

Abstract
A method has been developed for screening glyceollins and their metabolites based upon precursor ion scanning. Under higher-energy collision conditions employing a triple quadrupole mass spectrometer in the negative ion mode, deprotonated glyceollin precursors yield a diagnostic radical product ion at m/z 148. We propose this resonance-stabilized radical anion, formed in violation of the even-electron rule, to be diagnostic of glyceollins and glyceollin metabolites. Liquid chromatography-electrospray ionization tandem mass spectrometry (LC-ESI-MS/MS) established that scanning for precursors of m/z 148 can identify glyceollins and their metabolites from plasma samples originating from rats dosed with glyceollins. Precursor peaks of interest were found at m/z 337, 353, 355, 417, and 433. The peak at m/z 337 corresponds to deprotonated glyceollins, whereas the others represent metabolites of glyceollins. Accurate mass measurement confirmed m/z 417 to be a sulfated metabolite of glyceollins. The peak at m/z 433 is also sulfated, but it contains an additional oxygen, as confirmed by accurate mass measurement. The latter metabolite differs from the former likely by the replacement of a hydrogen with a hydroxyl moiety. The peaks at m/z 353 and 355 are proposed to correspond to hydroxylated metabolites of glyceollins wherein the latter additionally undergoes a double bond reduction.
Introduction
Soybean produces isoflavones that are known to have beneficial effects on human health.1,2,3,4,5 In recent years, much research has been conducted on genistein, an isoflavone that has been proposed to have anticancer activity.6,7,8 The potential chemopreventive effect of genistein has prompted researchers to investigate soybean further for anticancer agents. A more recently investigated type of isoflavone are the glyceollins that are produced by soybeans under stressed conditions. The stressful conditions may include exposure to UV light, or fungal (e.g., Aspergillus) or bacterial pathogens.9 Because they are produced by a defense mechanism in response to pathogen invasion, glyceollins may be referred to as phytoalexins.10
Glyceollins exist in three isomeric forms, Glyceollin I, II and III (Scheme 1). The isomers are derived from a daidzein precursor through several intermediate steps.11 Among the biological effects of glyceollins are potential human health benefits including anti-fungal, anti-oxidant, anti-inflammatory, anti-diabetic, and cancer cell anti-proliferative activity along with other beneficial properties.12,13,14 Recently, many studies have proposed glyceollins as prevention or therapy candidates for breast, ovarian, and prostate cancers. All three glyceollin isomers have exhibited anti-estrogenic effects on estrogen receptor function and estrogen-dependent tumor growth. 15, 16 Specifically, glyceollins bind to the estrogen receptor and they inhibit estrogen-induced tumor progression of breast cancer (MCF-7) and ovarian cancer (BG-1) cells.16 Among the three isomers, glyceollin I has the most potent anti-estrogenic activity.17,18 Another in vivo study examining post-menopausal monkeys suggested glyceollins reduce breast cancer biomarker expression.19 Human prostate cancer cell research demonstrated that glyceollins have multiple effects on prostate cancer cells (LNCaP).20 The inhibition of prostate cancer cell growth by glyceollins is similar to that exhibited by genistein, but the former also up-regulate cyclin-dependent kinase inhibitor and down-regulate mRNA levels for androgen-responsive genes through androgen-mediated pathways.20 In addition to the anti-estrogenic activity, glyceollins normalize glucose homeostasis and improve glucose utilization in adipocytes.13,21 The anti-diabetic potential was also noted in prediabetic21 and Type 2 diabetic rats21 and mice..22 These potential benefits of glyceollins have been well documented, but its metabolism is not well understood.23
Scheme 1.
Structures of Glyceollin isomers
Several mass spectrometric approaches that use various ionization techniques such as electron ionization (EI), fast atom bombardment (FAB), thermospray (TSP), atmospheric pressure chemical ionization (APCI), and electrospray ionization (ESI) have been used to investigate flavonoids.24,25,26,27,28 Tandem mass spectrometry using low-energy collision-induced dissociation (CID) to obtain structural information regarding mass-selected precursors has been shown to be advantageous in characterizing flavonoids28 and was recently applied in the analysis of glyceollins from soy bean extracts.29 Given the propensity for extensive metabolism of flavonoids following their ingestion30 and that no research has addressed identification of glyceollins’ metabolites in animal systems, the significance of the present research is that it investigates glyceollins in plasma and presents method development work designed for screening of glyceollins and glyceollin-related metabolites.
