

Abstract
Myotonic dystrophy type 1 (DM1) is an RNA dominant disease caused by expression of DM protein kinase (DMPK) transcripts that contain an expanded CUG repeat (CUGexp). The toxic mRNA localizes to nuclear foci and sequesters proteins involved in the regulation of alternative splicing, such as, muscleblind-like 1 (MBNL1). Here, we used synthetic short interfering RNAs (siRNAs) to target CUG repeats and test the concept that inhibiting the expression of CUGexp RNA can mitigate features of DM1 in transgenic mice. Intramuscular injection and electroporation of siRNA resulted in ~70–80% downregulation of CUGexp transcripts. A limited survey of endogenous mouse transcripts that contain nonexpanded CUG or CAG repeats showed that most were not affected, though Txlnb containing (CUG)9 was significantly reduced. By this strategy, the number and intensity of CUGexp nuclear foci were reduced and splicing of MBNL1-dependent exons was improved. These data suggest that the expanded CUG repeats are a potential target for allele-selective RNA interference.
Introduction
More than 20 human inherited diseases are caused by unstable expansions of simple repeat sequences.1 In several cases, the expansions are located in genomic regions that are transcribed but not translated, giving rise to RNA dominance, a process in which transcripts from the mutant allele have a direct deleterious effect on cell function.2,3 Although mechanisms for RNA dominance are not fully understood, one predominant effect is that toxic RNA can alter the processing and post-transcriptional regulation of transcripts from other genes.
Myotonic dystrophy type 1 (DM1) is an RNA dominant disorder caused by expansion of a CTG repeat in the 3′ untranslated region (UTR) of the DM protein kinase (DMPK) gene.4 Clinical features of DM1 are extremely variable but most commonly it presents in young adults with myotonia, weakness, and atrophy of skeletal muscle.5 Symptoms of smooth muscle, cardiac, and central nervous system involvement may also occur. Individuals with classical DM1 typically inherit alleles with hundreds of CTG repeats. However, the mutation is quite unstable in somatic cells and it continues to expand during postnatal life, reaching lengths of several thousand repeats in muscle and cardiac tissue.6 The DMPK transcripts containing these highly expanded CUG repeats (CUGexp) are retained in the nucleus in discrete foci.7 Their nuclear retention is partly a function of the interaction of CUGexp RNA with poly(CUG) binding proteins, such as, splicing factors in the muscleblind-like (MBNL) family.8,9 The pathogenic effects of CUGexp RNA are due in part to sequestration of MBNL proteins, which results in misregulated alternative splicing of transcripts that these proteins normally regulate.10 Another pathogenic effect of the mutant DMPK mRNA is to induce post-translational upregulation of another RNA-binding protein, CUG-BP1.11
Considering that the pathogenic gene product is a repeat-containing transcript, and that alterations of the transcriptome are its main effect,12 efforts to develop DM1 therapeutics have focused on drugs targeting RNA.13,14 In transgenic mice or cells, therapeutic oligonucleotides have been used to mitigate RNA dominance at several different levels. Morpholino oligomers that induce exon skipping have been used to correct the splicing abnormalities for individual genes.15 Morpholino oligomers composed of CAG repeats were able to hybridize to CUGexp RNA and block its deleterious protein interactions, thereby, improving a range of splicing and gene-expression abnormalities.16 CAG repeat morpholinos also had the unexpected effect of reducing the steady state level of CUGexp RNA. 2′-O-methyl antisense oligonucleotides composed of CAG repeats as well as lentiviral-overexpressed CAG repeat RNA that is fused with the U7 small nuclear RNA caused a marked reduction of mutant DMPK transcripts in cells. Their mechanisms of action have not yet been determined, and in both cases the effect was specific for transcripts from the mutant DMPK allele.13,17 In DM1 muscle cells, transfected siRNAs targeting the DMPK coding region caused reduction of wild-type DMPK transcripts in the cytoplasm.18 However, CUG-expanded DMPK transcripts in the nucleus were not reduced unless the siRNA was complexed with an amphipathic peptide to enhance cell uptake and transport to the nucleus. These results suggested that sensitivity of nuclear-retained transcripts to exogenous siRNA was low. By contrast, lentivirus-mediated expression of short hairpin RNAs targeting the DMPK coding region was able to reduce wild-type and mutant DMPK transcripts with similar efficiency.18
Here, we examined the effects of siRNAs in a transgenic mouse model of DM1. Human skeletal actin—long repeat (HSALR) mice show muscle-specific expression of an α (skeletal) actin transcript that contains ~220 CUG repeats in the 3′ UTR.19 The CUGexp transcripts are retained in the nucleus, they sequester MBNL1 protein, and they induce splicing changes and repetitive action potentials (myotonic discharges) similar to patients with DM1.10,20 As in human DM1, the myotonia in these mice results from misregulated alternative splicing of Clcn1, a transcript encoding the muscle-specific chloride ion channel, ClC-1.15 Because previous studies indicated that amphipathic (potentially immunogenic) peptides were required for activity of siRNAs targeting the DMPK coding region,18 we instead targeted the CUG-repeat tract with siRNA.
