

Abstract
Polymer-based nanoassemblies have emerged as viable platforms for the encapsulation and delivery of lipophilic molecules. Among the criteria that such carriers must meet, if they are to be effective, are the abilities to efficiently solubilize lipophilic guests within an assembled scaffold and to stably encapsulate the molecular cargo until desired release is achieved through the actions of appropriately chosen stimuli. The former feature, dictated by the inherent loading capacity of a nanocarrier, is well studied and it has been established that slight variations in assembly structure, such as introducing hydrophobic content, can improve miscibility with the lipophilic guests and increase the driving force for encapsulation. However, such clear correlations between assembly properties and the latter feature, nanocarrier encapsulation stability, are not yet established. For this purpose, we have investigated the effects of varying hydrophobic content on the loading parameters and encapsulation stabilities of self-crosslinked polymer nanogels. Through investigating this nanogel series, we have observed a fundamental relationship between nanoassembly structure, loading capacity and encapsulation stability. Furthermore, a combined analysis of data from different loading amounts suggests a model of loading-dependent encapsulation stability that underscores an important correlation between the principal features of noncovalent encapsulation in supramolecular hosts.
Keywords: polymeric nanogel, encapsulation stability, hydrophobicity, loading capacity, fluorescence resonance energy transfer
Introduction
A pervading challenge in the field of drug discovery concerns the bioavailability of several water-insoluble lead compounds that emerge from the pipeline.1 This complication inspires vast interest in the design and development of delivery systems that are capable of solubilizing these drugs and stably transporting them through circulation to a target site.2 Polymer-based supramolecular assemblies, such as micellar carriers formed from amphiphilic copolymers, are promising scaffolds for such systems.3 In addition to their ability to solubilize water-insoluble drug compounds within hydrophobic cores, these polymer micelles offer several advantages in their nanoscopic sizes, which allow for passive targeting of certain disease sites through the enhanced permeability and retention effect.4
The primary basis for encapsulation of lipophilic guest molecules within an amphiphilic assembly is miscibility with the hydrophobic interior.3,5 It is understandable that increasing the hydrophobicity of the assembly interior will provide a greater thermodynamic driving force for lipophilic encapsulation. This driving force defines the distribution coefficient of lipophilic guest molecules between the assembly interior and the bulk aqueous phase, which in turn dictates the loading capacity of the nanoassembly.6 Complementing loading capacity, encapsulation stability is an additional critical factor for a nanoassembly to serve effectively as a delivery platform for biological applications. Encapsulation stability can be defined as a measure of the dynamic exchange of guest molecules between the assembly interior and the surrounding environment, and is thus an indicator of potential guest molecule leakage.7 While increased hydrophobicity of the assembly interior will provide a shift in the thermodynamic distribution of lipophilic guest molecules to favor encapsulation, we were interested in probing the effect of assembly hydrophobicity on the dynamics of guest exchange. In this manuscript, we describe our concerted efforts towards this fundamental understanding.
Supramolecular hosts that can provide systematic variation in scaffold hydrophobicity, while consistently yielding an avenue for encapsulating lipophilic molecules, are relatively scarce.8 A nanoscale system that lends itself particularly well to this purpose is the crosslinked polymer nanogel.9 We have recently introduced a class of polymeric nanogels that provides a unique opportunity for such variation.10 In this system, we use an amphiphilic random copolymer to form nanoscale assemblies in which guest molecules are noncovalently encapsulated. This copolymer is based on a hydrophilic monomer containing oligoethylene glycol (OEG) units and a lipophilic monomer containing reactive pyridyldisulfide (PDS) moieties. Micelle-type aggregates formed by this copolymer are locked using a thiol-disulfide exchange based crosslinking reaction, which liberates the pyridyl units. While the relative hydrophobicity of the pyridyl unit forms the basis for initial amphiphilic self-assembly and lipophilic guest sequestration in this system, guest molecules remain encapsulated within the resulting polymer nanogels despite the release of the PDS units during crosslinking. This is likely due to an “incarceration” effect generated by the crosslinking disulfide bonds, which stabilize the structure and trap the initially sequestered guest molecules within the nanogel interior. This caging-assisted encapsulation provides a template for retaining reasonable lipophilic guest loading while varying the hydrophobicity of the nanogel host.
To probe the effect of hydrophobic variations on encapsulation and the dynamics of guest exchange, we were interested in varying the hydrophilic-lipophilic balance (HLB) in the assembled polymer architecture of our nanogel system. By modifying the OEG-PDS copolymer to contain hydrophobic alkyl chains (Figure 1), we explore variations in lipophilic loading parameters, afforded by altering the HLB of the precursor polymer. Additionally, we investigate the consequences of such alterations on the dynamic exchange of sequestered guest molecules between the nanogel container and the surrounding environment.
Figure 1.
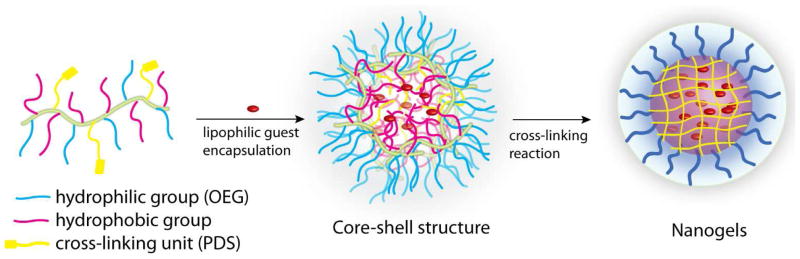
Cartoon representation of guest loaded nanogel formation from random copolymer precursors modified with alkyl chain derived monomers.
To monitor the dynamics of guest exchange we employ a recently introduced fluorescence resonance energy transfer (FRET)based assay.11 Briefly, in this method, nanogels separately encapsulating the lipophilic FRET pair dyes, DiOC18(3) (DiO) and DiIC18(3) (DiI), are prepared. When solutions of these dye containing carriers are mixed, two distinct outcomes (Figure 2) are possible. If the dye molecules are stably encapsulated within the nanogel interior, they will not exchange with the bulk solvent environment and will remain within their original carriers. If this is the case, no FRET development will be observed in the mixed solution, since the distance between DiI and DiO will remain greater than the dye pair’s Föster radius. However, if encapsulation stability is compromised, guest exchange will occur when the two solutions are mixed, causing time-dependent equilibration of the dye molecules among all the indistinguishable nanogel carriers in solution. This equilibration will cause DiI and DiO molecules to occupy the same interior, placing them within range of the Föster radius and leading to an increase in observed FRET. Thus, tracing the evolution of FRET in the mixed solutions, as measured by concomitant decrease in donor (DiO) emission intensity and increase in acceptor (DiI) emission intensity, provides insight into the stability of guest molecule encapsulation, the dynamics of guest exchange with the bulk environment and potential nanocarrier leakage.
