presence or absence of certain agents, and the number of fibroblast-like cells that appeared after transmigration was determined. This assay has proven useful in predicting the effects of several agents involved in monocyte-to-fibroblast differentiation, such as the effects of FcR gamma chain activation and its interaction with serum amyloid P, and Rho-kinase-1 deletion on interstitial fibrosis [21,26]. In the current study, we found that neither Ang-II nor TNF alone was sufficient to induce fibroblast maturation. However, we demonstrate that, if both agents were present, the number of fibroblasts maturing after MCP-1-driven transendothelial migration increased dramatically and addition of an AT1 receptor blocker prevented fibroblast formation. Thus, this assay was crucial in determining the synergistic effects of Ang-II and TNF, as both were required for generation of monocyte-derived fibroblasts.
To explore further the role of TNF in Ang-II-mediated fibrosis, we utilized mice that were deficient in TNF signaling and subjected these mice to Ang-II infusion for 1 week. We found that in the absence of both TNF receptors, TNFR1 and TNFR2, Ang-II did not induce the deposition of collagen in the heart. These in vivo data supported our conclusions derived from our in vitro assay in that Ang-II and TNF acted together on the maturation of myeloid-derived fibroblasts, the cells responsible for mediating interstitial cardiac fibrosis in the Ang-II infusion system. This provides a potential cellular target for the functional crosstalk between Ang-II and TNF in the regulation of cardiac structure and function as suggested by others [10,27]. This interaction, generated as part of the pathophysiology of cardiac hypertrophy and failure, exerts diverse actions on the cardiovascular system. For example, TNF was shown to stimulate as well as inhibit the renin-angiotensin system [28,29], and the deleterious effects of TNF overexpression in the heart were alleviated with AT1 receptor blockade [30]. A direct involvement of TNF in Ang-II-mediated fibrosis specifically has also been suggested, as inflammation is a key component of Ang-II-mediated cardiac hypertrophy and fibrosis. For instance, it was shown that TNF increased the AT1 receptor on cardiac fibroblasts, and that this sensitized cardiac fibroblasts to the pro-fibrotic actions of Ang-II [31]. Other studies have implicated an involvement of TNF in the Ang-II-mediated effects on fatty acid oxidation, hypertension, and hypertrophy [32,33]. In vitro, Ang-II and TNF have been shown to synergistically induce MCP-1 through activation of p38 and NF-κB [34]. The results of our previous and current studies enrich the evidence for a functional interplay between TNF and Ang-II, in that a) mice deficient in MCP-1 synthesis did not generate myocardial TNF and did not develop cardiac fibrosis in response to Ang-II infusion, b) mice deficient in TNFR1 signaling did not develop cardiac fibrosis in response to Ang-II infusion, and c) in vitro, more monocytes attracted to endothelial migration by MCP-1 matured into fibroblasts only if both AT1 and TNFR1 signaling were intact.
The TNF receptors differ considerably within their intracellular regions, and thus may elicit distinct molecular and cellular functions [17,18]. We found that signaling through TNFR1 was necessary for Ang-II-induced development of interstitial fibrosis in the heart. Ang-II infusion in mice lacking TNFR1 did not induce the presence of monocytic CD34+CD45+ fibroblast precursors in the heart, and obviated interstitial collagen deposition. On a molecular level, lack of TNFR1 signaling obviated the increase of inflammation-related genes, which was expected since TNFR1 contains an intracellular death domain region and is generally viewed as the pro-inflammatory receptor for TNF [17,18]. In particular, lack of MCP-1 expression may account for the absence of CD34+CD45+ cells in the heart, as we have shown previously that MCP-1 was obligatory for uptake of this fibroblast precursor population [12]. Thus, the initial inflammatory response to Ang-II may be necessary for the concurrent development of fibrosis (see Suppl. Fig. 5 describing our model). Importantly, absence of TNFR1 signaling obviated the increase of several fibrosis-related gene transcription such as collagen type I and III, α-SMA, osteopontin and TGF-β1, all of which have been implicated in perpetuating cardiac fibrosis [35]. The reduced levels of TGF-β1 may also affect the development of fibrosis, as it is typically associated with Ang-II-related fibrosis [35,36]. In addition, lack of TNFR1 may also protect cardiomyocytes from Ang-II-induced apoptosis, and together with the reduced amount of myeloid fibroblasts (but not overall number of macrophages), these processes may have additional protective effects on the Ang-II-exposed heart. Alternatively, lack of TNFR1 may affect the involvement of other cell types, e.g. lymphocytes, that secrete certain mediators that may have independent roles in the fibrotic development and/or may affect monocyte differentiation. The authors wish to emphasize that the current study focused on the early mechanisms of Ang-II-induced development of cardiac fibrosis since monocyte-derived fibroblasts are present most prominently in the heart only within the first 1–2 weeks while cardiac fibrosis persists thereafter [12]. Thus, this cell population may be most important in the initiation phase. Other mechanisms as described above may pertain to the modulation of fibrosis. It is possible that the effects of TNFR1 deletion are most profound during the early time points.