Results
siRNAs against CUG repeats downregulate CUGexp transcripts in vivo
The design of the (CAG)·(CUG) siRNA duplex targeting the CUGexp tract, designated here as “siCAG”, is shown in Figure 1a. Instead of using a pure (CAG)·(CUG) repeat, the sequence was altered in an effort to drive preferential loading of the CAG rather than CUG-repeat strand onto the RNA-induced silencing complex. As most microRNAs have U at the 5′ end of the guide strand, a U for C substitution was made at the 5′ end of the CAG-repeat strand. Opposite to this substitution, on the CUG-repeat strand, we made a C for G substitution, creating a U·C mismatch to reduce duplex stability. The CUG-repeat strand has either a G or C at the 5′ end, either of which is unfavorable for the active strand of siRNA. Nearest neighbor base-stacking parameters21 of the RNA structures shown in Figure 1a predicted that the CAG-repeat strand would be preferentially loaded as the guide strand (data not shown). The alternative version, siCAG tail, has the sequence 5′-AGUGUU-3′ incorporated at the 3′-end of the CAG-repeat strand. This hexanucleotide motif was previously shown to act as a nuclear localization signal for miR-29b.22 Of note, while the seed sequences at nucleotides 2–7 were uninterrupted CAG repeats, we expected that the base substitutions at other positions in the CAG-repeat strand, and the overall high G + C content of around 60%, would reduce the activity against native transcripts containing short CUG repeats. We postulated, however, that the multiplicity of binding sites may enhance activity against toxic RNAs containing the CUGexp tract.
Figure 1.

siRNA directed against CUG repeats (siCAG) causes knockdown of CUGexp expression and reduction of nuclear RNA foci in mouse skeletal muscle in vivo. (a) Sequences of siRNAs used in this study; gray box shows the putative siRNA nuclear localization signal in the siCAG-tail duplex. (b) Northern blot for CUGexp transcript in RNA from tibialis anterior (TA) muscle. RNA was isolated 7 days after electroporation of siCAG or siCAG tail (si), as compared with saline-electroporation control (sal). Results were normalized to endogenous mouse Acta1 for quantification on graph (mean ± SD, at least four independent experiments, * denotes P < 0.0001, t-test). (c) qRT-PCR for endogenous transcripts containing CUG repeats (Mapkap1, Txlnb, and Tnfrsf22) or CAG repeats (Nr3c1 and Dap). The number of repeats in each transcript is indicated. TA muscle from human skeletal actin—long repeat (HSALR) mice was taken 7 days after electroporation of saline, siCAG or siCAG tail (n = 3 mice per each group, *P < 0.0001, t-test). Expression was normalized to Gtf2b, a housekeeping transcript that shows low variance in wild type or HSALR muscle.12 WT, wild type.
To test activity in vivo, we injected 3 µg of siRNA into tibialis anterior (TA) muscle of HSALR mice. The TA in the opposite hindlimb was injected either with vehicle (saline) or control siRNA having no endogenous target. Following the injection, the muscle was electroporated as previously described15 to facilitate entry of siRNA into muscle fibers. Intramuscular injections and electroporation were performed under general anesthesia. Mice were killed for analysis 7 days after a single injection. Northern blots showed that siCAG caused a ~75% reduction of the CUGexp transcript in TA muscle (Figure 1b). The overall extent of knockdown with siCAG and siCAG tail were similar. By comparison, we did not observe any significant changes in transgene expression in HSALR mice injected with saline or control siRNA.
