

Abstract
Intestinal epithelial cells (IEC) play a role in mucosal inflammation by producing pro-inflammatory chemokines that may initiate or amplify local responses. IL-1 is a potent activator of IEC and its receptor localizes to focal adhesions. Since the Rho-associated kinase, ROCK, also localizes to focal adhesions, we examined the role of ROCK in IL-1-induced chemokine responses in IEC cell lines. Suppressing ROCK with the Y27632 inhibitor suppressed IL-1-stimulated Caco-2 cell CXCL8/IL-8 and IEC-6 cell CCL2/MCP-1 secretion and mRNA levels. ROCK inhibition also suppressed IL-1-induced JNK phosphorylation in both cell lines, but high levels of the inhibitor had no significant effect on IL-1-stimulated Caco-2 IκBα phosphorylation and degradation or IKK phosphorylation and kinase activity. Therefore, ROCK may exert an effect on IL-1-stimulated JNK signaling to AP-1 activation, with little effect on IKK/IκBα signaling, defining a potentially important mechanism for regulating IL-1 signaling in IEC that may be essential for optimal cytokine responses.
Keywords: IL-1, intestinal epithelial, CXCL8, JNK, CCL2, ROCK
1. Introduction
IL-1 is a potent pro-inflammatory cytokine with a known role in mucosal infections and inflammatory bowel disease (IBD). This cytokine has been shown to activate hundreds of genes including inflammatory cytokines, growth factors, receptors, proteases, signaling proteins and transcription factors [1, 2]. The intestinal epithelial cells (IEC) that line the surface of the intestine and colon are responsive to IL-1 and can produce several important immunoregulatory cytokines and chemokines, such as IL-6, CXCL8 (IL-8) and CCL2 (MCP-1) [3–6], to amplify or sustain the inflammatory response. This suggests that IEC have a role in intestinal inflammation and IBD by their inflammatory cytokine production.
IL-1 exerts its effect through a signaling pathway that results in the activation of the transcription factors, NF-κB and AP-1 [2]. IL-1 stimulation of human colonic epithelial cells and cell lines induces nuclear accumulation of NF-κB and CXCL8 secretion [7]. This results from the activation of the upstream serine kinases, IκB kinase (IKK) α and β, which are responsible for phosphorylating IκBα and initiating the activation of NF-κB [8]. IL-1 can also activate an intracellular signaling pathway that results in phosphorylation of the c-Jun NH2-Terminal Kinase, JNK [2]. Active JNK then phosphorylates c-Jun, which along with the Fos protein creates the archetypical AP-1 transcription factor [9]. High levels of phosphorylated JNK staining have been shown to localize to both leukocytes and epithelial cells in the inflamed mucosa of patients with IBD, as compared to normal controls [10]. These and other studies have suggested an important role for JNK signaling in IBD [11].
IEC attach to the extracellular matrix through cell surface integrins [12]. Integrin signaling can activate the Rho family of small GTPases, which are known to control the formation of actin stress fibers and actomyosin contractility in cell movement [13]. A major downstream effector for Rho is the Rho-associated protein kinase, ROCK, a serine/threonine kinase which phosphorylates myosin light chain phosphatase involved in actomyosin contractility [13]. ROCK exists as two isoforms, ROCK1 and ROCK2, which are highly homologous [14].
A recent study using rats with trinitrobenzene sulfonic acid induced colitis found that inhibiting the activity of ROCK with the chemical inhibitor, Y27632, reduced the symptoms of colitis suggesting that ROCK was involved in inflammatory bowel disease [15]. Then using peripheral blood mononuclear cells from Crohn’s disease patients or a macrophage cell line, they found that inhibiting ROCK could inhibit LPS induced IL-1 and TNF production, and IL-1 signaling through IKK/IκBα to NF-κB. These results suggested that IL-1 and LPS stimulation of macrophages and peripheral blood mononuclear cells resulted in the activation of ROCK, which had an important positive regulatory effect on cytokine responses and IL-1 induced intracellular signaling events. Therefore ROCK may play an important role in mucosal inflammatory responses mediated by IL-1. However, the effect of Y27632 treatment on IEC was not considered, despite the role that IEC play in the mucosal inflammatory response. Also it is not clear whether all cell types share the effect of ROCK seen above. In cervical stromal cells, Y27632 was found to block LPS, but not IL-1, induced CXCL8 production [16], while in astrocytes, IL-1 deactivates Rho-ROCK signaling to alter astrocyte function [17].
IEC activation by IL-1 and subsequent cytokine and pro-inflammatory responses may be very important in the early stages of infection or inflammatory disease as initiators of inflammation and may help to sustain or amplify ongoing inflammation. Yet cell migration and wound healing events may also alter IEC function through the activation of ROCK by integrin signaling pathways [18]. Therefore, it is essential that the relationship between IL-1 and ROCK activation in IEC be well understood. We have determined the role of ROCK in IEC cytokine responses to IL-1 stimulation and provide evidence that ROCK may play an important role in IL-1 signaling in these cells. However, unlike with peripheral blood mononuclear cells and macrophages [15], inhibiting ROCK was found to suppress IL-1 induced CXCL8 secretion and mRNA levels through an inhibition of JNK, but not in IKK/IκBα signaling in IEC. This novel finding provides important insights into the mechanisms that regulate IL-1 signaling in IEC.
2. Materials and Methods
2.1 Antibodies
Rabbit polyclonal antibodies against human phospho-IκBα (Ser 32 and 36), phospho-IKK (IKKα Ser 180/IKKβ Ser 181), or JNK; mouse monoclonal antibodies against human IκBα or phospho-JNK (Thr 183/Tyr 185); HRP-conjugated anti-rabbit and HRP-conjugated anti-mouse detection antibodies were obtained from Cell Signaling Technologies (Beverly, MA). The mouse monoclonal antibodies against human tubulin along with rabbit polyclonal antibody against human IKK were purchased from Santa Cruz Biotechnology (Santa Cruz, CA).
2.2 Cell Culture Conditions
The human colonic Caco-2 cell line (ATCC HTB37; American Type Culture Collection, Manassas, VA) was cultured in RPMI-1640 (Mediatech, Herndon, VA) with 10% fetal bovine serum (FCS; Hyclone Laboratories, Logan, UT), 2g/L sodium bicarbonate, 2mM L-glutamine (Lonza, Walkersville, MD), non-essential amino acids (Lonza), 25 IU penicillin and 25μg/ml streptomycin (Mediatech), referred to as % FCS-RPMI. The rat IEC-6 small intestine epithelial cell line (ATCC CRL1592) was cultured in DMEM (Hyclone) with 5% FCS (Hyclone), 2g/L sodium bicarbonate, 70 USP units bovine insulin (Sigma, St. Lois, MO), 2mM L-glutamine (Lonza), 25 IU penicillin and 25μg/ml streptomycin (Mediatech), referred to as 5% FCS-DMEM. All cell lines were maintained at 37°C in a 90% air-10% CO2 humid atmosphere.