To confirm our in vivo-derived conclusion, we again utilized our in vitro assay. Using an antibody that specifically bound and activated TNFR1, we showed that this agonist in combination with Ang-II was sufficient to increase the number of monocyte-derived fibroblasts after MCP-1-guided transendothelial migration.
Our results add a distinct mechanism to a body of literature suggesting a detrimental role for AT1 and TNFR1 signaling in the development of hypertrophy, apoptosis, adverse cardiac remodeling and cardiac dysfunction as they relate to fibrosis that occurs in the failing heart [13–15,18,37]. By contrast, engagement of TNFR2 was described to mediate protective mechanisms, including immuno-modulation and antagonizing inflammation [19,37,38]. The involvement of TNFR1 and TNFR2 signaling specifically in the development of cardiac fibrosis has also been investigated after ischemic events. Two studies, for instance, using murine cardiac infarction models showed that TNFR1 exacerbated, whereas TNFR2 ameliorated the effects of adverse LV remodeling and hypertrophy, but both receptors were necessary to induce diastolic dysfunction and oxidative stress [39,40]. In these studies, mice deficient in TNFR2 showed even greater hypertrophy and border zone fibrosis than wild-type mice. Taken together, the distinct involvement of the two different TNF receptors may depend on local and temporal TNF concentrations, and perhaps on the cell type involved (e.g. endothelial cells, monocytes, or myeloid-derived fibroblasts).
In our current study, TNF signaling not only mediated cardiac fibrosis, but was also involved in the Ang-II-mediated early changes in cardiovascular parameters. After 1 week of Ang-II infusion, TNFR1-KO mice developed less cardiac hypertrophy and smaller increases in systolic blood pressure. It is possible that the absence of a hypertensive response resulted in the observed lack of cardiac fibrosis in these mice. Although this would be a possibility, these processes seem to occur independently based on our observations: a) our in vitro experiments (Fig. 1) in which Ang-II and TNF signaling induced myeloid fibroblast formation, and b) our published data in mice deficient in MCP-1 in which cardiac fibrosis was absent despite no difference in blood pressure or hypertrophy after Ang-II exposure [12]. Therefore, the development of cardiac fibrosis may occur independently from the Ang-II-mediated hemodynamic changes while the TNFR1 deletion also reduced both hypertension and hypertrophy. The authors cannot suggest specific mechanisms for these latter observations; however, we can speculate that these changes may be due to TNF’s diverse effects on the cardiovascular system that may be Ang-II dependent or independent (as described above). For example, lack of pro-inflammatory TNFR1 signaling in endothelial cells may alter their response to Ang-II and protect from the development of hypertension. Indeed, an involvement of TNF in Ang-II-mediated cardiovascular changes has been suggested earlier, such as blockade of TNF with etanercept was shown to prevent renal damage and increase in blood pressure in genetically hypertensive rats as well as in Ang-II-infused rats [41,42]. A study using TNF-deficient mice demonstrated that TNF was integral to the development of hypertension, response to salt appetite, and cardiac hypertrophy [33]. Of note, another study using TNFR1-KO mice, although a different mouse strain, indicated that in these mice Ang-II induced even greater increases in blood pressure than in wild-type mice after 1 week of infusion [43]. The reasons for these contradicting results are unknown, but the difference in mouse strain, knockout strategy, or possible polymorphisms in the TNFR1 gene, as well as in blood pressure measurements (implantation of radiotelemetry probes vs tail-cuff plethysmography) may be accountable. The current study focused on the collaborative role of Ang-II and TNF and their receptors specifically on the molecular and cellular signaling mediating a direct fibrotic response resulting from the induction of myeloid fibroblasts.
5. CONCLUSION
In vitro, both TNFR1 and AT1 receptor engagement were required to enhance monocyte-to-fibroblast differentiation that occurred after transendothelial migration. In vivo, TNFR1 signaling was necessary for the Ang-II-induced transcriptional upregulation of several key fibrosis- and inflammation-related genes, uptake of fibroblast precursor cells, induction of myeloid fibroblasts and concurrent development of cardiac fibrosis. While synergy between Ang-II and TNF has been described with regard to multiple pathophysiologic outcomes in cardiovascular disease, this report is the first to define a specific signaling mechanism and cellular response by which this synergy occurs.