Figure 2.

Potential FRET outcomes observed upon mixing solutions of DiI/DiO encapsulating nanocarriers. Low encapsulation stability (top) permits dye leakage, equilibration and causes FRET development. High encapsulation stability (bottom) prevents dye exchange and negates FRET development.
Results and Discussion
Design and Syntheses of Polymers and Nanogels
In order to test the effects of varying hydrophobicity, amphiphilic random copolymer precursors of varying hydrophobic content were synthesized (Chart 1). Drawing from the OEG-PDS copolymer, which uses the PDS side chains as handles for chemically cross-linking the nanogel structure, the polymer precursors for these studies were designed to contain a constant percent composition of the PDS functionality. As the current study is aimed to develop a fundamental correlation between nanogel hydrophobicity and encapsulation stability, it was deemed necessary to keep crosslinking density low in these nanogels so that guest exchange would likely be observed and trends could be drawn from the data. Thus, the PDS-derived methacrylate monomer was selected to comprise only 20% of all the studied precursor polymers. The OEG methacrylate monomer was incorporated to provide a water soluble, charge-neutral side chain and constitute the hydrophilic content of the precursor polymers. To achieve polymers with varied hydrophobic content, lipophilic alkyl chain-derived monomers were added to this OEG-PDS composition. The relative hydrophobicities of the precursor polymers were varied in two ways: (i) by altering the percent composition of a comonomer containing an alkyl chain of constant length and (ii) by altering the alkyl chain length of an incorporated comonomer with constant percent composition.
Chart 1.
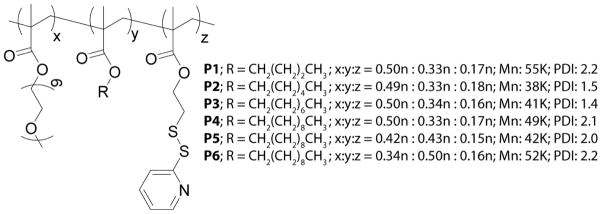
Structures and characteristics of the precursor polymers P1-P6. The relative hydrophobicities of these polymers are varied by altering the OEG:decyl chain ratio, while keeping the PDS amount constant.
For the series, six polymers (P1-P6) were prepared. For half the series, the polymers were prepared to contain 50% OEG methacrylate, 20% PDS-derived methacrylate and 30% alkyl chain-derived methacrylate, with the alkyl chain (R) length varied to generate polymers P1 (R = butyl), P2 (R = hexyl) and P3 (R = octyl). Polymers P4-P6 were prepared with varying percentages of incorporated decyl methacrylate comonomer. We varied the feed ratio of this hydrophobic unit to OEG, while keeping the PDS unit at a constant 20%. In synthesizing P4-P6, a polymer series was generated in which the targeted ratios of OEG methacrylate to decyl methacrylate to PDS methacrylate were made to equal 5:3:2, 4:4:2 and 3:5:2, respectively. It should be noted that P4 (R = decyl) can also be placed at the end of P1-P3, creating a continuous hydrophobicity variation within the P1-P6 series. The incorporated ratios of the monomers, as determined by integration of the 1H NMR spectra, were observed to closely match the feed ratios and are shown in Chart 1. Detailed synthetic protocols and polymer characteristics can be found in the supporting information. All precursor polymers were prepared by reversible addition-fragmentation chain transfer (RAFT) polymerization.
Containing both hydrophobic alkyl groups/PDS units and hydrophilic OEG chains, polymers P1-P6 were observed to form nanoscale aggregates upon self-assembly in aqueous solution. Aggregation of the amphiphilic copolymers generated assemblies with hydrophobic cores that could accommodate the sequestration of lipophilic guest molecules in water. For the studies presented here, nanogel samples were prepared by in situ loading of lipophilic dyes into these polymer aggregates. Dye encapsulation, initiated by addition from a dye stock solution to the polymer solution (both in acetone), was promoted by dilution with deionized water and gradual removal of acetone from the mixture. Subsequent addition of the reducing agent, dithiothreitol (DTT, 50 mol% with respect to the PDS groups in the precursor polymer), activated the intra/interchain disulfide bond formation that has been shown to generate the crosslinked polymer nanogel structure.10 The progress of this reaction was monitored by tracking the production of the pyridothione byproduct through its characteristic absorption at 343 nm. The efficiency of this reaction varied slightly between the polymers. With the formation of each crosslinking disulfide bond requiring two PDS groups, addition of 50 mol% DTT was expected to remove nearly all the PDS groups during the crosslinking procedure. The actual reaction efficiencies were observed to be near 50%, with around half of the available PDS groups consumed. From this reaction, dye loaded polymer nanogels NG1-NG6 were prepared from polymer precursors P1-P6 (Scheme 1), achieving noncovalent lipophilic guest encapsulation within the obtained crosslinked, water soluble nanogels.
Scheme 1.
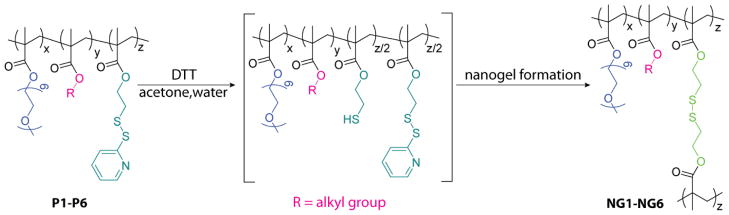
Synthesis of self-crosslinked polymer nanogels (NG1-NG6) from precursor polymers (P1-P6) containing OEG chains, PDS units, and hydrophobic alkyl groups.