The cells were removed from tissue culture flasks by brief treatment with a trypsin and EDTA solution (Sigma) and were resuspended in 5% FCS-DMEM. Wells of tissue culture plates (BD Labware, Franklin Lakes, NJ) were coated with 20μg/ml bovine plasma fibronectin (Sigma) in PBS (e.g. 200μl per well for 12-well plates) and incubated for 60 minutes at 37°C. The wells were then aspirated and allowed to dry under ultraviolet light for 30 minutes.
For experiments using the ROCK inhibitor, Caco-2 or IEC-6 cells were plated in FN coated wells and incubated for one hour at 37°C and 10% CO2. The culture supernatants were then removed and the cells treated with serum-free DMEM containing insulin, transferrin, and selenium (ITS; BD Biosciences, Bedford, MA) with or without the ROCK inhibitor, Y-27632 (Sigma), for a final concentration of 1–100μM. Cells were incubated for another two hours at 37°C and 10% CO2. At three hours after plating, the cells were treated with rhIL-1 (R&D Systems, Minneapolis, MN) at 1ng/ml (Caco-2) or 0.5ng/ml (IEC-6) as these were found to be optimal doses for CXCL8 or CCL2 secretion, respectively (data not shown).
2.3 Determining cytokine levels in culture supernatants
Caco-2 (1×105 cells/well in 96-well plates) and IEC-6 (2.5×105 cells/well in 24-well plates) cells were cultured and treated with the ROCK inhibitor and IL-1 as described above. Culture supernatants were harvested 24 hours after IL-1 stimulation and stored at −80°C. The total cells per well were determined by treating the cells with trypsin and EDTA (Sigma) and counting the cells using a hemacytometer. The amount of human CXCL8 or rat CCL2 present in the culture supernatants was determined with the DuoSet ELISA kit for human CXCL8/IL-8 (R&D Systems, Minneapolis, MN) or the Cytoscreen ELISA kit for rat CCL2/MCP-1 (Biosource, Camarillo, CA). The absorbances of the samples were measured using a Bio-Tek EL312e microplate reader (Bio-Tek Instruments Inc., Winooski, VT).
2.4 Reverse transcriptase-polymerase chain reaction (RT-PCR)
To determine the effect of the ROCK inhibitor on CXCL8 and CCL2 mRNA levels, Caco-2 and IEC-6 (1×105 cells/well) cells in 12 well plates were treated with the ROCK inhibitor (40μM and 20μM, respectively) and IL-1 as described above. Six hours after IL-1 stimulation, total RNA was harvested using TRIZOL reagent (Invitrogen) as per the manufacturer protocol. Samples were reverse transcribed using the ReactionReady First Strand cDNA Synthesis Kit (SuperArray, Frederick, MD) with annealing of random primers. PCR was performed using RT2 PCR Primer Sets for human glyceraldehyde-3-phosphate dehydrogenase (GAPD), human CXCL8, rat GAPD, and rat CCL2 (SuperArray). The PCR reaction with human primers was carried out at 95°C for 10 minutes followed by 26 cycles of 95°C for 15 seconds, 60°C for 30 seconds, and 72°C for 30 seconds and a final extension at 72°C for 7 minutes using a Perkin Elmer GeneAmp PCR System 2400 (Perkin Elmer, Waltham, MA). Amplification using rat primers were done as above for 29 (CCL2) or 32 (GAPD) cycles with an annealing temperature of 55°C for 45 seconds. The samples were then separated by agarose electrophoresis and stained with ethidium bromide. The images were captured using the Kodak 1D Image Analysis Software (Kodak, Memphis, TN) and band densities were quantified using the LabWorks 4.0 Image Analysis software (UVP Inc., Upland, CA).
2.5 Preparation of cell extracts
The adherent Caco-2 or IEC-6 cells were washed once in ice-cold PBS before isolation of cytoplasmic extracts as previously described by Li, et al. [19]. The cells were lysed in a cytoplasmic extract buffer with Calbiochem Protease Inhibitor Cocktail Set III (Calbiochem, San Diego, CA) for 10 minutes at 4°C. The extracts were centrifuged for 5 minutes at 13,000×g to remove the nuclei and debris. The protein concentrations were then determined using the Bio-Rad DC protein assay kit (Bio-Rad, Melville, NY) and the samples were stored at −80°C until further use.
2.6 Immunoblot analysis
Cytoplasmic extracts with equal amounts of protein or equal volumes of immunoprecipitates were separated with 8–15% polyacrylamide resolving gels and transferred to polyvinylidene difluoride membranes using the Mini Trans-blot system (Bio-Rad). The blots were blocked with 5% bovine serum albumin (BSA) in Tris buffered saline (TBS) with 0.1% Tween-20 (BSA-TBST) for 1 hour at room temperature and incubated overnight at 4°C with the primary antibody in BSA-TBST. The blots were washed with TBS-T followed by incubation with a secondary antibody in BSA-TBST of either an HRP-conjugated anti-rabbit or anti-mouse antibody (Cell Signaling Technologies, Danvers, MA). The blots were then washed three times with TBS-T and the protein bands were visualized using the Lumiglo detection kit (Cell Signaling Technologies) and exposure to x-ray film and scanning or images were collected using a Bio-Rad ChemiDoc System. The band densities of interest were then quantified using the LabWorks 4.0 Image Analysis software.
Blots were reprobed by washing twice with TBS-T followed by incubating with a stripping buffer of 62.5mM Tris-HCl, 2% SDS, and 100mM 2-mercaptoethanol at 50°C for 12–15 minutes. The blots were then washed four times with TBS-T and were reblocked with 5% BSA-TBST for one hour at room temperature with ROCK1ng before reprobing with the next primary antibody.
2.7 IKK kinase activity assay
A non-radioactive kinase assay was performed for IKKα/β activity as described in Li, et al. [19]. Cytoplasmic extracts with 200μg of protein in 0.5ml total volume were incubated with 1μg of anti-IKKα/β antibody overnight at 4°C with rocking. The samples were then incubated with 20μl of Protein A/G agarose (Santa Cruz Biotechnology) with rocking at 4°C before centrifuging at 5,000 rpm for 5 minutes and washing with kinase buffer. The immunoprecipitates of IKKα/β were suspended in the kinase buffer with ATP and 2μg of GST-IκBα target (Santa Cruz Biotechnology) for 15 minutes at 30°C. The reaction was stopped by adding 20μl of 2× Laemmli buffer and heating at 95–100°C for 5 minutes. Samples were separated on 4–20% Tris-HCl gradient gels (BioRad) and Western blotted with an anti-phospho-IκBα antibody. The blots were stripped and reprobed for IKKα/β as described above. Band density of phospho-IκBα was normalized with the corresponding band density of the IKKα/β.