A potential disadvantage of targeting the repeat sequence is the risk of reducing the expression of endogenous transcripts that contain the same targeting sequence. Examination of RefSeq sequences using the BLAST algorithm identified 43 mouse transcripts having seven or more uninterrupted CUG repeats, of which 19 showed moderate expression in a previous microarray study of mouse skeletal muscle (Supplementary Table S1).12 To gauge the effects of siCAG on endogenous mouse transcripts that contain CUG or CAG repeats, we selected eight of these transcripts and determined their expression level by quantitative or semiquantitative Real Time PCR (RT-PCR). The selected mRNAs contain 8–25 CUG or CAG repeats, located in the coding region, 5′ UTR or 3′ UTR. Five transcripts were assessed by real-time qRT-PCR (Figure 1c), and six were assessed by semiquantitative RT-PCR (Supplementary Figure S1), including three transcripts assessed by both methods. A single transcript showed significant reduction in siCAG and siCAG-tail treated muscle. The expression of Txlnb, an mRNA with (CUG)9 in the 5′ UTR was significantly downregulated (P < 0.0001) (Figure 1c). The level of another mRNA, Mllt3, containing (CUG)8 in 5′ UTR but also (CAG)28 in coding region, was reduced 2.2-fold (P = 0.036) in muscle treated with siCAG-tail, but not in muscle treated with siCAG. For Tnfrsf22, significant silencing (P = 0.041) in muscles treated with siCAG-tail was suggested by semiquantitative RT-PCR but not confirmed by qRT-PCR assay. We also analyzed two transcripts, Dap and Nr3c1, that contain 11 and 17 CAG repeats, respectively, to assess activity of CUG repeats as the guide strand. Neither mRNA showed altered expression by qRT-PCR.
Patients with DM1 and HSALR transgenic mice exhibit aggregation of CUGexp RNA in discreet foci in muscle nuclei. We used fluorescence in situ hybridization (FISH) to determine the effects of siRNA on nuclear foci. For this experiment, the siRNA was injected and electroporated into TA or flexor digitorum brevis (FDB) muscle. Individual whole muscle fibers isolated from FDB (Figure 2a and Supplementary Figure S2), or frozen sections of TA (Figure 2b), were examined using 5′-Texas Red-labeled (CAG)7 2′-O-methyl oligoribonucleotide (Figure 2a,b) or 5′-Cy5-labeled human-specific skeletal actin DNA oligonucleotide probe (Supplementary Figure S2). In FDB muscle, 7 days after a single injection of 2 µg of siCAG, the nuclei showed an absence of CUGexp foci or single foci, using either type of FISH probe (Figure 2a and Supplementary Figure S2). The sections of TA muscle showed reduction of CUGexp foci, but with some regional variation. Foci were absent in ~50% of TA fibers, whereas other fibers showed small numbers of inclusions (Figure 2a and Supplementary Figure S3). The variability of foci observed in the TA muscles probably reflects a regional variability of uptake of the injected siCAG. By comparison, muscles treated with saline consistently showed dozens of foci per muscle nucleus.
Figure 2.
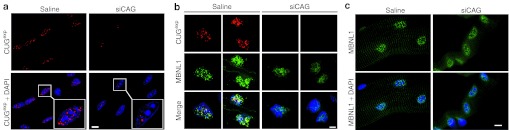
Nuclear foci of CUGexp RNA and MBNL1 protein are dispersed in muscles treated with siCAG. (a) Fluorescence in situ hybridization (FISH) shows reduction of CUGexp foci (red) in nuclei (blue) in isolated flexor digitorum brevis (FDB) muscle fibers. Muscle was taken 7 days after electroporation of siCAG or saline. Inset shows magnification of selected nuclei. Nuclei are counterstained blue using 4,6 diamino-2-phenylindole dihydrochloride (DAPI). (b) Combined FISH for CUGexp RNA (red) and immunofluorescence for MBNL1 protein (green) in transverse sections of tibialis anterior muscle from human skeletal actin—long repeat mice. Muscle was taken 7 days after electroporation of siCAG or saline. The CUGexp foci are dispersed and the MBNL1 distribution is more diffuse in siCAG-treated muscle. Nuclei are counterstained blue (DAPI). (c) Immunofluorescence of isolated FDB muscle fibers shows redistribution of MBNL1 (green) from punctate to diffuse localization in the nucleus (blue), 7 days after electroporation of siCAG.Scale bars = 5 µm. Objective: 100× Plan Apo, 1.4 NA oil.