Supplementary Material
HIGHLIGHTS.
Joint AT1 and TNFR1 signaling is required for monocyte-to-fibroblast maturation.
TNFR1 is involved in Ang-II-induced inflammatory and fibrotic gene transcription.
TNFR1 is involved in Ang-II-induced non-adaptive cardiac fibrosis.
TNFR1 is involved in Ang-II-induced cardiac remodeling and hypertension.
Acknowledgments
We thank summer students Beena Shah and Mark Dougherty, as well as Dorellyn B. Lee, Thuy Pham, Alberto Alonso, Adam Dean, and Jennifer Pocius for expert technical assistance.
FUNDING SOURCES
This work was supported by the American Heart Association grants 10SDG4280031 (SBH) and 12POST11830003 (ER), the ARCO Foundation Young Teacher-Investigator Award (SBH), and the Naman Family Fund for Basic Research (SBH), the Marshall Plan Scholarship (CD), the Medallion Foundation, the Hankamer Foundation, and the NIH grant HL089792 (MLE).
APENDICES
More detailed Material and Methods associated with this article can be found in the Supplementary Data section.
Footnotes
DISCLOSURES
None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Porter KE, Turner NA. Cardiac fibroblasts: at the heart of myocardial remodeling. Pharmacol Ther. 2009;123:255–78. doi: 10.1016/j.pharmthera.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 2.Creemers EE, Pinto YM. Molecular mechanisms that control interstitial fibrosis in the pressure-overloaded heart. Cardiovasc Res. 2011;89:265–72. doi: 10.1093/cvr/cvq308. [DOI] [PubMed] [Google Scholar]
- 3.Weber KT, Brilla CG, Janicki JS. Myocardial fibrosis: functional significance and regulatory factors. Cardiovasc Res. 1993;27:341–8. doi: 10.1093/cvr/27.3.341. [DOI] [PubMed] [Google Scholar]
- 4.Souders CA, Bowers SL, Baudino TA. Cardiac fibroblast: the renaissance cell. Circ Res. 2009;105:1164–76. doi: 10.1161/CIRCRESAHA.109.209809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Frangogiannis NG. Chemokines in the ischemic myocardium: from inflammation to fibrosis. Inflamm Res. 2004;53:585–95. doi: 10.1007/s00011-004-1298-5. [DOI] [PubMed] [Google Scholar]
- 6.Kania G, Blyszczuk P, Eriksson U. Mechanisms of cardiac fibrosis in inflammatory heart disease. Trends Cardiovasc Med. 2009;19:247–52. doi: 10.1016/j.tcm.2010.02.005. [DOI] [PubMed] [Google Scholar]
- 7.Westermann D. Does inflammation trigger fibrosis in hypertrophic cardiomyopathy: a burning question? Heart. 2012;98:965–6. doi: 10.1136/heartjnl-2012-301730. [DOI] [PubMed] [Google Scholar]
- 8.Sciarretta S, Paneni F, Palano F, Chin D, Tocci G, Rubattu S, et al. Role of the renin-angiotensin-aldosterone system and inflammatory processes in the development and progression of diastolic dysfunction. Clin Sci (Lond) 2009;116:467–77. doi: 10.1042/CS20080390. [DOI] [PubMed] [Google Scholar]
- 9.Schluter KD, Wenzel S. Angiotensin II: a hormone involved in and contributing to pro-hypertrophic cardiac networks and target of anti-hypertrophic cross-talks. Pharmacol Ther. 2008;119:311–25. doi: 10.1016/j.pharmthera.2008.05.010. [DOI] [PubMed] [Google Scholar]
- 10.Mann DL. Angiotensin II as an inflammatory mediator: evolving concepts in the role of the renin angiotensin system in the failing heart. Cardiovasc Drugs Ther. 2002;16:7–9. doi: 10.1023/a:1015355112501. [DOI] [PubMed] [Google Scholar]
- 11.Lopaschuk GD. Cardiac energy metabolism alterations in angiotensin II induced hypertrophy. J Mol Cell Cardiol. 2006;41:418–20. doi: 10.1016/j.yjmcc.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 12.Haudek SB, Cheng J, Du J, Wang Y, Hermosillo-Rodriguez J, Trial J, et al. Monocytic fibroblast precursors mediate fibrosis in angiotensin-II-induced cardiac hypertrophy. J Mol Cell Cardiol. 2010;49:499–507. doi: 10.1016/j.yjmcc.2010.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meldrum DR. Tumor necrosis factor in the heart. Am J Physiol. 1998 Mar;274(3 Pt 2):R577–R595. doi: 10.1152/ajpregu.1998.274.3.R577. [DOI] [PubMed] [Google Scholar]