2.9 Statistics
Results from three or more experiments were analyzed for significant differences using the Student’s t Test for comparison of two samples or ANOVA and Fisher’s protected least significant difference test with a level of significance set at p < 0.05 for comparing three or more samples. Analyses were done using Statview 4.01 (SAS Institute).
3. Results
3.1 Effect of inhibiting ROCK on IL-1 induced CXCL8 and CCL2 responses
As ROCK may have a potential role in regulating IL-1 responses in IEC and we have found that IEC cell lines express protein for both isoforms of ROCK (data not shown), the effect of inhibiting ROCK was determined on two IEC cell lines, the human Caco-2 and the rat IEC-6 cell lines. The cells were plated onto FN coated wells and incubated for one hour to allow cell attachment before treating the cells for two hours with different doses of the Y27632 ROCK inhibitor. The Y27632 ROCK inhibitor (RI) is a competitive inhibitor of ATP binding and inhibits equally both ROCK1 and ROCK2 isoforms [20, 21]. This inhibitor is specific for suppressing the ROCK kinase activity by up to 87% while showing little or no effect on other 23 other kinases [21]. Y27632 also has an affinity for ROCK that is 20 to 30 times higher than for the other Rho effector kinases like the citron kinase and PKN (also known as PRK) [20]. Although 20μM of Y27632 is commonly used for studies inhibiting ROCK [20], higher levels up to 100mM and 300mM have been used to show some effects such as G1 to S phase progression in cell cycle [20] and bronchial smooth muscle contraction studies [22]. After two hours, the cells were treated with IL-1β and incubated for 24 hours. CCL2 was chosen as a representative chemokine for the rat cell lines as rats do not produce CXCL8. Treatment of the Caco-2 or IEC-6 cells with the ROCK inhibitor had no significant effect at any of the concentrations used on the total number of cells counted at 24 hours at the end of the experiments (p > 0.16; data not shown). Treatment of the IEC cell lines with the ROCK inhibitor alone had no effect on cytokine secretion by either cell line (Fig. 1). However, inhibiting ROCK resulted in a significant 70% suppression of IL-1-stimulated Caco-2 CXCL8 secretion (Fig. 1A; 100μM) and an 84% (with 100μM) or 60% (with 10μM) suppression of IEC-6 CCL2 secretion. Of note, the IEC-6 cells were found to be somewhat more sensitive to ROCK inhibition than the Caco-2 cells.
Fig. 1.
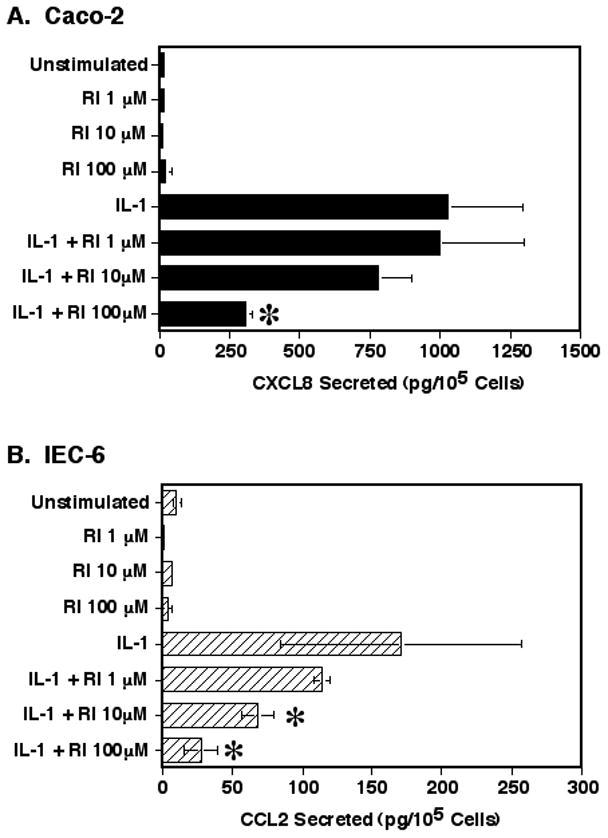
The effect of the ROCK inhibition on IL-1 induced CXCL8 or CCL2 secretion. (A) Caco-2 cells or (B) IEC-6 cells were cultured in FN coated wells for 1 hour. Cells were then treated with the Y27632 ROCK inhibitor (RI) in ITS-RPMI as indicated and cultured for 2 hours. Cultures were then stimulated with IL-1β for 24 hours. The culture supernatants were harvested and analyzed for cytokine levels by specific ELISA and the adherent cells were removed and counted. The data represents the mean ± SEM from 3 separate experiments. Asterisk indicates a significant difference when compared to the IL-1 only sample (p < 0.05).
Next, the effect of inhibiting ROCK on IL-1 induced cytokine mRNA levels was determined. Pre-treating the cells with the 40μM ROCK inhibitor as above and culturing with IL-1 stimulation for 6 hours resulted in a significant 57% suppression of IL-1 induced CXCL8 mRNA levels in the Caco-2 cells, as compared to that of cells stimulated with IL-1 alone (Fig. 2A). A similar 62% suppression of IL-1 induced CCL2 mRNA levels with ROCK inhibition was seen with the IEC-6 cells (Fig. 2B) confirming the effect. Taken together, these experiments suggest that ROCK may play a significant role in the IL-1 activation of IEC to produce important pro-inflammatory cytokines.
Fig. 2.