MBNL1 spicing regulation activity is restored in siCAG-treated muscles
Previous studies have shown that HSALR transgenic mice, like human patients with DM1, have sequestration of MBNL1 protein in nuclear foci of CUGexp RNA. We used immunofluorescence and FISH to assess the distribution of MBNL1 relative to CUGexp foci in the nucleus (Figure 2b). The overall distribution of nuclear MBNL1 was more diffuse, and less coincident with CUGexp foci, in siCAG-treated TA muscle (Figure 2b). A similar redistribution of MBNL1 was observed in isolated FDB fibers following siCAG injection (Figure 2c).
To determine the effect of siRNA on MBNL1 activity, we used RT-PCR to examine the alternative splicing of MBNL1-regulable exons. For this analysis, we selected six splice events that show similar misregulation in patients with DM1, HSALR transgenic mice, and MBNL1 knockout mice (ref. 10 and Sobczak et al., unpublished data). Included in this analysis were three exons whose inclusion is promoted by MBNL1 (Atp2a1 exon 22, Arfgap2 exon 8, and Camk2b exon 13) and three exons that are repressed by MBNL1 (M-line region of Titin exon 5, Nfix exon 7, and Ldb3 exon 11) (Figure 3a,b). One exon on the panel is known to be a direct target of MBNL1 (exon 22 of Atp2a1), MBNL1 regulation for the other exons is unstudied and may be direct or indirect. For all exons tested, we observed improved splicing regulation for both siCAG and siCAG-tail treated muscles, with near normalization of splicing for several exons (Figure 3c). We did not observe any significant splicing changes of the same exons in saline or control siRNA-treated TA muscles of HSALR mice (Figure 3c and Supplementary Figure S4a), or in siCAG-treated TA muscles of wild-type mice (Supplementary Figure S4b).
Figure 3.

Effect of siCAG and siCAG tail on MBNL1 splicing regulatory activity. (a) RT-PCR analysis of alternative splicing for exon 22 (ex.22) of Atp2a1, exon 8 (ex.8) of Arfgap2, and exon 13 (ex.13) of Camk2b, three exons whose splicing is promoted by MBNL1. Splice products in wild-type mice are shown on the right side of gel. In response to siCAG, partial correction of alternative splicing was observed for each exon. RNA was extracted from tibialis anterior (TA) muscle of human skeletal actin—long repeat mice, 7 days after electroporation of siCAG or siCAG tail (si). The contralateral TA was electroporated with saline (sal). (b) Same as in (b), except that RT-PCR analysis was performed on exons whose alternative splicing is normally repressed by MBNL1: M-line region exon 5 (Mex.5) of Titin, exon 7 (ex.7) of Nfix, and exon 11 (ex.11) of Ldb3. (c) Quantification of results in (b) and (c), based on three independent experiments (mean ± SD). Results are expressed as the percentage of splice products including the alternative exons for four groups of animals (*P < 0.0001, **P < 0.05, Student's t-test for comparison with saline-treated muscles). HSALR, human skeletal actin—long repeat mouse; WT, wild type mouse.
Previous studies have suggested that transfection of single-stranded oligoribonucleotides can lead to RNAi-like effects.23 However, when we electroporated 3 µg of single-strand RNA, identical to the CAG-repeat strand of siCAG (ssCAG), we observed a modest reduction of CUGexp RNA (64 ± 8% of untreated muscles) but no significant correction of alternative splicing for Atp2a1 and Titin (Supplementary Figure S5), indicating that duplex siRNA is more active in this experimental system.