The particle sizes of polymers P1-P6 in water prior to cross-linking were investigated by dynamic light scattering (DLS), as it was hypothesized that the sizes of these micelle-type aggregates would dictate the sizes of the obtained nanogels. Aqueous solutions of P1-P6 (1 mg mL−1) exhibited assembly sizes with diameters centered at 8–14 nm by DLS measurement (Figure 3a). While nanogels NG2-NG6 (1 mg mL−1) formed from these polymers were observed to have similar size distributions, butylmethacrylate containing NG1 exhibited a much larger nanogel size centered at a 190 nm diameter (Figure 3b). It is hypothesized that this discrepancy may be due to a lack of stable polymer aggregation in solutions of P4, which contains the shortest lipophilic side chain used in the present work. Considering the large contribution to the overall aggregate HLB from the 50% of hydrophilic OEG segments, it is likely that this butyl chain does not sufficiently stabilize the observed ~9 nm polymer aggregates. Such instability may increase the occurrence of interchain crosslinking during the DTT initiated reaction, resulting in a larger nanogel than those formed from the more stable aggregates of the other polymers.
Figure 3.
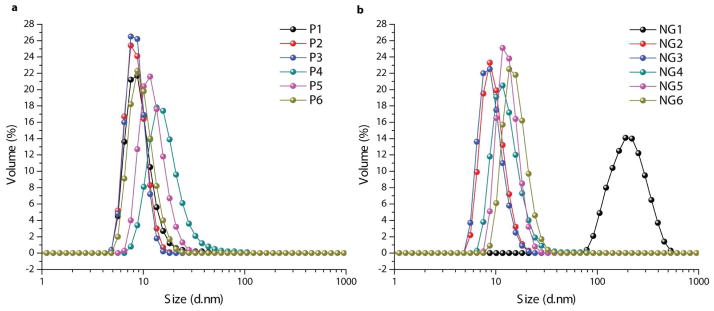
Size distributions as measured by volume based DLS for aqueous solutions (1 mg mL−1) of a) polymer aggregates of P1-P6 and b) self-crosslinked polymer nanogels NG1-NG6.
Non-covalent Encapsulation of Lipophilic Guest Molecules
It was hypothesized that increasing the hydrophobic content of the precursor polymer would result in increasing hydrophobicity of the corresponding nanogel container. To probe this possibility, nanogels NG1-NG6 were prepared in the presence of 10 wt% pyrene. After purification of the nanogel samples by filtering off unencapsulated dye and dialysis against deionized water, the fluorescence emission spectra of the samples were recorded. A sample fluorescence spectrum of NG6 is shown in Figure 4a with the first and third emission maxima highlighted in red and blue, respectively. The ratio of the first and third emission intensities of pyrene, commonly referred to as the I1/I3 ratio, has been shown to correlate with the polarity of the dye’s environment, with values ranging from 1.9 in polar solvents to 0.6 in certain hydrocarbon solvents.12 Thus, the I1/I3 values for pyrene encapsulated in NG1-NG6 were calculated to probe the relative polarities of the nanogel carriers. Going through NG1-NG6, a trend of decreasing value in the pyrene I1/I3 ratio was observed, indicating the dye’s encapsulation in an increasingly nonpolar environment (Figure 4b). The I1/I3 values of the nanogel samples range from 1.52 (NG1) to 1.31 (NG6).This suggests that as the hydrophobic content of the precursor polymer is increased, either through longer alkyl chain incorporation or increased percent composition of the decyl chain-containing monomer, the resulting nanogel container indeed becomes more hydrophobic. It is noteworthy that the I1/I3 value for NG1 is much higher than those of the rest of the nanogels. Containing the shortest alkyl chain and having a much larger size, NG1 is likely a more swelled and water filled nanogel structure, creating the most polar encapsulation microenvironment for pyrene. Due to this significant size difference and its apparent effect on encapsulation, NG1 was not investigated further in this comparative study.
Figure 4.
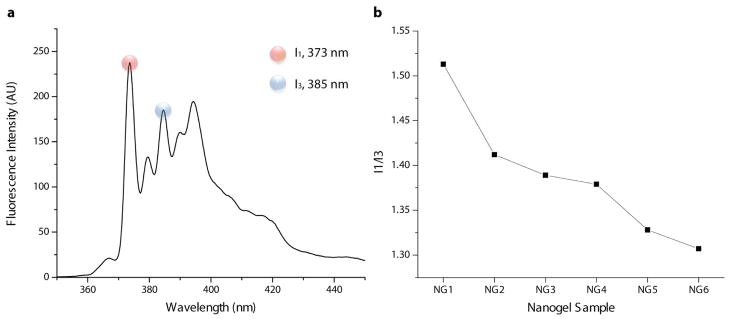
a) Fluorescence emission spectrum recorded for NG6 prepared with 10 wt% pyrene feeding and b) calculated I1/I3 ratios for pyrene encapsulated in the NG1-NG6 samples. All pyrene emission spectra were recorded at 0.05 mg.mL−1 nanogel concentration.
In addition to this trend of decreasing I1/I3 values, analyses of the pyrene absorbance spectra (Figure S4) suggest that the increasing hydrophobicities of the containers from NG2-NG6 may generate more favorable environments for lipophilic guest encapsulation and afford enhanced loading capacities. To investigate the extent of guest loading in these nanogels, samples of NG2-NG6 were prepared by the DTT-initiated crosslinking reaction in the presence of both 10 wt% DiI and 10 wt% DiO to afford noncovalent encapsulation of the FRET pair dyes within the nanogel interiors. After filtering off excess dye and removing water-soluble byproducts of the crosslinking reaction by dialysis, guest encapsulation in these nanogels was measured by UV-visible spectroscopy. The absorption spectra of the dye loaded nanogels, shown in Figure 5, reveal a trend of increasing dye encapsulation as the hydrophobic content of the nanogel is increased from NG2-NG6. A more detailed analysis of this loading capacity trend, with calculated loading efficiencies, is provided in a later section (Table 1).
Figure 5.

UV-vis spectra of NG2-NG6 prepared with 10 wt% dye feeding (spectra recorded at 0.05 mg.mL−1 nanogel) showing the effect of nanogel hydrophobicity on DiI/DiO loading.
Table 1.