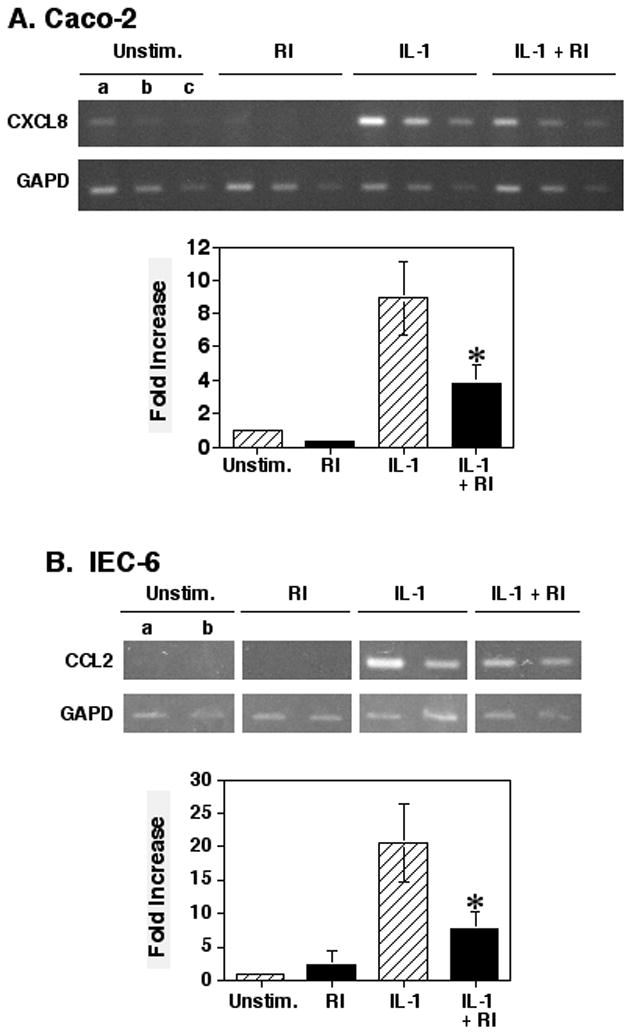
Treatment of cells with the ROCK inhibitor suppressed IL-1 induced CXCL8 or CCL2 mRNA production. (A) Caco-2 cells were cultured for 1 hour and then treated with the Y27632 ROCK inhibitor for 2 hours. Cultures then were stimulated with IL-1β for 6 hours before isolating total RNA for reverse transcription and PCR amplification for CXCL8 mRNA. For PCR, (a) 0.5μl, (b) 0.25μl, or (c) 0.125μl of cDNA was used each sample. Graph shows a comparison of the means ± SEM of the 0.5μl cDNA samples as the fold-increase of the ratios of the CXCL8 to GAPD band densities from 4 separate experiments. (B) IEC-6 cells were cultured as described with the ROCK inhibitor and IL-1β. RNA extracts were analyzed by RT-PCR for CCL2 mRNA. Graph shows a comparison of the means ± SEM of the 0.25μl cDNA samples as the fold-increase of the ratios of the CXCL8 to GAPD band densities from 4 separate experiments. Asterisk indicates a significant difference when compared to the IL-1 only sample (p < 0.05).
3.2 Effect of inhibiting ROCK on IL-1 signaling events through IκBα/NF-κB
A major downstream signaling pathway from IL-1 stimulation is the activation of the IKK complex, which results in the phosphorylation and eventual degradation of IκBα to release active NF-κB [2]. Therefore, the effect of inhibiting ROCK with the Y27632 inhibitor was first determined on the phosphorylation and degradation of IκBα. Treatment of the Caco-2 cells with a high level of the ROCK inhibitor (100μM) resulted in no significant effect on IκBα phosphorylation or degradation at any time point (p = 0.1 to 0.98) after IL-1 stimulation as compared to cells treated with IL-1 alone (Fig. 3). Although the IL-1 induced IκBα phosphorylation levels at 15 minutes were lower with the ROCK inhibitor treated cells, this level was not significantly different from the IL-1 only treated cells (p=0.1).
Fig. 3.
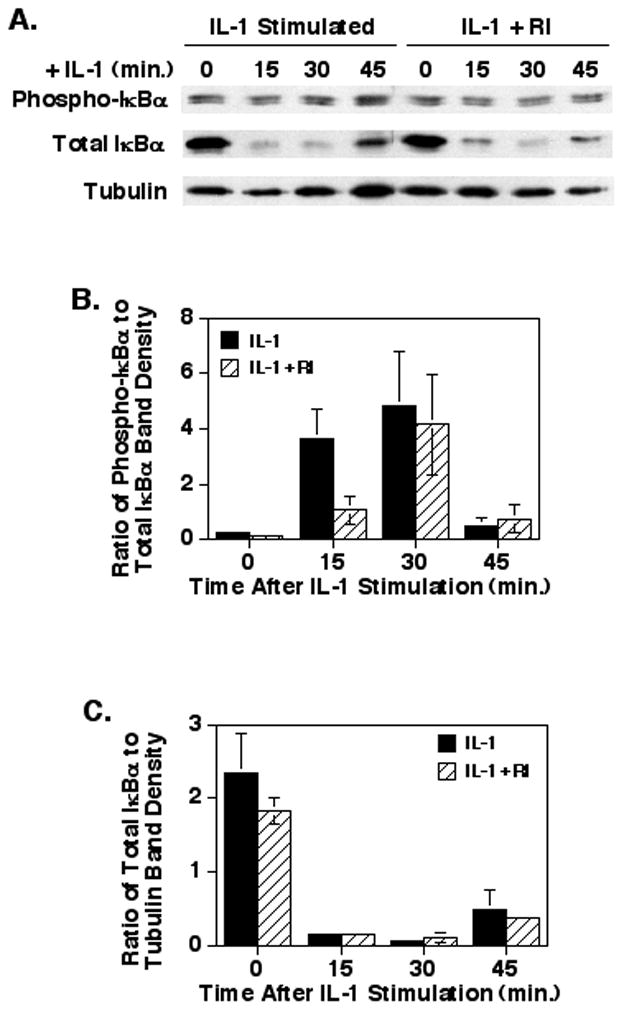
Treatment with the Y27632 ROCK inhibitor does not affect IL-1 induced IκBα phosphorylation or degradation in Caco-2 cells. Caco-2 cells were treated with the ROCK inhibitor (100μM) and IL-1β as described. At the times indicated, cytoplasmic extracts were harvested and separated by SDS-PAGE for Western blotting. (A) A representative blot probed for phospho-IκBα, total IκBα, and tubulin. (B) A graph of the ratios of the phospho-IκBα to total IκBα band density values. (C) A graph of the ratios of the total IκBα to tubulin band density values. The data represents the mean ± SEM of 3 separate experiments and no significant differences were seen between samples treated with or without the ROCK inhibitor.
Since the phosphorylation of IκBα is dependent upon the phosphorylation and activation of the upstream kinase, IKKα/β, the phosphorylation status and kinase activity of IKK was determined. As shown in Fig. 4, inhibiting ROCK had no significant effect on the IL-1 induced phosphorylation of IKKα/β at any time-point after stimulation (p = 0.21 to 0.88). In addition, inhibiting ROCK had no significant effect on the IL-1 induced specific activity of immunoprecipitated IKK toward an IκBα target (Fig. 4B; p = 0.26 to 0.94). These results with IKK activation confirm the findings that the ROCK inhibitor had little effect on IκBα phosphorylation and degradation and suggest that ROCK may play no role in the IKK/IκBα/NF-κB signaling in IEC, even with relatively high levels of the Y27632 ROCK inhibitor (100μM).