Experimental
Extraction of glyceollin isomers
A mixture of glyceollins I, II, and III was obtained using a procedure developed at the Southern Regional Research Center (ARS, USDA, New Orleans, LA). Briefly, soybean seeds (1 kg) were sliced and inoculated with Aspergillus sojae. After 3 days, the glyceollins were extracted from the inoculated seeds with 1 L methanol (Aldrich Chemical Co., St. Louis, MO). The glyceollins were isolated using preparative scale HPLC using two Waters (Milford, MA) 25 × 100 mm, 10 μm particle size μBondapak C18 radial compression column segments that were combined using an extension tube. HPLC was performed on a Waters 600E System Controller combined with a Waters UV-Vis 996 detector scanning from 210-400 nm. Elution was carried out at a flow rate of 8.0 mL/min with the following solvent system: A = acetonitrile (Aldrich Chemical Co.), B = water (Millipore system, Billerica, MA) 5% A for 10 min, then 5% A to 90% A in 60 min followed by holding at 90% A for 20 min. The injection volume was 20 mL. The fraction containing the glyceollins was concentrated under vacuum and freeze-dried. Confirmation of individual glyceollins was based on matching of HPLC retention times and matching of UV-Vis absorbance spectra with those of authentic standards isolated at SRRC17. UV-Vis spectrophotometry at 285 nm allowed an estimation of mixture contents used in all experiments: glyceollin I (68%), glyceollin II (21%), and glyceollin III (11%).
Glyceollin dosing of rats and plasma sample collection
The administration of glyceollins to rats and subsequent sample collection have been previously described.21 Briefly, glyceollins were dissolved in poloxamer to administer 90 mg/kg via oral gavage (3 mL) to male ZDSD rats (PreClinOmics, Indianapolis, IN) that were housed in a suspended wire cage and maintained on a 12:12 hr light-dark cycle. Three hr after administration, rats (approx. 500 g wt.) were euthanized at various time points by decapitation and trunk blood was collected into EDTA-coated tubes supplemented with aprotinin. Plasma was separated and stored at −80 °C until analysis. Glyceollins were extracted for mass spectrometry analysis by thawing the samples and transferring 125 μL of plasma into a microcentrifuge tubes. An equivalent volume of acetonitrile was then added. The mixture was vortexed and centrifuged at 10,000 rpm for 20 min. 100 μL of supernatant was transferred into a clean microcentrifuge tube for analysis.
Liquid Chromatography-Mass Spectrometry
LC-MS and LC-MS/MS analyses were conducted on an Agilent 1200 series LC system (Agilent, Santa Clara, CA) coupled with a 3200 QTrap triple quadrupole mass spectrometer (Applied Biosystems/MDS SCIEX, Foster, City, CA). Separation was performed on an Agilent Eclipse - XDB C18 column (4.6 × 150 mm ID, 5 Fm). 10 μL was injected onto the column held at 25 °C. Mobile phase A was water with 0.1% formic acid whereas mobile phase B was acetonitrile with 0.1% formic acid. The gradient was 0-2 min 3% B, 2-7 min 3% to 60% B, 7-14 min 60% to 100% B, 14-20 min 100% B, 20-30 min 100% to 3% B. Flow rate was 0.500 mL/min. The UV absorbance detector was set at 210 and 282 nm.
All mass spectrometry experiments were performed in the negative ion mode. For LC-ESI-MS and LC-ESI-MS/MS analyses, electrospray parameters were set at: curtain gas 10 psi, ionspray voltage - 4000 V, GAS1 60 psi, GAS2 60 psi, source temperature 600 °C, CAD gas pressure 6 psi, entrance potential −10 V, collision cell exit potential −3 V. Declustering potential and collision energy were optimized to be −75 V and −34 eV, respectively. The information-dependent acquisition (IDA) method was employed to perform full scan, tandem MS and precursor ion scans sequentially (1 sec/scan).
Direct Infusion ESI-MS
Direct infusion parameters were the same as above except for GAS1, GAS2 and source temperature, which were set at 20 psi, 0 psi, and 50 °C, respectively. Samples were infused for 2 min at a flow rate of 4 μL/min. Direct infusion data was acquired using multiple-channel acquisition (MCA).
Accurate mass measurements by ESI-FT-ICR-MS
Accurate mass measurements to determine metabolite empirical formulas were performed on a Solarix 7T fourier transform ion cyclotron resonance (FT-ICR) mass spectrometer (Bruker Daltonics, Bremen, Germany). An ESI voltage of 4500 V was used with 2 Bar nebulizer gas pressure; drying gas was delivered at 4 L/min and 200 °C drying temperature. Peaks at m/z 113, 432, and 602 from ESI Tuning Mix (Agilent Technologies, Santa Clara, CA) were used as internal standards in mass spectra consisting of 40 averaged acquisitions.