Dye encapsulation efficiencies for NG2-NG6 prepared with 10, 2 and 0.5 wt% DiI/DiO.
| Samplea | 10 wt% Dye Feedingb | 2 wt% Dye Feedingc | 0.5 wt% Dye Feedingd | |||
|---|---|---|---|---|---|---|
| DiI | DiO | DiI | DiO | DiI | DiO | |
| NG2 | 34% | 13% | 82% | 41% | 82% | 63% |
| NG3 | 39% | 29% | 84% | 73% | > 95% | 86% |
| NG4 | 43% | 34% | 91% | 76% | > 95% | 87% |
| NG5 | 57% | 47% | > 95% | 83% | > 95% | 89% |
| NG6 | 65% | 54% | > 95% | 80% | > 95% | 90% |
All nanogels were prepared with 2 mg polymer total scale at 1 mg mL−1. Reported encapsulation efficiencies were calculated based on dye feedings of
0.225 mg DiI/0.205 mg DiO ;
0.045 mg DiI/0.041 mg DiO ;
0.011 mg DiI/0.010 mg DiO.
Effect of Hydrophobicity on Encapsulation Stability
Having demonstrated that the loading capacity for lipophilic guest molecules can be tuned in our nanogel system by introducing hydrophobic variations into the precursor polymer, we were next interested in investigating the consequences that such variations would have on the encapsulation stabilities of the guest-loaded nanocarriers. For this purpose, we first used the FRET-based assay (see Figure 2) to compare the encapsulation stabilities exhibited by the 10 wt% DiI/DiO loaded decyl chain-containing nanogels, NG4-NG6. Following this method, nanogel samples containing encapsulated DiI were mixed with those containing encapsulated DiO, with both nanogels having equal concentrations of 0.1 mg mL−1 in the resulting mixture. The evolution of FRET between the dye pair in mixed solutions of NG4-NG6, indicated by concomitant decrease in donor (DiO) emission intensity at 506 nm and increase in acceptor (DiI) emission intensity at 569 nm, was recorded over a six hour period.
It was hypothesized that increasing the hydrophobicity of the nanogels would enhance not only the lipophilic loading capacities, but also the encapsulation stabilities of the obtained nanocarriers. By providing an increasingly hydrophobic core, nanogels with a higher density of decyl side chains would create a more preferable environment for the solubilization of lipophilic guests, enhancing the ability of the nanogel to hold onto its dye molecules and limiting exchange with the surrounding environment.
The time-dependent FRET evolution, as observed in the fluorescence spectra of these mixed nanogel solutions, is plotted in Figure 6. As seen in the figure, these solutions exhibit faster rates of FRET development for the more hydrophobic nanogel containers, suggesting that increasing hydrophobicity actually decreases the encapsulation stability of the nanocarrier. However, it is important to note that there is significant variation in the loading amounts of DiI and DiO among the 10 wt% loaded NG4-NG6 samples. It is possible that this difference in dye loading, a consequence of the variations in nanogel hydrophobicity, influences the observed dye exchange rates. NG4 exhibited the lowest dye encapsulation with 10 wt% feeding, but also displayed the most gradually increasing FRET over the tested six hour period (Figure 6a). NG5 encapsulated an intermediate amount of dye and exhibited much faster FRET development (Figure 6b). Finally, NG6 encapsulated the highest amount of dye and displayed a rapid burst of FRET evolution, with the majority of observable exchange occurring within the first hour of the experiment (Figure 6c).
Figure 6.
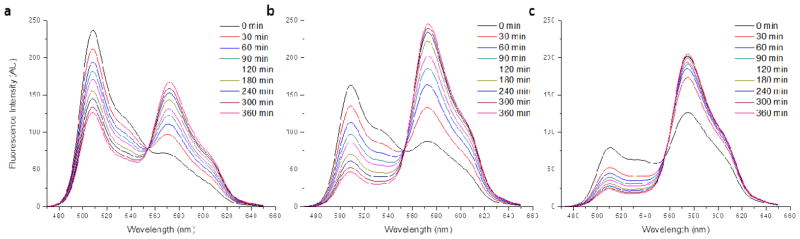
Fluorescence spectra of mixed solutions of NG4-NG6 (left to right) prepared with 10 wt% DiI/DiO tracing the development of FRET between the dyes over time.
This apparent inverse relationship between loading amount and encapsulation stability is clearly seen in the plots of FRET ratio vs. time for the 10 wt% NG4-NG6 series (Figure 7a). The FRET ratio Ia/(Id+Ia), where Ia and Id are the fluorescence intensities of the acceptor (DiI) and donor (DiO), respectively, can be used as a measure of the degree of FRET between the dye pair and, thus, the extent of dye exchange observed in each mixed nanogel solution. Note that in all the prepared nanogel samples, the amount of encapsulated DiI exceeded that of DiO (Figure 5, Table 1). An important consequence of this is that the maximum allowed FRET ratio for each mixed solution approaches 1.0, achieved only when every DiO molecule can transfer energy to a sufficiently proximal DiI molecule. From Figure 7a, it is observed that NG5 and NG6 are nearing this theoretical plateau at the end of the six hour test period. Additionally, it is noted that there are large differences in the initial FRET ratios of these mixed solutions, with the values increasing from NG4 (0.23) to NG6 (0.61). While self-quenching of DiO emission may contribute to this observation, the trend indicates that, with the rapidly exchanging NG5 and NG6, a significant amount of dye exchange occurs immediately upon mixing of the solutions before the first emission spectrum is recorded.
Figure 7.
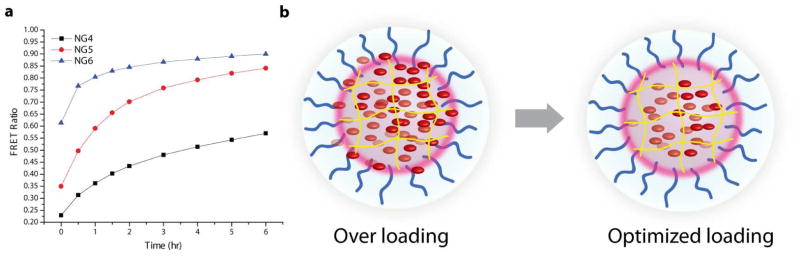
(a) FRET ratio vs. time plot of the mixed solutions of NG4-NG6 prepared with 10 wt% DiI/DiO and (b) a cartoon representation of an overloaded nanogel with low encapsulation stability and an optimally loaded nanogel with high encapsulation stability.