Fig. 4.
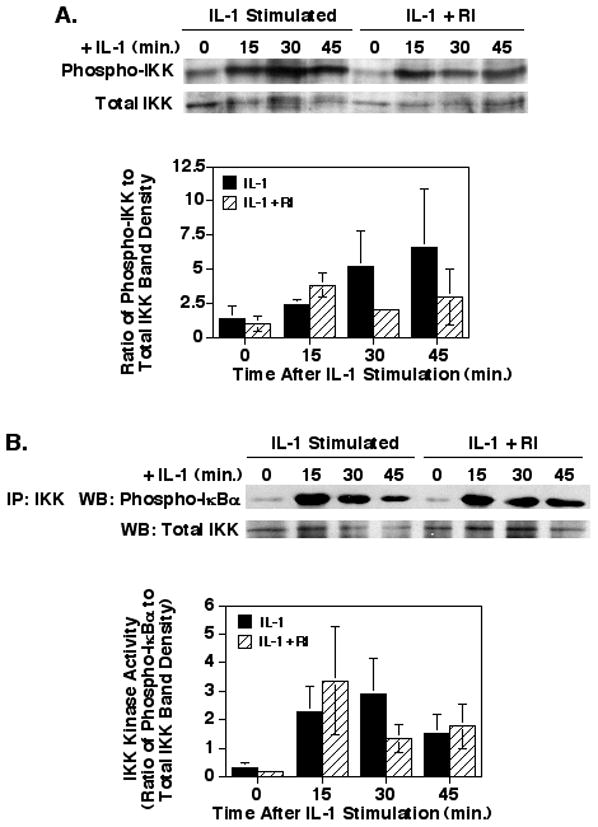
Inhibition of ROCK does not affect IL-1-stimulated IKK phosphorylation or specific kinase activity in Caco-2 cells. Caco-2 cells were cultured, treated with the Y27632 ROCK inhibitor and IL-1, and cytoplasmic extracts were collected as described in Fig. 3. (A) Shown is representative blot and a graph of the ratios of the phospho-IKKα/β to total IKKα/β band density values. (B) Shown is a representative blot of immunoprecipitated IKKα/β subjected to a kinase assay with GST-IκBα as the target. A graph of the ratios of the phospho-IκBα to total IKKα/β band density values shows no effect of the ROCK inhibitor on IKK kinase activity. The data shown represent the mean ± SEM for 3 separate experiments.
3.3 Inhibiting ROCK results in an inhibition of IL-1 induced JNK phosphorylation
IL-1 signaling events downstream of the TAK1 kinase split into the IKK/IκBα/NF-κB signaling pathway and the JNK/c-Jun/AP-1 signaling pathway [2]. Therefore, the effect of inhibiting ROCK with the Y27632 inhibitor was determined on IL-1 induced JNK phosphorylation, a step necessary for the activation of this key kinase. Pre-treatment of the Caco-2 cells with 100μM Y27632 (Fig. 5A) resulted in a significant 87% suppression of p46 JNK phosphorylation at 30 minutes (p<0.01) and a significant 94% suppression of p54 JNK phosphorylation at 15 minutes (p<0.05). However, as this was a high dose of the Y27632, we also examined the effect of treating the Caco-2 cells with 40μM Y27632 ROCK inhibitor prior to IL-1 stimulation (Fig. 5B), which resulted in a significant 50% suppression of IL-1 induced p46 JNK phosphorylation at 30 minutes (p<0.05). Yet, treatment with this lower level of the ROCK inhibitor had no significant effect on IL-1 induced p54 JNK phosphorylation at any time point (p = 0.15 to 0.5). To confirm these results, the IEC-6 cells were treated with 20μM Y27632 inhibitor prior to IL-1 stimulation which yielded a significant 86% suppression of IL-1 induced p46 JNK phosphorylation at 15 minutes (p = 0.02) and 80% and 64% suppression of IL-1 induced p54 JNK phosphorylation at 15 (p = 0.009) and 30 minutes (p = 0.02), respectively (Fig. 5C). A lower level of Y27632 ROCK inhibitor was used with the IEC-6 cells as the results in Fig. 1 indicated that these cells were more sensitive to the inhibitor. These experiments suggest that ROCK may play a significant role in the IL-1 signaling pathway leading to JNK activation.
Fig. 5.
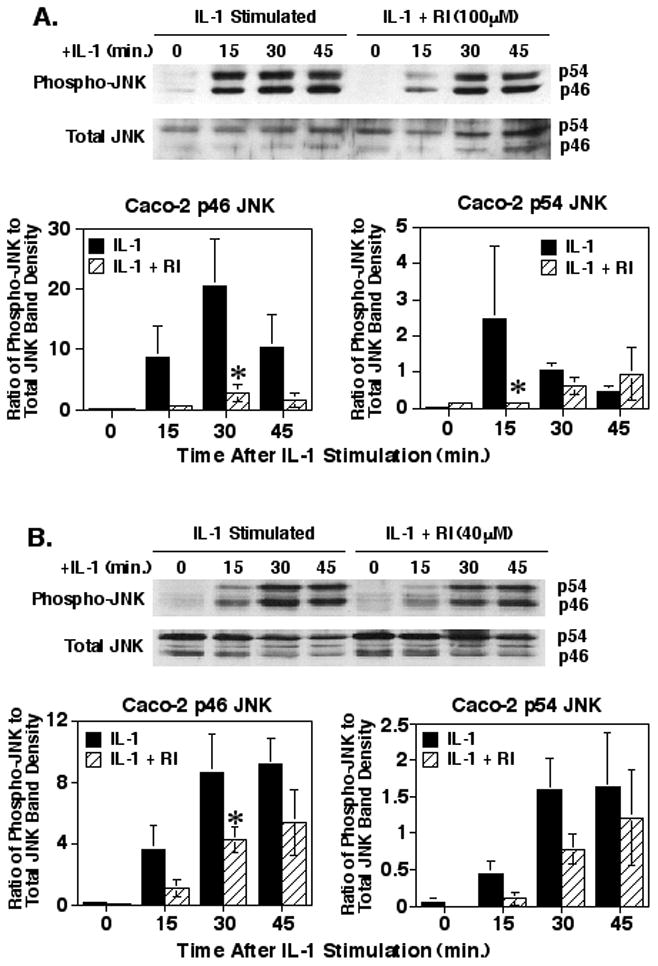
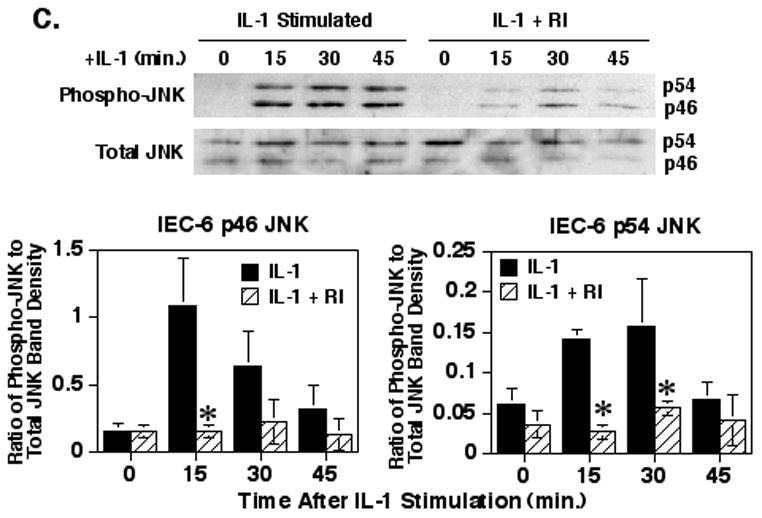
Inhibition of ROCK significantly decreases IL-1-stimulated JNK phosphorylation. Caco-2 cells were cultured and treated with 100μM (A) or 40μM (B) of the Y27632 ROCK inhibitor and IL-1 similar to as in Fig. 3. Shown are representative blots of Caco-2 cytoplasmic extracts probed for phospho-JNK and total JNK and a graph of the means ± SEM of the ratios of the Caco-2 phospho-JNK to total JNK band density values for the p46 and p54 isoform bands from four (A) or three (B) separate experiments. (C) IEC-6 cells were cultured and treated with the ROCK inhibitor and IL-1β to as in Fig. 3. Shown is a representative blot of IEC-6 cytoplasmic extracts probed for phospho-JNK and total JNK. The graph is the means ± SEM of the ratios of the IEC-6 phospho-JNK to total JNK band density values for the p46 and p54 isoform bands from three separate experiments. Asterisk indicates a significant difference compared to IL-1 treated samples at the corresponding time points (p<0.05).
4. Discussion
IL-1 is an important component of the inflammatory response at mucosal surfaces and understanding the mechanism by which IL-1 activates cells at the intestinal mucosa, especially the IEC, is essential to understanding the inflammation and devising potential therapies to combat this inflammation. The results of this study indicate that the Rho-associated kinase, ROCK, plays a necessary role in IL-1 responses in IEC. Inhibiting ROCK resulted in a suppression of IL-1 induced pro-inflammatory cytokine responses by IEC, and this effect was mediated through an effect of ROCK on IL-1 induced JNK activation. However, IKK/IκBα signaling was generally not affected. Therefore, ROCK, and an unknown upstream Rho small GTPase, must have a function