Results and Discussion
Collision-Induced Dissociations of Glyceollins
Isomeric glyceollin I, II, and III standards and plasma samples obtained from rats dosed with glyceollins were analyzed and characterized by negative ion MS and tandem MS. [M-H]− peaks corresponding to deprotonated forms of glyceollin isomers were observed at m/z 337. Low-energy CID of these [M-H]− precursors (Figure 1) produced product ion peaks similar to those reported by Gruppen et al.29 For instance, m/z 319, 215, 187, 175, 161, 149 were all present in high abundances. In addition, low abundance peaks reported by Gruppen et al.29 (m/z 322, 309, and 293) were also observed. Surprisingly, under higher-energy conditions in the negative ion mode, a previously unreported CID peak emerged at m/z 148 with such a strong signal that it became the base peak in the product ion spectrum (Figure 1).31 We attribute the dominance of this open-shell fragment in the product ion mass spectrum of glyceollins to its exceptional stability as supported by the extensive delocalization of both the charge and the radical over the aromatic ring system (see inset, Figure 1). Indeed, fragmentation to form this product constitutes a violation of the even-electron rule,32 i.e., normally formation of a radical product ion plus a radical neutral is forbidden from decompositions of an even-electron precursor. However, homolytic cleavage of even-electron ions has been documented to occur especially in cases where exceptional stability is acquired by the formed radical ion and radical neutral.32,33,34,35 Because the m/z 148 product ion is formed from the portion of the molecule that is conserved in all of the glyceollin isomers, we propose to use the appearance of this unique radical ion in MS/MS spectra as a product ion diagnostic of all glyceollins.31 Thus, the presence of this product ion can be used as a signature to identify glyceollins and their related metabolites. As a single caveat, in cases where the D or B rings become modified, the m/z 148 ion could be shifted to another even m/z value with a possible decrease in intensity. Notably, this product ion was also observed during low-energy sustained off-resonance irradiation (SORI) - CID in a FT-ICR mass spectrometer. The empirical formula (C8H4O3) of the resonance stabilized m/z 148 ion was confirmed by accurate mass measurement.
Figure 1.

Negative ion electrospray product ion mass spectrum (−75 eV CID ) of [M-H]− precursor of glyceollin (m/z 337). Under these higher-energy conditions, the product ion at m/z 148 appears in high abundance.
Optimization of CID conditions
CID conditions for the triple quadrupole, including the “declustering potential” (V, also known as nozzle-skimmer potential) and the collision energy (eV, Elab) were serially adjusted to maximize the intensity of the diagnostic product ion at m/z 148 formed from glyceollin [M-H]− precursors. Other MS conditions such as ion spray voltage, curtain, nebulizing and turbo gases were maintained at constant values (see Experimental section). The declustering potential value for precursor m/z 337 was optimized (by ramping up its voltage) to obtain the highest intensity of m/z 148 in product ion mode. The optimum declustering potential yielding the maximum intensity of m/z 148 was −75 V. Tandem MS of m/z 337 was then performed using a fixed declustering potential of −75 V, while ramping up the collision energy. The highest signal for m/z 148 was observed at an Elab value of −34 eV. Figure 2 shows the automated infusion “quantitative optimization” of the declustering potential (Figure 2a, m/z 148 only) and Elab (Figure 2b, 3 highest intensity fragments: m/z 148, m/z 149 and m/z 319). The breakdown curves shown in Figure 2b clearly indicate that upon decomposition of m/z 337, m/z 149 has a lower appearance energy than m/z 148. This finding can be rationalized by considering that for these closely related fragments, production of even-electron m/z 149 (with formation of an additional carbon-hydrogen bond and resulting stabilization) requires less energy than formation of odd-electron m/z 148. However, as the internal energy uptake is increased, the rate constant of m/z 148 formation becomes more favorable and the curves for formation of the respective products cross (Figure 2b inset). This curve-crossing is a classic example36,37 of competitive ion formation where the fragment with the lower appearance threshold dominates under lower energy collisions (e.g., conditions used by Gruppen et al.29, m/z 149 is favored), whereas the fragment formed by the higher frequency factor32 process dominates at higher collision energies (m/z 148, see Figure 1).
Figure 2.

(a) The optimum declustering potential value to produce m/z 148 from CID of m/z 337 precursors was determined by ramping the declustering potential in product ion mode. The maximum intensity of m/z 148 was observed at −75 V. (b) Optimization of collision energy for m/z product ion formation at fixed declustering potential of −75 V. Abundances of three strong peaks: m/z 148, 149, 319 are shown as a function of collision energy. The highest signal for m/z 148 was observed at −34 eV (Elab). The inset schematically shows the rates of formation of m/z 148 and 149 as a function of internal energy of m/z 337.