To further examine the mechanism of CUGexp silencing by siCAG, we used HT1080-800R cells that are stably transfected with a construct expressing the DMPK 3′ UTR with 800 CUG repeats.24 Transfection of siCAG produced significant knockdown of DMPK-(CUG) 800 transcripts in HT1080-800R cells. However, the knockdown was abrogated by cotransfection of siRNA directed against Argonaute 2 (AGO2) (Supplementary Figure S6). These results provide further support for the involvement of RNAi pathways in siCAG action.
Dose response and duration of action
We examined the dose response of siCAG after electroporation in TA muscles of HSALR mice. Analysis 7 days after the injection showed that 1 and 3 µg doses caused reduction of CUGexp transcripts by >50%, accompanied by strong correction of alternative splicing for Atp2a1, Titin, Nfix, and Clcn1 (Figure 4a,b). Previous studies have shown that misregulated alternative splicing for Clcn1 causes loss of function for the muscle-specific chloride ion channel, resulting in repetitive action potentials (myotonic discharges).20,25 Needle electrode recordings showed that improvement of Clcn1 splicing was accompanied by suppression of myotonic discharges (Figure 4c), consistent with studies showing that myotonia is rescued by morpholino oligomers that specifically correct the Clcn1 splicing defect.15 By comparison, the 0.1 µg dose had no effect, and effects of the 0.3 µg dose were intermediate. We also compared therapeutic effects at 7 and 14 days after a single injection of 3 µg siCAG. By 14 days, the knockdown of CUGexp transcripts was beginning to wane, and splicing defects were reverting to their basal state (Figure 5a,b). Electromyography-measured myotonia in mice 14 days after injection of siCAG was not significantly different from control treated mice (data not shown).
Figure 4.

Dose response relationship for siCAG. (a) The level of CUGexp transcript was determined by northern analysis in tibialis anterior (TA) muscle. Four doses of siCAG (3, 1, 0.3, or 0.1 µg) were electroporated into TA of human skeletal actin—long repeat mice (n = 2 per each group). Total RNA was isolated after 7 days. Saline electroporated TA serves as control. (b) RT-PCR analysis of alternative splicing for mice described in (a). The two higher siCAG doses produced strong splicing correction, while the lowest dose had no effect. (c) Reduction of myotonia 1 week after electroporation of siCAG into TA muscle. The contralateral TA was electroporated with saline alone (n = 2 mice for each siRNA dose; *P < 0.0001 for siCAG versus saline according to Student's t-test). Myotonic discharges were rated by a blinded examiner using extracellular needle electrode recordings (electromyography) under general anesthesia. Myotonia was significantly reduced in muscles treated with 3 and 1 µg of siCAG. HSALR, human skeletal actin—long repeat mouse; WT, wild type mouse. **P < 0.05.
Figure 5.

Duration of siCAG response. (a) Slot-blot hybridization assay to determine the levels of CUGexp accumulation in tibialis anterior muscle. Muscle was harvested 7 or 14 days after electroporating 3 µg of siCAG. Hybridization signal from (CAG)8 probe (CUGexp RNA) was normalized to mouse skeletal actin probe. Each bar represents the mean ± SD for indicated number of mice and bases on two independent hybridization experiments. *P < 0.0001, **P < 0.05 for siRNA versus untreated muscle (Student's t-test). (b) RT-PCR splicing assay for five MBNL1-dependent exons. Analyses were performed 7 or 14 days after electroporating 3 µg of siCAG (si) or saline (sal).