that positively affects only the JNK signaling arm of IL-1 signaling. A role for ROCK in JNK signaling in angiotensin II induction of vascular smooth muscle cell migration [23] and epithelial tight junction remodeling [24] has been suggested, but the selective role of ROCK in IL-1 induced JNK signaling in IEC is novel. The impact of finding that ROCK may be involved in epithelial JNK, but not IKK/IκBα/NF-κB, signaling could then be expanded to include an effect on the many other IL-1 responsive genes, such as other cytokines, proteases, receptors, and signaling protein/transcription factor genes which are controlled by NF-κB and AP-1. As discussed below, inhibiting ROCK could provide a potential therapeutic avenue to selectively allow IL-1 induced NF-κB activation while suppressing AP-1 mediated responses, or possibly enhancing JNK responses by enhancing ROCK activity.
That inhibiting ROCK had little effect on the IKK/IκBα signaling arm of IL-1 responses was confirmed at the level of IKK phosphorylation and activation and with IκBα phosphorylation and degradation providing strong evidence for the lack of an effect. This was in contrast to the findings of Segain and co-workers [15] who found that inhibiting ROCK lead to a suppression of IL-1-stimulated NF-κB reporter activity, IκBα phosphorylation and degradation, and IKK activation in the THP-1 macrophage cell line. Inhibiting ROCK has also been shown to have no effect on IL-1 induced CXCL8 responses in cervical stromal cells [16]. These results strongly suggest that the effect of ROCK inhibition, and hence the role of ROCK in IL-1 signaling, may be very cell-type specific.
Of note, a complete inhibition of IL-1-stimulated CXCL8/CCL2 secretion or mRNA levels with the Y27632 ROCK inhibitor was not seen. The significant decrease was probably due to the inhibition of JNK signaling, but not IKK/IκBα signaling, the latter which could be responsible for the significant residual CXCL8 responses. CXCL8 and CCL2 gene transcription is known to be regulated by both NF-κB and AP-1, which can synergize to greatly enhance cytokine secretion [25, 26]. Inhibition of JNK induced AP-1 activity would reduce the IL-1 induced transcription of these cytokine genes, but would not completely inhibit the response.
Now that ROCK has been shown to have an important role in the JNK signaling arm of IL-1 responses, a direct link between these two signaling pathways must be investigated. ROCK may exert an essential effect on the upstream kinases of JNK, such as MKK4 or MKK7, or even the kinase further upstream, which for IL-1 signaling is presumably TAK1 [9]. A possible effect on TAK1 would be interesting as this kinase is the pivotal kinase in the branch to IKK/IκBα and JNK signaling arms of the IL-1 pathway [2]. Of note, the ROCK1 isomer is known to be essential for focal adhesion formation [27] and the IL-1 Receptor Type I has been shown to localize to focal adhesions [28], suggesting that ROCK1 could very well be involved in IL-1 signaling events. In addition, we have found that IL-1 stimulation can induce a significant activation of ROCK1 kinase activity (data not shown and experiments addressing this avenue of research are now underway) providing a second link of ROCK1 to IL-1 responses.
Several recent reports have investigated the importance of JNK signaling and NF-κB signaling in IBD and animal models. High levels of phosphorylated JNK were shown to be localized to both leukocytes and epithelial cells in the inflamed mucosa of patients with IBD, as compared to normal controls [10]. Furthermore, two recent reports have shown that treatment of dextran sodium sulfate-induced colitic rats or mice treated with the specific JNK inhibitor, SP600125, resulted in reduced inflammation and wasting [11, 29]. Also, JNK inhibition resulted in significantly reduced levels of epithelial apoptosis [29]. These studies strongly suggest a role for JNK in the pathology of IBD. However, genetic knock-out of JNK1 or JNK2 did not prevent the development of colitis in dextran sodium sulfate-treated mice, and in fact the JNK2 knockout exacerbated the colitis [30]. This suggests that targeting only one JNK isoform may not be an appropriate therapy for IBD, but instead broader spectrum treatments may be needed.