We propose that the development of FRET observed by this method is dependent on two separate events. The first is leakage of dye molecules from the interior of the originally encapsulating nanogel. The second is re-sequestration of the leaked dye molecules into nanogels present in solution. While the mechanism (collision- or diffusion- based) of this dye transfer remains unknown, the inability of the leaked dye molecules to discriminate between the nanogels in solution causes the previously described equilibration and FRET evolution. The results obtained with the 10 wt% NG4-NG6 series suggest that the total amount of encapsulated dye may significantly influence the probabilities of these processes happening and the rates at which they occur. This observation led us to form the hypothesis of loading-dependent encapsulation stability.
It is apparent from the spectra in Figure 5 that the capacity for lipophilic encapsulation is enhanced as the hydrophobic content of the precursor polymer increases. The amphiphilic natures of polymers P2-P6 induce formation of nanoscale micelle-type aggregates, in which the majority of hydrophobic content is buried within the cores. A possible feature of lipophilic guest encapsulation in these aggregates is that initially sequestered guest molecules accumulate deep within the core and gradually move out towards the assembly periphery as the core volume is filled. In this case, the extent of such peripheral loading will likely depend on the nature (length and density) of the hydrophobic units dispersed throughout the copolymer network, the size of the nanocarrier core and the amount of dye used during nanogel preparation. Thus, as the percent composition of decyl chains is increased from NG4 to NG6, the observed increase in loading capacity may, in part, be due to an enhanced ability to load lipophilic dye molecules closer to the nanogel surface. However, it is also reasonable to propose that these non-optimally loaded dye molecules will be least stably encapsulated and most easily exchange with the surrounding environment. This could explain why the enhanced dye loading observed from NG4 to NG6 translates to more rapid guest exchange and faster FRET evolution. This situation, illustrated in Figure 7b, would suggest that a given nanogel carrier must be loaded below its maximum loading capacity in order to achieve optimal core loading and high encapsulation stability.
Loading Capacity and Encapsulation Stability
This hypothesis was tested by preparing NG2-NG6 samples with lower guest loading, i.e. in the presence of 2 wt% and 0.5 wt% DiI/DiO. The observed dye loadings, indicated by the calculated encapsulation efficiencies for these nanogels, are summarized in Table 1 (See SI for UV-vis spectra). Analysis of these loading data reveals two important observations: (i) as dye feeding decreases, the encapsulation efficiencies of both DiI and DiO increase and the observed dependence of loading capacity on nanogel hydrophobicity diminishes. (ii) This effect is most apparent with the dye loading efficiencies among the nanogel samples fed with 0.5 wt% DiI/DiO, which are nearly identical and exhibit complete DiI encapsulation and close to 90% DiO encapsulation for NG3-NG6. Hexyl-containing NG2 still exhibits comparatively poor encapsulation efficiency with 2 wt% and 0.5 wt% dye feeding, indicating that, even at these lower loading amounts, the relative lack of hydrophobic content in the core of this nanogel affords a less preferable encapsulation environment than its more hydrophobic octyl- and decyl-containing counterparts.
At these lower dye loadings, similar encapsulation efficiencies were observed for DiI and DiO among the nanogel carriers. With such normalized loading, we hypothesized that a larger percentage of the dye content in these samples would be stably encapsulated in the nanogel cores. In this case, the observed rates of guest exchange in these nanogel carriers should provide a true correlation between hydrophobicity of the supramolecular host’s interior and encapsulation stability for lipophilic guest molecules.
Indeed, analysis of the fluorescence spectra from the mixing experiments performed with these nanogels suggests that increasing the hydrophobicity of the nanogel enhances the carrier’s encapsulation stability. In the case of the 2 wt% samples (Figure 8), dye molecule exchange evidenced by FRET evolution is observed to decrease continuously from NG2 to NG6, presumably due to the more preferable encapsulation environment afforded by the increasingly hydrophobic nanogel core. This trend is most clearly seen in the plots of FRET ratio vs. time for the mixed solutions (Figure 8f). With 2 wt% dye feeding, both NG2 and NG3 exhibit faster guest exchange than the NG1-NG3 series. While exchange is more rapid in NG5, both samples are observed to be approaching a FRET ratio plateau of around 0.7 by the end of the six hour test period.
Figure 8.

Fluorescence spectra of the mixed solutions of NG2-NG6 (a–e) prepared with 2 wt% DiI/DiO tracing the development of FRET between the dyes over time. (f) FRET ratio vs. time plots generated from the 2 wt% mixing data.
With the nanogel samples prepared with 0.5 wt% dye (Figure 9), the rates of guest exchange are significantly reduced. However, they remain much higher for NG2 and NG3 than for NG4-NG6, all of which exhibit nearly complete shutdown of guest leakage at this low dye loading. Independently, these mixing experiments performed with the 2 wt% and 0.5 wt% dye samples reveal the same trend; as the hydrophobic content of the nanogel container is increased from NG2 to NG6 the likelihood of guest exchange with the surrounding environment, and thus the rate of observed FRET evolution, is decreased.
Figure 9.

Fluorescence spectra of the mixed solutions of NG2-NG6 (a–e) prepared with 0.5 wt% DiI/DiO tracing the development of FRET between the dyes over time. (f) FRET ratio vs. time plots generated from the 0.5 wt% mixing data.
To compare the rates of guest exchange exhibited by the NG2-NG6 series, the slopes of the linear fits to the plots of FRET ratio vs. time were calculated. We have previously defined this slope value as the leakage coefficient (Λ), a quantitative relation to the rate of guest exchange between the assembly interior and the bulk solvent exhibited by a mixed nanocarrier solution.11 However, it must be noted that the plots used in this previous work were of normalized FRET ratio vs. time. Unlike the nanogel samples used in previous studies, which were prepared with 1 wt% DiI/DiO, some of the samples in the current work exhibit significant dye exchange immediately upon mixing. This is especially true for the NG4-NG6 series prepared with 10 wt% dye. To account for this initial exchange, we have not normalized these values and have reported the FRET ratios calculated directly from the obtained fluorescence data. From plotting these FRET ratios vs. time, we have determined the slope of the portion of this plot that can be accurately fit by a linear regression (R2 ≥ 0.95). We have defined this slope value as the corrected leakage coefficient (Λc), and have used this value to compare the dynamics of guest exchange exhibited by the studied nanogels.