Discussion
Our results indicate that siRNA targeting CUG-expanded transcripts were effective for reducing muscle accumulation of toxic RNA. To our knowledge, these are the first few studies showing nuclear activity of synthetic siRNA in mammalian tissue in vivo. By reducing the burden of CUGexp RNA, the distribution and activity of MBNL1 was improved, assessed by immunolocalization of the protein and splicing regulation for its physiological targets. As expected, correction of alternative splicing for Clcn1 was accompanied by marked reduction of myotonia. Of note, the addition of a hexanucleotide nuclear localization signal to the active strand of the siRNA duplex did not enhance the knockdown efficiency in this system. It is possible that this approach could be applied in other disorders that involve transdominant RNA toxicity due to expression of highly expanded tandem repeats.26,27,28,29,30
Although our CAG·CUG duplex was designed for RNAi activity, the mechanism for CUGexp silencing in our study is uncertain. Previous studies have shown that single-strand CAG-repeat antisense, including morpholino 25-mers,16 fully-substituted 2′-O-methyl 21-mers,13 locked nucleic acid 16-mixmers,24 and viral vector-mediated expression of hU7-snRNA-(CAG)17 can reduce the cellular levels of CUGexp RNA. The mechanism for knockdown by these single-stranded CAG-repeat oligomers has not been determined, but appears to be independent of RNase H or RNA-induced silencing complex, and may result from accelerated decay by cellular pathways once the CUGexp transcript is released from nuclear foci and transported to the cytoplasm. Nevertheless, siRNA activity is suggested by our results based on observations that (i) siCAG was significantly more active than ssCAG; (ii) potency was higher than we observed with morpholino or 2′-O-methyl CAG-repeat antisense oligonucleotides (~10-fold lower molar dose of siCAG for equivalent effect); (iii) the extent of target knockdown was more complete than we observed using morpholino or 2′-O-methyl CAG-repeat antisense oligonucleotides in the same model (75% versus ≤ 50% knockdown); and (iv) activity of siCAG against CUGexp transcripts in transfected cells was reduced by depletion of AGO2 mRNA.
The RNAi strategy was tested previously in DM1 myoblasts by targeting a nonrepetitive gene-specific sequence in DMPK,18 and comparing the relative activity against wild-type DMPK transcripts that are predominantly in the cytoplasm with mutant DMPK transcripts that are retained in the nucleus. In DM1 myoblasts expressing short hairpin RNAs (precursors of the DICER ribonuclease-processed siRNAs) both mutant and wild-type transcripts were efficiently downregulated in nuclear and cytoplasmic fractions. In contrast, synthetic siRNAs delivered via cationic lipids did not silence the expanded repeat-containing transcripts in the nucleus. The differences between cell-based experiments and our in vivo study may relate to the delivery methods (lipofection versus electroporation) or differences in siRNA metabolism and activity between cultured cells and multinucleate muscle fibers. The previous studies also showed that activity of synthetic siRNA against nuclear-retained DMPK transcripts could be enhanced by delivering a ribonucleoprotein complex of siRNA and fusion peptide containing the SV40 nuclear localization signal.18
The major concern for targeting a tandem repeat is that the specificity advantage of RNAi—knockdown of a unique transcript according to its targeting sequence—is partially surrendered. In the case of siCAG, there is potential impact on the wild-type DMPK allele (which we cannot evaluate in HSALR transgenic mice because CTG repeats are not conserved at the murine DMPK locus), but more importantly a group of several dozen endogenous transcripts having CUG repeats that, while not pathologically expanded, have sufficient length to allow full complementarity with siCAG. We examined a subset of these, selected for being informative in skeletal muscle. Our results are consistent with previous studies using non-RNAi strategies, in that targeting of N-any nucleotide trinucleotide repeats was selective for pathologically expanded repeats.16,31 Although the biochemical basis of target selectivity in this and previous studies is unknown, we suggest that in this study, it results from low-intrinsic potency of siCAG against the general population of CUG-repeat-containing transcripts, coupled with heightened sensitivity of the toxic transcripts with expanded repeats. The sensitization may result from the multiplicity of binding sites and localization of transcripts in foci, creating a high local concentration of targeting sites. Of note, in previous studies of Huntingtin and ATX3, only slight differences in knockdown efficiency were observed for RNAi knockdown of transcripts containing expanded versus nonexpanded CAG repeats.31,32,33 These CAG-expanded transcripts have shorter repeat tracts (usually 42–80 repeats) than those that exist in DM1, and they presumably have a diffuse location in the cytoplasm. However, when mutations were introduced in the siRNA seed sequence, to obtain miRNA-like suppression of translation, a large selectivity for the expanded allele was observed.32 Notably, in our study, the selectivity of siCAG was not absolute, in that significant knockdown was observed for one of eight CUG-containing transcripts that we examined, Txlnb. As Txlnb is uncharacterized and its postdevelopmental requirement is unknown, it is unclear whether its downregulation would be deleterious. Using siCAG tail, we observed significant (similar to 50%) reduction of two additional endogenous CUG-containing transcripts, Mllt3 and Tnfrsf22. Whether the current version of siCAG is sufficiently selective, or whether additional optimization is required, will require further study.