On the other hand, impairment of the gene for NEMO (IKKγ), a regulatory component of the IKK complex, and hence impaired signaling to NF-κB, in IEC of mice resulted in a rapid development of colitis and a compromised integrity of the epithelium [31]. Furthermore, genetic knockout or specific inhibition of IKKβ in dextran sodium sulfate-treated mice resulted in delayed healing of the colitis, suggesting a role for NF-κB in acute colitis [32]. Studies have also shown that NF-κB is expressed by IEC in wounded monolayers and NF-κB activation is essential for wound healing [33]. These studies emphasize the importance of epithelial cell NF-κB responses in inflammation and maintaining normal mucosal homeostasis [34, 35].
Therefore, treatments that may blunt JNK mediated signaling, while allowing NF-κB signaling, especially in IEC, may provide an important mechanism to treat acute inflammation at the level of the IEC. This becomes more significant when one considers the accessibility of oral treatments for targeting epithelial cells in the intestine and colon. To this end, treatment of trinitrobenzene sulfonic acid induced colitic rats with the Y27632 ROCK inhibitor has been shown to reduce inflammation in these rats [15], and although they suggested an effect of ROCK inhibition on peripheral blood mononuclear cells and macrophages, a protective effect could have occurred on IEC through inhibiting JNK mediated responses while allowing IKK/IκBα/NF-κB responses to occur. This, coupled with our findings that inhibiting ROCK suppresses IL-1 induced pro-inflammatory cytokine responses by inhibiting JNK, but not IKK/IκBα signaling, may provide a mechanism for the treatment of the epithelial specific effects of acute colitis.
Highlights.
Inhibiting ROCK suppressed IL-1-stimulated secretion and mRNA levels for Caco-2 cell CXCL8 and IEC-6 cell CCL2 responses.
Suppressing ROCK inhibited IL-1-induced JNK phosphorylation, but had no significant effect on IKK or IκBα responses.
Suggests a role for ROCK in IL-1-stimulated JNK signaling, but not IKK/IκBα signaling, in intestinal epithelial cells.
Acknowledgments
The authors would like to thank Sayantan Banerjee for his help with these studies. This work was supported by U.S. PHS Grant DK089459 and a grant from the Binghamton University Harpur College.
Abbreviations
- FN
fibronectin
- GAPD
glyceraldehyde-3-phosphate dehydrogenase
- IBD
inflammatory bowel disease
- IEC
intestinal epithelial cell
- IKK
IκB kinase
- IL
interleukin
- ITS
insulin, transferrin, selenium
- RI
Y27632 ROCK inhibitor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Vincenti MP, Brinkerhoff CE. Early response genes induced in chondrocytes stimulated with the inflammatory cytokine interleukin-1β. Arthritis Res. 2001;3:381–388. doi: 10.1186/ar331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weber A, Wasiliew P, Kracht M. Interleukin-1 (IL-1) pathway. Sci Signal. 2010;3:1–6. doi: 10.1126/scisignal.3105cm1. [DOI] [PubMed] [Google Scholar]
- 3.Jung HC, Eckmann L, Yang S-K, Panja A, Fierer J, Morzycka-Wroblewska E, Kagnoff MF. A distinct array of proinflammatory cytokines is expressed in human colon epithelial cells in response to bacterial invasion. J Clin Invest. 1995;95:55–65. doi: 10.1172/JCI117676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McGee DW, Beagley KW, Aicher WK, McGhee JR. Transforming growth factor-β and IL-1β act in synergy to enhance IL-6 secretion by the intestinal epithelial cell line, IEC-6. J Immunol. 1993;151:970–978. [PubMed] [Google Scholar]
- 5.Eckmann L, Jung HC, Schurer-Maly C, Panja A, Morzycka-Wroblewska E, Kagnoff MF. Differential cytokine expression by human intestinal epithelial cell lines: Regulation of interleukin-8. Gastroenterology. 1993;105:1689–1697. doi: 10.1016/0016-5085(93)91064-o. [DOI] [PubMed] [Google Scholar]
- 6.Reinecker H-C, Loh EY, Ringler DJ, Mehta A, Rombeau JL, MacDermott RP. Monocyte-chemoattractant protein 1 gene expression in intestinal epithelial cells and inflammatory bowel disease. Gastroenterology. 1995;108:40–50. doi: 10.1016/0016-5085(95)90006-3. [DOI] [PubMed] [Google Scholar]
- 7.Jobin C, Haskill S, Mayer L, Panja A, Sartor RB. Evidence for altered regulation of IκBα degredation in human colonic epithelial cells. J Immunol. 1997;158:226–234. [PubMed] [Google Scholar]
- 8.Jobin C, Sartor RB. The IκB/NF-κB system: A key determinant of mucosal inflammation and protection. Am J Physiol Cell Physiol. 2000;278:C451–C462. doi: 10.1152/ajpcell.2000.278.3.C451. [DOI] [PubMed] [Google Scholar]
- 9.Weston CR, Davis RJ. The JNK signal transduction pathway. Curr Op Cell Biol. 2007;19:142–149. doi: 10.1016/j.ceb.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 10.Mitsuyama K, Suzuki A, Tomiyasu N, Tsuruta O, Kitazaki S, Takeda T, Satoh Y, Bennett BL, Toyonaga A, Sata M. Pro-inflammatory signaling by Jun-N-terminal kinase in inflammatory bowel disease. Int J Mol Med. 2006;17:449–455. [PubMed] [Google Scholar]
- 11.Roy PK, Rashid F, Bragg J, Ibdah JA. Role of the JNK signal transduction pathway in inflammatory bowel disease. World J Gastroenterol. 2008;14:200–202. doi: 10.3748/wjg.14.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beaulieu J-F. Integrins and human intestinal cell functions. Frontiers Biosci. 1999;4:d310–d321. doi: 10.2741/beaulieu. [DOI] [PubMed] [Google Scholar]
- 13.Gilcrease MZ. Integrin signaling in epithelial cells. Cancer Lett. 2006;247:1–25. doi: 10.1016/j.canlet.2006.03.031. [DOI] [PubMed] [Google Scholar]