The calculated Λc values for the NG2-NG6series prepared with 10 wt%, 2 wt% and 0.5 wt% DiI/DiO are recorded in Table 2, revealing the clear difference in trend observed with the 10 wt% samples. The significant increase in Λc value from NG4 to NG6 in this series lends support to the claim that the 10 wt% samples fit the overloaded nanogel model previously described. With greater numbers of decyl units, the more hydrophobic NG5 and NG6 exhibit higher capacity for guest loading in regions that do not provide sufficient encapsulation stability to prevent leakage into the bulk exterior. Thus, an inverse relationship between nanogel hydrophobicity and encapsulation stability is observed.
Table 2.
Calculated Λc values for the NG2-NG6 nanogel series prepared with varying dye amounts.
| Dye Feeding | NG2 | NG3 | NG4 Λc (h−1) | NG5 | NG6 |
|---|---|---|---|---|---|
| 10 wt% | ----- | ----- | 0.100 | 0.173 | 0.308 |
| 2 wt% | 0.434 | 0.126 | 0.051 | 0.036 | 0.024 |
| 0.5 wt% | 0.050 | 0.018 | 0.005 | 0.005 | 0.000 |
In contrast, the NG2-NG6 samples prepared with 2 wt% and 0.5 wt% dye may more closely fit the optimally loaded nanogel model, in which the majority of encapsulated dye molecules are stably sequestered within the cores. As a result, the expected trend of enhanced encapsulation stability with increasing nanogel hydrophobicity is observed with these samples. Additionally, it is noted that among nanogel samples of the same composition (e.g. all NG6), the corrected leakage coefficient value is observed to decrease with lower dye loading. Taken together, these results support the loading-dependent encapsulation stability hypothesis and suggest that optimal encapsulation, at which little to no guest exchange between containers in solution and the surrounding environment exists, is achieved well below the maximum loading capacity of a nanocarrier.
Conclusions
We have designed and synthesized a series of amphiphilic copolymer precursors and corresponding disulfide crosslinked nanogels. These nanogels were used to understand the effects on lipophilic guest encapsulation characteristics when hydrophobic variations are introduced into a supramolecular host. In performing this systematic study, we have shown that: (i) the encapsulation microenvironment of lipophilic guest molecules sequestered within the nanogels can be systematically altered by either tuning the percentage of lipophilic alkyl methacrylate co-monomer or varying the length of the alkyl chain in that co-monomer; (ii) the loading capacities and efficiencies, as probed with noncovalent encapsulation of lipophilic dye molecules, are dependent on nanogel hydrophobicity; (iii) at high dye feeding, the more hydrophobic nanogels exhibit significantly higher guest encapsulation. However, this enhanced loading does not necessarily translate to encapsulation stability; (iv) the observed trend in encapsulation stability at high loading is attributed to the possibility that a smaller percentage of the guest molecules are stably/optimally encapsulated within the nanogel core. This hypothesis has been substantiated by the fact that, at low loading, encapsulation stability follows an excellent correlation with nanogel hydrophobicity.
While these results provide insight into the relationships between guest loading, nanocarrier hydrophobicity and encapsulation stability, there remains a question regarding these particular nanogels: do the results here simply suggest that the stable encapsulation of guest molecules in this class of nanogel is restricted to low loading percentages? It is important to recognize that this is not the case, because: (i) The nanogels used in this work are small and have inherently lower loading capacities than larger nanogels that can be generated by this method10; (ii) In the studied systems, the percent crosslinking was intentionally limited to afford leaky nanogels that allowed us to develop the fundamental structure-property relationship studies we have outlined. Based on our previously reported results, the stability of guest encapsulation in these nanogels can be significantly enhanced by increasing the crosslinking density.11
Consideration of the dynamic guest exchange between a supramolecular host and the bulk solvent environment is critical for several applications. For example, if a host is used as a drug delivery vehicle, any exchange of guest molecules during circulation will likely cause leakage of non-covalently encapsulated cargo, accumulation at off-target sites and give rise to adverse side effects.13 Stable encapsulation of non-covalently bound guest molecules and specifically triggered release in response to a chosen stimulus are thus crucial design criteria for supramolecular hosts. Given our observations, we propose that inherently high loading capacities and efficiencies in a supramolecular host are not sufficient qualifying criteria for applications such as targeted drug delivery. It is equally, if not more, important to evaluate the percentage of this loading that is stably encapsulated within the nanocarrier. Considering that variations in hydrophilic-lipophilic balance form the basis for several amphiphilic supramolecular assemblies and that encapsulation stability is a critical parameter in several applications, we believe that the findings in this work are likely to impact the design and evaluation of next generation supramolecular hosts, especially those intended for use in biological applications such as drug delivery and theranostics.
Supplementary Material
Acknowledgments
This work was partially supported by DARPA (53271-MS-DRP) and NIGMS of the National Institutes of Health (GM-065255). We are also supported by the NSF-MRSEC facility at the University of Massachusetts for the infrastructure used in these characterizations.