Materials and Methods
siRNA oligoribonucleotides. siRNA duplexes were purchased from Dharmacon (Lafayette, CO). All were 2′-deprotected, desalted, and high-performance liquid chromatography purified. The sequence of siCAG sense strand is: 5′-UAGCAGCAGCAGCAGCAGCTT-3′ and antisense strand: 5′-GCUGCUGCUGCUGCUGCUCTT-3′. The sequence of siCAG-tail sense strand is: 5′-UAGCAGCAGCAGCAGCAGUGUU-3′ and antisense strand: 5′-CACUGCUGCUGCUGCUGCUCTT-3′. The sequence for the sense strand of siRNA against human AGO2 is 5′-GCACGGAAGUCC AUCUGAAUU-3′ and antisense strand: 5′-P-UUCAGAUGGACUUCC GUGCUU-3′. The control siRNA of unrelated sequence (Silencer Negative Control #1 siRNA) was purchased from Applied Biosystems/Ambion (Austin, TX).
Experimental mice and siRNA treatment. Homozygous HSALR transgenic mice (line 20b) were previously described.19 FVB wild-type mice served as controls. All animal experiments were approved by the University of Rochester Institutional Animal Care and Use Committee. Injection and electroporation was performed under general anesthesia (intraperitoneal injection of 100 mg/kg ketamine, 10 mg/kg xylazine, and 3 mg/kg acepromazine) as described previously.15 TA or FDB muscles of adult (2–5 month) mice were pretreated by intramuscular injection of bovine hyaluronidase (15 µl, 0.4 U/µl) (Sigma-Aldrich, St. Louis, MO) and after 2 hours injected with 0.1–3 µg of siCAG or control siRNA (doses are specified on figure legends) in 20 µl of saline, or with saline alone. Immediately following muscle injection, electroporation was performed using electrodes placed parallel to the long axis of the muscle. Electroporation parameters were 100 V/cm, 10 pulses at 1 Hz, and 20 ms duration per pulse.
siCAG and siAGO2 treatment of HT1080-800R cells. HT1080-800R cells stably expressing human DMPK-3′ UTR with ~800 CUG repeats24 were either mock treated or transfected with 100 nmol/l of siCAG and/or 5 nmol/l of siAGO2 simultaneously using Lipofectamine RNAiMAX (Invitrogen, Carlsbad, CA). Total RNA was harvested 48 hours after transfection using RNeasy Mini Kit (Qiagen, Valencia, CA) followed by treatment with Turbo DNA-free (Ambion) to remove genomic DNA. Half microgram of total RNA was primed with random hexamers plus oligo dT (Promega, Madison, WI) and reverse transcribed using Superscript II (Invitrogen). Quantitative real-time PCR of AGO2 and DMPK-3′ UTR normalized to 18S rRNA was performed using a StepOnePlus Real Time PCR system (Applied Biosystems), analyzed using the Quantitative-comparative CT (ΔΔCT) method, and displayed as relative quantity. DMPK-3′ UTR primers and probe sequences were described previously.24 The TaqMan gene expression assays for AGO2 (Hs01085579_m1) and 18S rRNA (proprietary sequences) were from Applied Biosystems.
Northern and slot-blot analysis of CUGexp transcripts were performed as described previously.16 Briefly, 3 µg of the total RNA isolated with use of Tri-Reagent (Molecular Research Center, Cincinnati, OH) from TA muscles was separated on 1.2% agarose gels containing 1.5% formaldehyde and transferred onto Hybond-N+ membrane (Amersham Pharmacia Biotech, Buckinghamshire, UK). Blots were hybridized with 5′-end [32P]-labeled oligo-DNA probes specific either for human skeletal actin transgene (5′-CTGTGTCAGTTTACGATGGCAGCAAC-3′) or endogenous mouse skeletal actin (5′-ACCCTGCAACCACAGCACGATTGTCGATTG-3′, loading control). For slot-blot analysis, the 5′-end-labeled (CAG)8 DNA-oligonucleotide was used as a probe.