- 14.Nakagawa O, Fujisawa K, Ishizaki T, Saito Y, Nakao K, Narumiya S. ROCK-I and ROCK-II, two isoforms of Rho-associated coiled-coil forming protein serine/threonine kinase in mice. FEBS Lett. 1996;392:189–193. doi: 10.1016/0014-5793(96)00811-3. [DOI] [PubMed] [Google Scholar]
- 15.Segain JP, De La Bletiere DR, Sauzeau V, Bourreille A, Hilaret G, Cario-Toumaniantz C, Pacaud P, Galmiche JP, Loirand G. Rho kinase blockade prevents inflammation via nuclear factor κB inhibition: evidence in Crohn’s Disease and experimental colitis. Gastroenterology. 2003;124:1180–1187. doi: 10.1016/s0016-5085(03)00283-x. [DOI] [PubMed] [Google Scholar]
- 16.Shimizu S, Tahara M, Ogata S, Hashimoto K, Morishige K, Tasaka K, Murata Y. Involvement of nuclear factor-κB activation through RhoA/Rho-kinase pathway in LPS-induced CXCL8 production in human cervical stromal cells. Mol Hum Reprod. 2007;13:181–187. doi: 10.1093/molehr/gal113. [DOI] [PubMed] [Google Scholar]
- 17.John GR, Chen L, Rivieccio MA, Menendez-Vasquez CV, Hartley A, Brosnan CF. Interleukin-1β induces a reactive astroglial phenotype via deactivation of the Rho GTPase-Rock axis. J Neurosci. 2004;24:2837–2845. doi: 10.1523/JNEUROSCI.4789-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou H, Kramer RH. Integrin engagement differentially modulates epithelial cell motility by RhoA/ROCK and PAK1. J Biol Chem. 2005;280:10624–10635. doi: 10.1074/jbc.M411900200. [DOI] [PubMed] [Google Scholar]
- 19.Li G, Lubin FD, McGee DW. α3β1 Integrin induced suppression of the Caco-2 epithelial cell IL-1 signaling pathway leading to NF-κB activation. Cell Immunol. 2004;231:30–39. doi: 10.1016/j.cellimm.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 20.Ishizaki T, Uehata M, Tamechika I, Keel I, Nonomura K, Maekawa M, Narumiya S. Pharmacological properties of Y-27632, a specific inhibitor of Rho-associated kinases. Mol Pharmacol. 2000;57:976–983. [PubMed] [Google Scholar]
- 21.Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000;351:95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yoshii A, Iizuka K, Dobashi K, Horie T, Harada T, Nakazawa T, Mori M. Relaxation of contracted rabbit tracheal and human bronchial smooth muscle by Y-27632 through inhibition of Ca+ sensitization. Am J Respir Cell Mol Biol. 1999;20:1190–1200. doi: 10.1165/ajrcmb.20.6.3441. [DOI] [PubMed] [Google Scholar]
- 23.Ohtsu H, Mifune M, Frank GD, Saito S, Inagami T, Kim-Mitsuyama S, Takuwa Y, Sasaki T, Rothstein JD, Suzuki H, Nakashima H, Wollfolk EA, Motley ED, Eguchi S. Signal-crosstalk between Rho/ROCK and c-Jun NH2-terminal kinase mediates migration of vascular smooth muscle cells stimulated by angiotensin II. Arterioscler Thromb Vasc Biol. 2005;25:1831–1836. doi: 10.1161/01.ATV.0000175749.41799.9b. [DOI] [PubMed] [Google Scholar]
- 24.Naydenov NG, Hopkins AM, Ivanov AI. c-Jun N-terminal kinase mediates disassembly of apical junctions in model intestinal epithelia. Cell Cycle. 2009;8:2110–2121. doi: 10.4161/cc.8.13.8928. [DOI] [PubMed] [Google Scholar]
- 25.Roebuck KA. Regulation of interleukin 8 gene expression. J Interferon Cytokine Res. 1999;19:429–438. doi: 10.1089/107999099313866. [DOI] [PubMed] [Google Scholar]
- 26.Martin T, Cardarelli PM, Parry GCN, Felts KA, Cobb ER. Cytokine induction of monocyte chemochemoattractant protein-1 gene expression in human endothelial cells depends on the cooperative action of NF-kB and AP-1. Eur J Immunol. 1997;27:1091–1097. doi: 10.1002/eji.1830270508. [DOI] [PubMed] [Google Scholar]
- 27.Yoneda A, Multhaupt HAB, Couchman JR. The Rho kinases I and II regulate different aspects of myosin II activity. J Cell Biol. 2005;170:443–454. doi: 10.1083/jcb.200412043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arora PD, Ma J, Min W, Cruz T, McCulloch CA. Interleukin-1-induced calcium flux in human fibroblasts is mediated through focal adhesions. J Biol Chem. 1995;270:6042–6049. doi: 10.1074/jbc.270.11.6042. [DOI] [PubMed] [Google Scholar]
- 29.Assi K, Pillai R, Gomez-Munoz A, Owen D, Salh F. The specific JNK inhibitor SP600125 targets tumor necrosis factor-α production and epithelial cell apoptosis in acute murine colitis. Immunology. 2006;118:112–121. doi: 10.1111/j.1365-2567.2006.02349.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chromik AM, Müller AM, Körner JJ, Belyaev O, Holland-Letz T, Schmitz F, Herdegen T, Uhl W, Mittelkötter U. Genetic deletion of JNK1 and JNK2 aggravates the DSS-induced colitis in mice. J Invest Surg. 2007;20:23–33. doi: 10.1080/08941930601126140. [DOI] [PubMed] [Google Scholar]
- 31.Nenci A, Becker C, Wullaert A, Gareus R, van Loo G, Danese S, Huth M, Nikolaev A, Neufert C, Madison B, Gumucio D, Neurath MF, Pasparakis M. Epithelial NEMO links innate immunity to chronic intestinal inflammation. Nature. 2007;446:557–561. doi: 10.1038/nature05698. [DOI] [PubMed] [Google Scholar]
- 32.Eckmann L, Nebelsiek T, Fingerle AA, Dann SM, Mages J, Lang R, Rovine S, Kagnoff MF, Schmid RM, Karin M, Arkan MC, Greten FR. Opposing functions of IKKβ during acute and chronic intestinal inflammation. Proc Natl Acad Sci USA. 2008;105:15058–15063. doi: 10.1073/pnas.0808216105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Egan LJ, de Lecea A, Lehrman ED, Myhre GM, Eckmann L, Kagnoff MF. Nuclear factor-κB activation promotes restitution of wounded intestinal epithelial monolayers. Am J Physiol Cell Physiol. 2003;285:C1028–C1035. doi: 10.1152/ajpcell.00167.2003. [DOI] [PubMed] [Google Scholar]
- 34.Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007;448:427–434. doi: 10.1038/nature06005. [DOI] [PubMed] [Google Scholar]
- 35.Atreya I, Atreya R, Neurath MF. NF-κB in inflammatory bowel disease. J Int Med. 2008;263:591–596. doi: 10.1111/j.1365-2796.2008.01953.x. [DOI] [PubMed] [Google Scholar]