Precursor Ion Scan of m/z 148
Upon optimization of CID conditions, m/z 148 was observed as the base peak in the product ion mass spectra of [M-H]− precursors from each purified standard of glyceollin I, II, and III. To follow through on the idea of using m/z 148 as a diagnostic product ion for glyceollins, the above optimized CID conditions maximizing m/z 148 production were used to obtain precursor ion scans of m/z 148 using glyceollin mixtures. As expected, [M-H]− at m/z 337 was detected in high intensity, thus confirming that precursor ion scans of the diagnostic radical m/z 148 can be used to detect glyceollins and glyceollin related compounds. In principle, one could also consider using m/z 149 for precursor ion scans, but in practice, this would be a poor choice because a wide variety of phtahlate esters (ubiquitous plasticizers) produce m/z 149 upon ESI-MS/MS.38
LC-ESI-MS and LC-ESI-MS/MS analyses of glyceollins and their metabolites in rat plasma
To test the validity of this method to identify glyceollins and their metabolites, it was applied to the analysis of plasma samples derived from rats dosed with glyceollins. For these LC-ESI-MS and LC-ESI-MS/MS experiments, the MS data acquisition method cycled through a full MS scan, a CID precursor ion scan of m/z 148, and a CID product ion spectrum of m/z 337. Once the precursors of m/z 148 were identified, in subsequent runs, the CID product ion scans of those precursors were added to the acquisition scan cycle. In a plasma sample taken at 20 min following a 90 mg/kg dose of glyceollins, the precursor ion scan of m/z 148 showed eluting peaks at 10.5 and 12.5 min (Figure 3a). The peak at 12.5 min corresponds to (unmetabolized) deprotonated glyceollins (m/z 337) as shown in the averaged mass spectrum corresponding to this chromatographic peak (Figure 3 inset). The earlier eluting peak (10.5 min) corresponds to an apparent metabolite of glyceollins which gave a base peak at m/z 417 (Figure 3 inset). Notably, the employed CID conditions produced enough internal energy uptake to cause consecutive decomposition of this metabolite to produce m/z 337 (deprotonated glyceollins). In a different rat plasma sample taken at 4 hrs following the dose of glyceollins (90 mg/kg), precursor ion scanning of m/z 148 resulted in the appearance of glyceollin metabolites eluting at 3.9, 4.2 and 9.6 min (Figure 4). These chromatographic peaks correspond to metabolites of m/z 451, 433, and 417, respectively (see inset Figure 4). Again, consecutive decomposition of the m/z 417 metabolite was observed to produce m/z 337. It appears that metabolism was extensive in this case because unmodified glyceollins were not detected in this sample.
Figure 3.

LC-MS/MS precursor ion scans showing total ion current of all precursors of m/z 148 from glyceollins dosed rats. The insets show the precursor ion spectra of m/z 148 averaged over the width of the peaks eluting at 10.5 and 12.5 min showing the presence of a metabolite and unmetabolized glyceollins, respectively.
Figure 4.

LC-MS/MS precursor ion scan showing total ion current of all precursors of m/z 148 from glyceollins dosed rat plasma. Inset are the averaged mass spectra obtained across the three observed chromatographic peaks.
Identification of glyceollins’ metabolites using LC-ESI-MS and LC-ESI-MS/MS
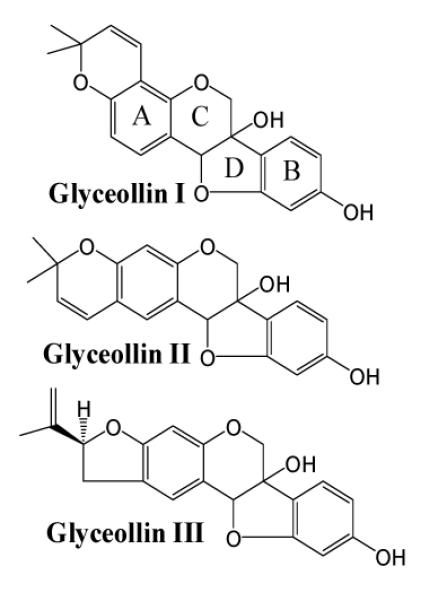
The CID product ion spectrum of the m/z 417 precursor (Figure 5) shows many of the same fragments as observed in the CID product ion spectrum of m/z 337 (Figure 1). In considering the mass increase of this metabolite, in general, an addition of 80 Da in a biological medium may correspond to either a sulfation or a phosphorylation process. The metabolism of glyceollins in the digestive tract is not well documented23 and to our knowledge there have not been any reports of glyceollins’ metabolites. However, another isoflavone, genistein, has been extensively studied, and it has been reported to undergo phase II metabolism by glucuronidation, sulfation, and methylation in small intestine and liver.30,39,40 Based on these reports of genistein metabolism, and interpretation of the obtained CID spectrum of m/z 417 (Figure 5), the peak was tentatively assigned as the sulfated metabolite of glyceollins. However, because m/z 417 could correspond to either the sulfated or the phosphorylated metabolite (deprotonated forms), to definitively identify m/z 417, accurate mass analysis was performed on this ion. The addition of a phosphate group implies a molecular formula of C20H18O8P with an exact m/z of 417.074478, whereas sulfated glyceollins have the molecular formula C20H17O8S with an exact m/z of 417.064962. The accurate mass measurement by FT-ICR gave m/z 417.064812 confirming the assignment of m/z 417 as a sulfated metabolite of glyceollins (0.360 ppm error). Because the CID product ion spectrum of m/z 417 (Figure 5) shows similar fragments as the CID product ion spectrum of m/z 337 (Figure 1), and because there are no fragment ions that are shifted by 80 m/z units in the former spectrum relative to the latter, we conclude that the sulfate group is the most labile moiety of the m/z 417 metabolite. Thus, the first step in decompostion of m/z 417 is loss of SO3 neutral to form deprotonated glyceollins at m/z 337. All of the lower m/z fragments in Figure 5 are proposed to be formed by consecutive decompositions of m/z 337. Notably, at the constant Elab collision energy (− 34 eV) employed throughout this paper, energy is consumed in the decomposition of m/z 417 leading to m/z 337. The m/z 337 ions thereby formed have less internal energy available to induce consecutive decompositions as compared to m/z 337 precursors that are subjected directly to −34 eV collisions in the central quadrupole. This loss of internal energy in the first step of decomposition results in more favorable kinetics for m/z 149 formation as compared to m/z 148 (see Figure 2b inset) in consecutive decompositions.