Footnotes
Supporting Information Available: Experimental details (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv Drug Delivery Rev. 1997;23:3–25. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]; (b) Lipinski CA. Drug-like Properties and the Causes of Poor Solubility and Poor Permeability. J Pharmacol Toxicol Methods. 2000;44:235–249. doi: 10.1016/s1056-8719(00)00107-6. [DOI] [PubMed] [Google Scholar]; (c) Lipinski CA. Poor Aqueous Solubility-An Industry Wide Problem in Drug Discovery. Am Pharm Rev. 2002;5:82–85. [Google Scholar]
- 2.(a) Allen TM, Cullis PR. Drug Delivery Systems: Entering the Mainstream. Science. 2004;303:1818–1822. doi: 10.1126/science.1095833. [DOI] [PubMed] [Google Scholar]; (b) Lee CC, MacKay JA, Fréchet JMJ, Szoka FC. Designing Dendrimers for Biological Applications. Nat Biotechnol. 2005;23:1517–1526. doi: 10.1038/nbt1171. [DOI] [PubMed] [Google Scholar]; (c) Haag R, Kratz F. Polymer Therapeutics: Concepts and Applications. Angew Chem, Int Ed. 2006;45:1198–1215. doi: 10.1002/anie.200502113. [DOI] [PubMed] [Google Scholar]; (d) Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nanocarriers as an Emerging Platform for Cancer Therapy. Nat Nanotechnol. 2007;2:751–760. doi: 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]; (e) Davis ME, Chen Z, Shin DM. Nanoparticle Therapeutics: An Emerging Treatment Modality for Cancer. Nat Rev Drug Discovery. 2008;7:771–782. doi: 10.1038/nrd2614. [DOI] [PubMed] [Google Scholar]; (f) Gillies ER, Fréchet JMJ. Dendrimers and Dendritic Polymers in Drug Delivery. Drug Discov Today. 2005;10:35–43. doi: 10.1016/S1359-6446(04)03276-3. [DOI] [PubMed] [Google Scholar]; (g) Koo OM, Rubinstein I, Onyuksel H. Role of Nanotechnology in Targeted Drug Delivery and Imaging: A Concise Review. Nanomedicine: NBM. 2005;1:193–212. doi: 10.1016/j.nano.2005.06.004. [DOI] [PubMed] [Google Scholar]; (h) Oh JK, Drumright R, Siegwart DJ, Matyjaszewski K. The Development of Microgels/Nanogels for Drug Delivery Applications. Prog Polym Sci. 2008;33:448–477. [Google Scholar]; (i) Farokhzad OC, Langer R. Impact of Nanotechnology on Drug Delivery. ACS Nano. 2009;3:16–20. doi: 10.1021/nn900002m. [DOI] [PubMed] [Google Scholar]
- 3.(a) Jeong B, Bae YH, Lee DS, Kim SW. Biodegradable Block Copolymers as Injectable Drug-Delivery Systems. Nature. 1997;388:860–862. doi: 10.1038/42218. [DOI] [PubMed] [Google Scholar]; (b) Savić R, Luo L, Eisenberg A, Maysinger D. Micellar Nanocontainers Distribute to Defined Cytoplasmic Organelles. Science. 2003;300:615–618. doi: 10.1126/science.1078192. [DOI] [PubMed] [Google Scholar]; (c) Haag R. Supramolecular Drug-Delivery Systems Based on Polymeric Core-Shell Architectures. Angew Chem, Int Ed. 2004;43:278–282. doi: 10.1002/anie.200301694. [DOI] [PubMed] [Google Scholar]; (d) Kabanov AV, Nazarova IR, Astafieva IV, Batrakova EV, Alakhov VY, Yaroslavov AA, Kabanov VA. Micelle Formation and Solubilization of Fluorescent Probes in Poly(oxyethylene-boxypropylene-b-oxyethylene) Solutions. Macromolecules. 1995;28:2303–2314. [Google Scholar]; (e) Wooley KL. Shell Crosslinked Polymer Assemblies: Nanoscale Constructs Inspired from Biological Systems. J Polym Sci Part A: Polym Chem. 2000;38:1397–1407. [Google Scholar]; (f) Torchilin VP. Targeted Polymeric Micelles for Delivery of Poorly Soluble Drugs. Cell Mol Life Sci. 2004;61:2549–2559. doi: 10.1007/s00018-004-4153-5. [DOI] [PubMed] [Google Scholar]; (g) Yoo HS, Park TG. Folate Receptor Targeted Biodegradable Polymeric Doxorubicin Micelles. J Controlled Release. 2004;96:273–283. doi: 10.1016/j.jconrel.2004.02.003. [DOI] [PubMed] [Google Scholar]; (h) O’Reilly RK, Hawker CJ, Wooley KL. Cross-linked Block Copolymer Micelles: Functional Nanostructures of Great Potential and Versatility. Chem Soc Rev. 2006;35:1068–1083. doi: 10.1039/b514858h. [DOI] [PubMed] [Google Scholar]; (i) Kale TS, Klaikherd A, Popere B, Thayumanavan S. Supramolecular Assemblies of Amphiphilic Homopolymers. Langmuir. 2009;25:9660–9670. doi: 10.1021/la900734d. [DOI] [PubMed] [Google Scholar]; (j) Liu S, Maheshwari R, Kiick KL. Polymer-Based Therapeutics. Macromolecules. 2009;42:3–13. doi: 10.1021/ma801782q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Matsumura Y, Maeda H. A New Concept for Macromolecular Therapeutics in Cancer Chemotherapy: Mechanism of Tumoritropic Accumulation of Proteins and the Antitumor Agent Smancs. Cancer Res. 1986;46:6387–6392. [PubMed] [Google Scholar]; (b) Baban DF, Seymour LW. Control of Tumour Vascular Permeability. Adv Drug Delivery Rev. 1998;34:109–119. doi: 10.1016/s0169-409x(98)00003-9. [DOI] [PubMed] [Google Scholar]; (c) Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. Tumor Vascular Permeability and the EPR Effect in Macromolecular Therapeutics: A Review. J Controlled Release. 2000;65:271–284. doi: 10.1016/s0168-3659(99)00248-5. [DOI] [PubMed] [Google Scholar]; (d) Duncan R. The Dawning Era of Polymer Therapeutics. Nat Rev Drug Discovery. 2003;2:347–360. doi: 10.1038/nrd1088. [DOI] [PubMed] [Google Scholar]