- 14.Mann DL. Recent insights into the role of tumor necrosis factor in the failing heart. Heart Fail Rev. 2001;6:71–80. doi: 10.1023/a:1011449708842. [DOI] [PubMed] [Google Scholar]
- 15.Kleinbongard P, Heusch G, Schulz R. TNFalpha in atherosclerosis, myocardial ischemia/reperfusion and heart failure. Pharmacol Ther. 2010;127:295–314. doi: 10.1016/j.pharmthera.2010.05.002. [DOI] [PubMed] [Google Scholar]
- 16.Clark J, Vagenas P, Panesar M, Cope AP. What does tumour necrosis factor excess do to the immune system long term? Ann Rheum Dis. 2005;64(Suppl 4):iv70–iv76. doi: 10.1136/ard.2005.042523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wajant H, Pfizenmaier K, Scheurich P. Tumor necrosis factor signaling. Cell Death Differ. 2003;10:45–65. doi: 10.1038/sj.cdd.4401189. [DOI] [PubMed] [Google Scholar]
- 18.Cabal-Hierro L, Lazo PS. Signal transduction by tumor necrosis factor receptors. Cell Signal. 2012;24:1297–305. doi: 10.1016/j.cellsig.2012.02.006. [DOI] [PubMed] [Google Scholar]
- 19.Higuchi Y, McTiernan CF, Frye CB, McGowan BS, Chan TO, Feldman AM. Tumor necrosis factor receptors 1 and 2 differentially regulate survival, cardiac dysfunction, and remodeling in transgenic mice with tumor necrosis factor-alpha-induced cardiomyopathy. Circulation. 2004;109:1892–7. doi: 10.1161/01.CIR.0000124227.00670.AB. [DOI] [PubMed] [Google Scholar]
- 20.Schulz R, Heusch G. Tumor necrosis factor-alpha and its receptors 1 and 2: Yin and Yang in myocardial infarction? Circulation. 2009;119:1355–7. doi: 10.1161/CIRCULATIONAHA.108.846105. [DOI] [PubMed] [Google Scholar]
- 21.Haudek SB, Trial J, Xia Y, Gupta D, Pilling D, Entman ML. Fc receptor engagement mediates differentiation of cardiac fibroblast precursor cells. Proc Natl Acad Sci USA. 2008;105:10179–84. doi: 10.1073/pnas.0804910105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haudek SB, Xia Y, Huebener P, Lee JM, Carlson S, Crawford JR, et al. Bone marrow-derived fibroblast precursors mediate ischemic cardiomyopathy in mice. Proc Natl Acad Sci USA. 2006;103:18284–9. doi: 10.1073/pnas.0608799103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, et al. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem. 2009;55:611–22. doi: 10.1373/clinchem.2008.112797. [DOI] [PubMed] [Google Scholar]
- 24.Karatzis EN, Giannakopoulou AT, Papadakis JE, Karazachos AV, Nearchou NS. Myocardial performance index (Tei index): evaluating its application to myocardial infarction. Hellenic J Cardiol. 2009;50:60–5. [PubMed] [Google Scholar]
- 25.Distler JH, Schett G, Gay S, Distler O. The controversial role of tumor necrosis factor alpha in fibrotic diseases. Arthritis Rheum. 2008;58:2228–35. doi: 10.1002/art.23645. [DOI] [PubMed] [Google Scholar]
- 26.Haudek SB, Gupta D, Dewald O, Schwartz RJ, Wei L, Trial J, et al. Rho kinase-1 mediates cardiac fibrosis by regulating fibroblast precursor cell differentiation. Cardiovasc Res. 2009;83:511–8. doi: 10.1093/cvr/cvp135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sekiguchi K, Li X, Coker M, Flesch M, Barger PM, Sivasubramanian N, et al. Cross-regulation between the renin-angiotensin system and inflammatory mediators in cardiac hypertrophy and failure. Cardiovasc Res. 2004;63:433–42. doi: 10.1016/j.cardiores.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 28.Brasier AR, Li J, Wimbish KA. Tumor necrosis factor activates angiotensinogen gene expression by the Rel A transactivator. Hypertension. 1996;27:1009–17. doi: 10.1161/01.hyp.27.4.1009. [DOI] [PubMed] [Google Scholar]
- 29.Todorov V, Muller M, Schweda F, Kurtz A. Tumor necrosis factor-alpha inhibits renin gene expression. Am J Physiol Regul Integr Comp Physiol. 2002;283:R1046–R1051. doi: 10.1152/ajpregu.00142.2002. [DOI] [PubMed] [Google Scholar]