Figure 5.
LC-MS/MS product ion mass spectrum of m/z 417 precursors from rats dosed with glyceollins. The m/z 417 metabolite was assigned as a sulfated form of glyceollins.
The hydroxyl sites of glyceollins are preferred sites of sulfation. We assign sulfation to the hydroxyl site of the phenol group (see inset, Figure 5) based upon the observed facile loss of SO3 neutral which, in this case, leaves a resonance stabilized phenoxy anion (corresponding to deprotonated glyceollins at m/z 337). The loss of SO3 neutral from glyceollins sulfated at the alkyl hydroxide would be expected to be less favorable. As glyceollins are known to exhibit competitive behavior with estrogens, this assignment is supported by the documented preferential sulfation of the 3-phenolic hydroxyl relative to the 16-aliphatic hydroxyl in estriol.41
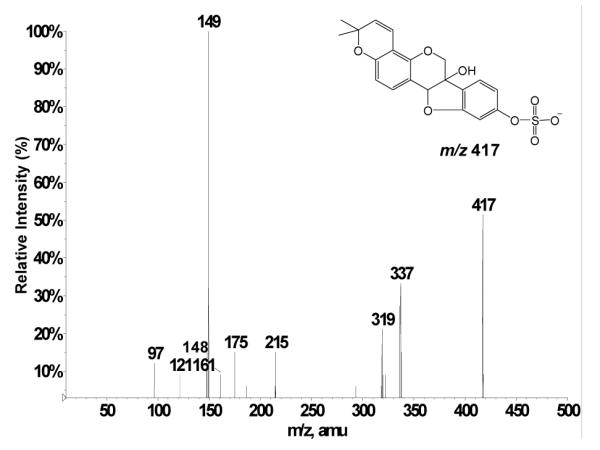
Another metabolite appearing in Figure 4 (retention time 4.2 min corresponding to m/z 433) was investigated further. The composition of m/z 433 was established to be C20H17O9S by accurate mass measurement (m/z 433.059649, 0.524 ppm error). This peak at m/z 433 is proposed to correspond to a (deprotonated) sulfated metabolite of glyceollins that contains one additional oxygen relative to m/z 417 discussed above. The LC-ESI-MS/MS low-energy CID product ion spectrum of the m/z 433 precursor (Figure 6) reveals the B fragment peaks (m/z 148, 149) to be the same as those in the product ion spectrum of deprotonated glyceollins (m/z 337, Figure 1) whereas A fragments are shifted by 16 m/z units (i.e., m/z 175 and 227 are shifted to 191 and 243, respectively), plus m/z 337 is shifted to 353. This different behavior of A and B fragments allows us to narrow down the potential sites of hydrogen replacement with a hydroxyl group that accounts for the 16 Da shift in the mass of the metabolized molecule. All possible hydroxylation sites are marked with asterisks on the structure of m/z 433 shown in Figure 6.
Figure 6.
Negative ion low-energy CID product ion mass spectrum of m/z 433 from rats dosed with glyceollins. Potential sites of replacement of a hydrogen with a hydroxyl group are marked with asterisks. The labeling of the ring system has been adopted from Gruppen et al.29
Screening of glyceollins’ metabolites by direct infusion
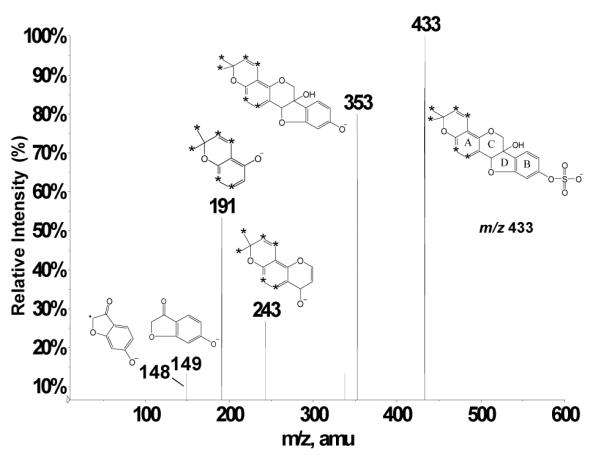
LC separation prior to ESI-MS or ESI-MS/MS analyses offers the theoretical advantage that each analyte species may enter the ESI source of the mass spectrometer free from interferences. Even so, the sequential isolation and analysis steps of tandem mass spectrometry can enable direct mixture analyses in the absence of chromatographic separation. When very complex mixtures are under investigation, however, analyte desorption behavior may be affected by competing sample species. It is well documented that in ESI-MS, surface-active species tend to be desorbed most efficiently, whereas the less surface-active molecules tend to experience signal suppression.42 To compare the performance of our precursor ion scanning approach in the presence and absence of chromatographic separation, the same rat plasma mixture as used above was employed, except that this time precursor ion scans of m/z 148 were performed directly on the complex mixture with no prior LC separation. Direct infusion of the acetonitrile extracts of a rat plasma mixture resulted in the detection of m/z 148 precursor ions at m/z 337, 353, 355, 417, 433, 451, and 469 (Figure 7). The peak at m/z 337 once again represents unmetabolized deprotonated glyceollins. The peaks at m/z 417 and 433 were previously assigned as the sulfated metabolite of glyceollins, and the sulfated metabolite of glyceollins with one additional oxygen, respectively. It is fair to say that neither the m/z 417 nor the m/z 433 signal was suppressed during ESI-MS/MS direct mixture analysis. This may be rationalized by considering that the sulfated conjugates of glyceollins have substantial surfactant character, which can explain the favorable signal response relative to other components of the complex mixture.