- 5.(a) Kataoka K, Harada A, Nagasaki Y. Block Copolymer Micelles for Drug Delivery: Design, Characterization and Biological Significance. Adv Drug Delivery Rev. 2001;47:113–131. doi: 10.1016/s0169-409x(00)00124-1. [DOI] [PubMed] [Google Scholar]; (b) Lin WJ, Juang LW, Lin CC. Stability and Release Performce of a Series of Pegylated Copolymeric Micelles. Pharm Res. 2003;20:668–673. doi: 10.1023/a:1023215320026. [DOI] [PubMed] [Google Scholar]; (c) Liu J, Xiao Y, Allen C. Polymer-Drug Compatibility: A Guide to the Development of Delivery Systems for the Anticancer Agent, Ellipticine. J Pharm Sci. 2004;93:132–143. doi: 10.1002/jps.10533. [DOI] [PubMed] [Google Scholar]; (d) Gaucher G, Dufresne MH, Sant VP, Kang N, Maysinger D, Leroux JC. Block Copolymer Micelles: Preparation, Characterization and Application in Drug Delivery. J Controlled Release. 2005;109:169–188. doi: 10.1016/j.jconrel.2005.09.034. [DOI] [PubMed] [Google Scholar]; (e) Aliabadi HM, Lavasanifar A. Polymeric Micelles for Drug Delivery. Expert Opin Drug Deliv. 2006;3:139–162. doi: 10.1517/17425247.3.1.139. [DOI] [PubMed] [Google Scholar]
- 6.(a) Lavasanifar A, Samuel J, Kwon GS. Poly(ethylene oxide)-block-poly(L-amino acid) Micelles for Drug Delivery. Adv Drug Delivery Rev. 2002;54:169–190. doi: 10.1016/s0169-409x(02)00015-7. [DOI] [PubMed] [Google Scholar]; (b) Savić R, Eisenberg A, Maysinger D. Block Copolymer Micelles as Delivery Vehicles of Hydrophobic Drugs: Micelle-Cell Interactions. J Drug Target. 2006;14:343–355. doi: 10.1080/10611860600874538. [DOI] [PubMed] [Google Scholar]; (c) Letchford K, Liggins R, Burt H. Solubilization of Hydrophobic Drugs by Methoxy Poly(ethylene glycol)-block-polycaprolactone Diblock Copolymer Micelles: Theoretical and Experimental Data and Correlations. J Pharm Sci. 2008;97:1179–1190. doi: 10.1002/jps.21037. [DOI] [PubMed] [Google Scholar]; (d) Kim SW, Shi Y, Kim JY, Park K, Cheng JX. Overcoming the Barriers in Micellar Drug Delivery: Loading Efficiency, in vivo Stability, and Micelle-Cell Interaction. Expert Opin Drug Deliv. 2010;7:49–62. doi: 10.1517/17425240903380446. [DOI] [PubMed] [Google Scholar]
- 7.(a) Bae YH, Yin H. Stability Issues of Polymeric Micelles. J Controlled Release. 2008;131:2–4. doi: 10.1016/j.jconrel.2008.06.015. [DOI] [PubMed] [Google Scholar]; (b) Chen H, Kim S, He W, Wang H, Low PS, Park K, Cheng J-X. Fast Release of Lipophilic Agents from Circulating PEG-PDLLA Micelles Revealed by in Vivo Föster Resonance Energy Transfer Imaging. Langmuir. 2008;24:5213–5217. doi: 10.1021/la703570m. [DOI] [PubMed] [Google Scholar]
- 8.For examples, see: Bütün V, Billingham NC, Armes SP. Synthesis of Shell Cross-Linked Micelles with Tunable Hydrophilic/Hydrophobic Cores. J Am Chem Soc. 1998;120:12135–12136.Tang Y, Liu SY, Armes SP, Billingham NC. Solubilization and Controlled Release of a Hydrophobic Drug Using Novel Micelle-Forming ABC Triblock Copolymers. Biomacromolecules. 2003;4:1636–1645. doi: 10.1021/bm030026t.Danquah M, Fujiwara T, Mahato R. Self-Assembling Methoxypoly(ethylene glycol)-b-poly(carbonate-co-L-lactide) Block Copolymers for Drug Delivery. Biomaterials. 2010;31:2358–2370. doi: 10.1016/j.biomaterials.2009.11.081.Li F, Danquah M, Mahato R. Synthesis and Characterization of Amphiphilic Lipopolymers for Micellar Drug Delivery. Biomacromolecules. 2010;11:2610–2620. doi: 10.1021/bm100561v.
- 9.(a) Kabanov AV, Vinogradov S. Nanogels as Pharmaceutical Carriers: Finite Networks of Infinite Capabilities. Angew Chem Int Ed. 2009;48:5418–5429. doi: 10.1002/anie.200900441. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chacko R, Ventura J, Zhuang J, Thayumanavan S. Polymer Nanogels: A Versatile Nanoscopic Drug Delivery Platform. Adv Drug Deliv Rev. 2012;64:836–851. doi: 10.1016/j.addr.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Raemdonck K, Demeester J, Smedt SD. Advanced Nanogel Engineering for Drug Delivery. Soft Matter. 2009;5:707–715. [Google Scholar]; (d) Vinogradov S. Nanogels in the Race for Drug Delivery. Nanomedicine. 2010;5:165–168. doi: 10.2217/nnm.09.103. [DOI] [PubMed] [Google Scholar]; (e) Zha L, Banik B, Alexis F. Stimulus Responsive Nanogels for Drug Delivery. Soft Matter. 2011;7:5908–5916. [Google Scholar]
- 10.(a) Ryu JH, Chacko RT, Jiwpanich S, Bickerton S, Babu RP, Thayumanavan S. Self-Cross-Linked Polymer Nanogels: A Versatile Nanoscopic Drug Delivery Platform. J Am Chem Soc. 2010;132:17227–17235. doi: 10.1021/ja1069932. [DOI] [PubMed] [Google Scholar]; (b) Ryu JH, Jiwpanich S, Chacko R, Bickerton S, Thayumanavan S. Surface-Functionalizable Polymer Nanogels with Facile Hydrophobic Guest Encapsulation Capabilities. J Am Chem Soc. 2010;132:8246–8247. doi: 10.1021/ja102316a. [DOI] [PubMed] [Google Scholar]
- 11.Jiwpanich S, Ryu JH, Bickerton S, Thayumanavan S. Noncovalent Encapsulation Stabilities in Supramolecular Nanoassemblies. J Am Chem Soc. 2010;132:10683–10685. doi: 10.1021/ja105059g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kalyanasundaram K, Thomas JK. Environmental Effects on Vibronic Band Intensities in Pyrene Monomer Fluorescence and their Application in Studies of Micellar Systems. J Am Chem Soc. 1977;99:2039–2044. [Google Scholar]; (b) Wilhelm M, Zhao C-L, Wang Y, Xu R, Winnik M, Mura JL, Riess G, Croucher M. Poly(styrene-ethylene oxide) Block Copolymer Micelle Formation in Water: A Fluorescence Probe Study. Macromolecules. 1991;24:1033–1040. [Google Scholar]
- 13.(a) Chen H, Kim S, Li L, Wang S, Park K, Cheng JX. Release of Hydrophobic Molecules from Polymer Micelles into Cell Membranes Revealed by Föster Resonance Energy Transfer Imaging. Proc Natl Acad Sci USA. 2008;105:6596–6601. doi: 10.1073/pnas.0707046105. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Xu P, Gullotti E, Tong L, Highley CB, Errabelli DR, Hasan T, Cheng JX, Kohane DS, Yeo Y. Intracellular Drug Delivery by Poly(lactic-co-glycolic acid) Nanoparticles, Revisted. Mol Pharmaceutics. 2009;6:190–201. doi: 10.1021/mp800137z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.