- 30.Flesch M, Hoper A, Dell’Italia L, Evans K, Bond R, Peshock R, et al. Activation and functional significance of the renin-angiotensin system in mice with cardiac restricted overexpression of tumor necrosis factor. Circulation. 2003;108:598–604. doi: 10.1161/01.CIR.0000081768.13378.BF. [DOI] [PubMed] [Google Scholar]
- 31.Gurantz D, Cowling RT, Varki N, Frikovsky E, Moore CD, Greenberg BH. IL-1beta and TNF-alpha upregulate angiotensin II type 1 (AT1) receptors on cardiac fibroblasts and are associated with increased AT1 density in the post-MI heart. J Mol Cell Cardiol. 2005;38:505–15. doi: 10.1016/j.yjmcc.2004.12.015. [DOI] [PubMed] [Google Scholar]
- 32.Pellieux C, Montessuit C, Papageorgiou I, Lerch R. Angiotensin II downregulates the fatty acid oxidation pathway in adult rat cardiomyocytes via release of tumour necrosis factor-alpha. Cardiovasc Res. 2009;82:341–50. doi: 10.1093/cvr/cvp004. [DOI] [PubMed] [Google Scholar]
- 33.Sriramula S, Haque M, Majid DS, Francis J. Involvement of tumor necrosis factor-alpha in angiotensin II-mediated effects on salt appetite, hypertension, and cardiac hypertrophy. Hypertension. 2008;51:1345–51. doi: 10.1161/HYPERTENSIONAHA.107.102152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Takahashi M, Suzuki E, Takeda R, Oba S, Nishimatsu H, Kimura K, et al. Angiotensin II and tumor necrosis factor-alpha synergistically promote monocyte chemoattractant protein-1 expression: roles of NF-kappaB, p38, and reactive oxygen species. Am J Physiol Heart Circ Physiol. 2008;294:H2879–H2888. doi: 10.1152/ajpheart.91406.2007. [DOI] [PubMed] [Google Scholar]
- 35.Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol. 2008;214:199–210. doi: 10.1002/path.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weber KT. Fibrosis, a common pathway to organ failure: angiotensin II and tissue repair. Semin Nephrol. 1997;17:467–91. [PubMed] [Google Scholar]
- 37.Van HF, Vandenbroucke RE, Libert C. Treatment of TNF mediated diseases by selective inhibition of soluble TNF or TNFR1. Cytokine Growth Factor Rev. 2011;22:311–9. doi: 10.1016/j.cytogfr.2011.09.004. [DOI] [PubMed] [Google Scholar]
- 38.Faustman D, Davis M. TNF receptor 2 pathway: drug target for autoimmune diseases. Nat Rev Drug Discov. 2010;9:482–93. doi: 10.1038/nrd3030. [DOI] [PubMed] [Google Scholar]
- 39.Monden Y, Kubota T, Inoue T, Tsutsumi T, Kawano S, Ide T, et al. Tumor necrosis factor-alpha is toxic via receptor 1 and protective via receptor 2 in a murine model of myocardial infarction. Am J Physiol Heart Circ Physiol. 2007;293:H743–H753. doi: 10.1152/ajpheart.00166.2007. [DOI] [PubMed] [Google Scholar]
- 40.Hamid T, Gu Y, Ortines RV, Bhattacharya C, Wang G, Xuan YT, et al. Divergent tumor necrosis factor receptor-related remodeling responses in heart failure: role of nuclear factor-kappaB and inflammatory activation. Circulation. 2009;119:1386–97. doi: 10.1161/CIRCULATIONAHA.108.802918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Elmarakby AA, Quigley JE, Pollock DM, Imig JD. Tumor necrosis factor alpha blockade increases renal Cyp2c23 expression and slows the progression of renal damage in salt-sensitive hypertension. Hypertension. 2006;47:557–62. doi: 10.1161/01.HYP.0000198545.01860.90. [DOI] [PubMed] [Google Scholar]
- 42.Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, et al. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med. 2007;1;204:2449–60. doi: 10.1084/jem.20070657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen CC, Pedraza PL, Hao S, Stier CT, Ferreri NR. TNFR1-deficient mice display altered blood pressure and renal responses to ANG II infusion. Am J Physiol Renal Physiol. 2010;299:F1141–F1150. doi: 10.1152/ajprenal.00344.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.