Figure 7.
Direct infusion negative ion electrospray −75 V CID precursor ion scan of m/z 148 from rat plasma derived from rats.
Notably, peaks at m/z 353, 355, and 469 were detected in direct infusion mixture analysis that were absent in LC-ESI-MS/MS. The precursor peaks at m/z 353 and 355 are most likely hydroxylated metabolites of glyceollins, wherein the latter contains an additional reduced double bond. The improved detectability in direct infusion ESI-MS/MS may be rationalized if one considers the additional acquisition time afforded by direct infusion that serves to improve signal-to-noise (S/N) ratios. Moreover, in LC-MS/MS experiments, the (S/N) ratios may not be maximized if spectra are averaged across the entire width of the chromatographic peak (due to weaker signals away from the center of the peak).43 A literature example reports a 20-fold gain in sensitivity observed with direct infusion of LC fractions using multichannel acquisition, as compared to LC/MS.44 Therefore, we suspect that the conditions employed for LC-ESI-MS/MS resulted in a detection limit just above the threshold for observation of these m/z 353, 355, and 469 signals.
Conclusion
A precursor ion scan method based on m/z 148 product ion formation was developed and optimized to screen for glyceollins and glyceollin related compounds, including metabolites. LC-ESI-MS/MS analyses with both precursor ion and product ion scans were carried out on a triple quadrupole to identify glyceollins and their metabolites in rat plasma. Precursor ion scanning of m/z 148 allowed the characterization of glyceollins’ metabolites at m/z 417, 433, 353, and 355. After inspection and interpretation of obtained tandem mass spectra, accurate mass measurements of m/z 417 and 433 were performed, thus, confirming the in vivo sulfation, and sulfation plus oxygen addition, respectively, of glyceollins in rats. In addition, metabolites at m/z 353 and 355 were observed and were proposed to represent hydroxylated forms of glyceollins wherein the latter has one less double bond. Along with developing a method to screen for glyceollins and its related compounds, this study is the first to establish hydroxylation and sulfation metabolic pathways of glyceollins in dosed animals.
Acknowledgments
We thank Dr. Mark L. Heiman of NuMe Health for his expertise and input in designing the rat study, including identification of the vehicle. We thank Mr. Xiaohua Liu for performing accurate mass analyses by FT-ICR-MS. Financial support for this research was provided by the National Science Foundation through CHE-1058764. Additional financial support was provided by the MetaboHub project, by the Louisiana Cancer Research Consortium, and by NIH-RCMI grant #5G12RR026260 from the National Institute on Minority Health and Health Disparities. The contents are solely the responsibility of the authors and do not necessarily represent the official views of the Louisiana Cancer Research Consortium or the NIH.
References
- 1.Radhakrishnan G, Rashmi, Agarwal N, Vaid NB. Internet Journal of Gynecology and Obstetrics. 2009;11(1):12. [Google Scholar]
- 2.Oergaard A, Jensen L. Experimental Biology and Medicine. 2008;233(9):1066–1080. doi: 10.3181/0712-MR-347. [DOI] [PubMed] [Google Scholar]
- 3.Miyanaga N, Akaza H, Hinotsu S, Fujioka T, Naito S, Namiki M, Takahashi S, Hirao Y, Horie S, Tsukamoto T, Mori M, Tsuji H. Cancer Science. 2012;103(1):125–130. doi: 10.1111/j.1349-7006.2011.02120.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kingsley K, Truong K, Low E, Hill CK, Chokshi SB, Phipps D, West A, Keiserman MA, Bergman CJ. Journal of Dietary Supplements. 2011;8(2):169–188. doi: 10.3109/19390211.2011.571656. [DOI] [PubMed] [Google Scholar]
- 5.Sahin K, Tuzcu M, Sahin N, Akdemir F, Ozercan I, Bayraktar S, Kucuk O. Nutrition and Cancer. 2011;63(8):1279–1286. doi: 10.1080/01635581.2011.606955. [DOI] [PubMed] [Google Scholar]
- 6.Merchant K, Kumi-Diaka J, Rathinavelu A, Esiobu N, Zoeller R, Hartmann J, Johnson M. Functional Foods in Health and Disease. 2011;1(3):91–105. [Google Scholar]
- 7.Majid S, Kikuno N, Nelles J, Noonan E, Tanaka Y, Kawamoto K, Hirata H, Li LC, Zhao H, Okino ST, Place RF, Pookot D, Dahiya R. Cancer Research. 2008;68(8):2736–2744. doi: 10.1158/0008-5472.CAN-07-2290. [DOI] [PubMed] [Google Scholar]
- 8.Barnes S, Sfakianos J, Coward L, Kirk M. Advances in Experimental Medicine and Biology. Vol. 401. Dietary Phytochemicals in Cancer Prevention and Treatment; 1996. pp. 87–100. [DOI] [PubMed] [Google Scholar]
- 9.Boue SM, Carter CH, Ehrlich KC, Cleveland TE. Journal of Agricultural and Food Chemistry. 2000;48(6):2167–2172. doi: 10.1021/jf9912809. [DOI] [PubMed] [Google Scholar]
- 10.Darvill AG, Albersheim P. Annual Review of Plant Physiology. 1984;35:243–75. doi: 10.1104/pp.75.2.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tilghman SL, Boue SM, Burow ME. Molecular and Cellular Pharmacology. 2010;2(4):155–160. [Google Scholar]
- 12.Kim HJ, Suh H, Kim J, Park S, Joo Y, Kim J. Journal of Agricultural and Food Chemistry. 2010;58(22):11633–11638. doi: 10.1021/jf102829z. [DOI] [PubMed] [Google Scholar]
- 13.Park S, Ahn IS, Kim JH, Lee MR, Kim JS, Kim HJ. Journal of Agricultural and Food Chemistry. 2010;58(3):1551–1557. doi: 10.1021/jf903432b. [DOI] [PubMed] [Google Scholar]
- 14.Rhodes LV, Tilghman SL, Boue SM, Wang S, Khalili H, Muir SE, Bratton MR, Zhang Q, Wang G, Burow ME, Collins-Burrow, Bridgette M. Oncology Letters. 2012;3(1):163–171. doi: 10.3892/ol.2011.460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Burow ME, Boue SM, Collins-Burow BM, Melnik LI, Duong BN, Carter-Wientjes CH, Li, Shuanfang W, Thomas E, Cleveland TE, McLachlan JA. Journal of Clinical Endocrinology and Metabolism. 2001;86(4):1750–1758. doi: 10.1210/jcem.86.4.7430. [DOI] [PubMed] [Google Scholar]
- 16.Salvo VA, Boue SM, Fonseca JP, Elliott S, Corbitt C, Collins-Burow BM, Curiel TJ, Srivastav SK, Shih BY, Carter-Wientjes C, Wood CE, Erhardt PW, Beckman BS, McLachlan JA, Cleveland TE, Burow ME. Clinical Cancer Research. 2006;12(23):7159–7164. doi: 10.1158/1078-0432.CCR-06-1426. [DOI] [PubMed] [Google Scholar]
- 17.Zimmermann C, Tilghman SL, Boue SM, Salvo VA, Elliott S, Williams KY, Skripnikova EV, Ashe H, Payton-Stewart F, Vanhoy-Rhodes L, Fonseca JP, Corbitt C, Collins-Burow BM, Howell MH, Lacey M, Shih BY, Carter-Wientjes C, Cleveland TE, McLachlan JA, Wiese TE, Beckman BS, Burow ME. Journal of pharmacology and experimental therapeutics. 2010;332(1):35–45. doi: 10.1124/jpet.109.160382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Payton-Stewart F, Khupse RS, Boue SM, Elliott S, Zimmermann MC, Skripnikova EV, Ashe H, Tilghman SL, Beckman BS, Cleveland TE, McLachlan JA, Bhatnagar D, Wiese TE, Erhard P, Burow ME. Steroids. 2010;75(12):870–878. doi: 10.1016/j.steroids.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 19.Wood CE, Clarkson TB, Appt SE, Franke AA, Boue SM, Burow ME, McCoy Thomas, Cline J. Mark. Nutrition and Cancer. 2006;56(1):74–81. doi: 10.1207/s15327914nc5601_10. [DOI] [PubMed] [Google Scholar]
- 20.Payton-Stewart F, Schoene NW, Kim YS, Burow ME, Cleveland TE, Boue SM, Wang TY. Molecular Carcinogenesis. 2009;48(9):862–871. doi: 10.1002/mc.20532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boue SM, Isakova IA, Burow ME, Cao H, Bhatnagar D, Sarver JG, Shinde KV, Erhardt PW, Heiman ML. Journal of Agricultural and Food Chemistry. 2012;60(25):6376–6382. doi: 10.1021/jf301057d. [DOI] [PubMed] [Google Scholar]
- 22.Park S, Kim DS, Kim JH, Kim JS, Kim HJ. Nutrition. 2012;28(2):204–211. doi: 10.1016/j.nut.2011.05.016. [DOI] [PubMed] [Google Scholar]
- 23.Kim HJ, Lim J, Kim W, Kim J. Proceedings of the Nutrition Society. 2012;71(1):166–174. doi: 10.1017/S0029665111003272. [DOI] [PubMed] [Google Scholar]
- 24.Hedin PA, Phillips VA. Journal of Agricultural and Food Chemistry. 1992;40(4):607–11. [Google Scholar]
- 25.Crow FW, Tomer KB, Looker JH, Gross ML. Analytical Biochemistry. 1986;155(2):286–307. doi: 10.1016/0003-2697(86)90438-0. [DOI] [PubMed] [Google Scholar]
- 26.Kiehne A, Engelhardt UH. Zeitschrift fur Lebensmittel-Untersuchung und – Forschung. 1996;202(1):48–54. doi: 10.1007/BF01229684. [DOI] [PubMed] [Google Scholar]
- 27.Jia-Ping L, Heng L, Jin S, Han-Ming S, Nam O. Journal of chromatography B. 2007;848(2):215–25. [Google Scholar]
- 28.Ma YL, Heuvel HV, Claeys M. Rapid Communications in Mass Spectrometry. 1999;13(19):1932–1942. doi: 10.1002/(SICI)1097-0231(19991015)13:19<1932::AID-RCM735>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 29.Simons R, Vincken J, Bohin MC, Kuijpers TFM, Verbruggen MA, Gruppen H. Rapid Communications in Mass Spectrometry. 2011;25(1):55–65. doi: 10.1002/rcm.4826. [DOI] [PubMed] [Google Scholar]
- 30.Liu Y, Hu M. Drug Metabolism and Disposition. 2002;30(4):370–377. doi: 10.1124/dmd.30.4.370. [DOI] [PubMed] [Google Scholar]
- 31.Quadri SS, Stratford RE, Boué SM, Cole RB. A New Mass Spectrometric Approach for Screening of Glyceollins and Their Metabolites. Proceedings of the 60th ASMS Conference on Mass Spectrometry and Allied Topics; Vancouver, BC. May 22, 2012. [Google Scholar]
- 32.Karni M, Mandelbaum A. Journal of Mass Spectrometry. 1980;15(2):53–64. [Google Scholar]
- 33.Bowen RD, Clifford P, Gallagher RT. European Mass Spectrometry. 1996;2(4/5):233–246. [Google Scholar]
- 34.Chen K, Rannulu NS, Cai Y, Lane P, Liebl AL, Rees BB, Corre C, Challis GL, Cole RB. Journal of the American Society for Mass Spectrometry. 2008;19(12):1856–1866. doi: 10.1016/j.jasms.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 35.Cai Y, Mo Z, Rannulu NS, Guan B, Kannupal S, Gibb BC, Cole RB. Journal of Mass Spectrometry. 2010;45(3):235–240. doi: 10.1002/jms.1706. [DOI] [PubMed] [Google Scholar]
- 36.Wahrhaftig AL. In: Gaseous ion Chemistry and MS. Futrell JH, editor. Wiley; New York: 1986. pp. 7–24. [Google Scholar]
- 37.McLafferty FW, Turecĕk F. Interpretation of Mass Spectra. 4th ed University Science Books; California: 1993. pp. 115–134. [Google Scholar]
- 38.Li Z, Xue F, Xu L, Peng C, Kuang H, Ding T, Xu C, Sheng C, Gong Y, Wang L. Journal of Chromatographic Science. 2011;49(4):338–343. doi: 10.1093/chrsci/49.4.337. [DOI] [PubMed] [Google Scholar]
- 39.Bolling BW, Court MH, Blumberg JB, Chen C.Y. Oliver. Journal of Nutritional Biochemistry. 2010;21(6):498–503. doi: 10.1016/j.jnutbio.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhu W, Xu H, Wang S, Hu M. AAPS Journal. 2010;12(4):525–536. doi: 10.1208/s12248-010-9209-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Levitz M, Kadner S, Young BK. Steroids. 1976;27(2):287–294. doi: 10.1016/0039-128x(76)90105-7. [DOI] [PubMed] [Google Scholar]
- 42.Cech N, Enke C. In: Electrospray and MALDI Mass Spectrometry. Cole Richard B., editor. Wiley; New Jersey: 2010. pp. 49–74. [Google Scholar]
- 43.Zhang Z, McElvain JS. Analytical Chemistry. 1999;71(1):39–45. doi: 10.1021/ac980724h. [DOI] [PubMed] [Google Scholar]
- 44.Hopfgartner G, Varesio E, Tschaeppaet V, Grivet C, Bourgogne E, Leuthold LA. Journal of Mass Spectrometry. 2004;39(8):845–855. doi: 10.1002/jms.659. [DOI] [PubMed] [Google Scholar]