Quantitative analysis of mouse endogenous transcripts containing CUG and CAG repeats. TaqMan PCR analyses were performed according to manufacturer recommendations with reagents purchased from Roche (Mannheim, Germany) using TaqMan assays specific for following transcripts: Mapkap1 (Mm 01327444_m1), Txlnb (Mm 01193251_m1), Tnfrsf22 (Mm 00445826), Dap (Mm 00524774_m1), and Nr3c1 (Mm 01260498_m1). Data were normalized to the level of general transcription factor 2b (Gtf2b) mRNA. Semiquantitative RT-PCRs were carried out as described previously16 for seven mouse transcripts: Mapkap1, Fgd4, Pcolce, Tnfrsf22, Mllt3, Nr3c1, and Gapdh control (Supplementary Table S2).
RT-PCR analysis of alternative splicing. Total RNA was isolated from TA muscles 1 and 2 weeks after injection with siCAG, siCAG tail, control siRNA, or saline only. cDNA synthesis and alternative splicing analyses for Clcn1 (ClC-1), Atp2a1 (Serca-1), Titin M-line region exon 5 (Mex5, exon 362), and Ldb3 (Zasp) were performed as described previously.10 DNA amplification for remaining mRNA fragments was carried out with following primer pairs: Arfgap2 (Arfgap2-F, 5′- GACTCTGATTT CTTCACAGAACAT-3′ Arfgap2-R, 5′-TGCTCTCGGAGCTTCTCTGCC A-3′), Camk2b (Camk2b-F, 5′-CAGGAGACTGTGGAATGTCTG-3′ Camk2b-R, 5′-GGCATCTTCATCCTCTATGGTTG-3′), and Nfix (Nfix-F, 5′-GAGTCCAGTAGATGATGTGTTCTA-3′ Nfix-R, 5′-CTGCACAAACTCCTTCAGCGAGTC-3′).
FISH. Individual FDB muscle fibers were isolated and FISH was performed as described previously.16,34 Briefly, FDB fibers and cross-sections of TA were fixed in 3% paraformaldehyde for 15 minutes at room temperature (RT), washed in phosphate-buffered saline (PBS), and permeabilized in 0.5% Triton X-100/saline solution for 5 minutes at RT. Hybridization was for 3 hours at 37 °C using either 5′-Cy5-labeled HSA-specific DNA probe (5′-TGTGTCAGTTTACGATGGCAGCAAC-3′ IDT, Coralville, IA) or the 5′-Texas Red-labeled CUG repeat-specific 2′-O-methyl oligoribonucleotide (CAG)7 probe. Untreated, saline-treated, and siCAG-treated muscles were hybridized at the same time.
Immunofluorescence. Localization of MBNL1 protein in single FDB fibers and cryosections of TA muscles were performed as described previously.16,34 Briefly, freshly isolated FDB fibers were fixed in 2% paraformaldehyde/saline solution (pH 7.3) for 5 minutes at RT, washed in PBS/0.05% Tween-20, permeabilized in prechilled 50% methanol/50% acetone for 10 minutes at −20 °C, washed in PBS/0.05% Tween-20, blocked in 5% normal goat serum, and placed in primary antibody (anti-MBNL1 A2764 1:2000 in 1% bovine serum albumin/PBS) overnight at 4 °C. After washing in 1% bovine serum albumin/PBS, samples were placed in secondary antibody (goat antirabbit Alexa 488 1:400; Molecular Probes, Eugene, OR) in 1% bovine serum albumin/PBS with 4,6 diamino-2-phenylindole dihydrochloride; 1:20,000 1 hour at RT then washed twice in 1% bovine serum albumin/PBS before three final washes in PBS alone. For immunofluorescence of TA muscles, 10 µmol/l cryosections were fixed in 3% paraformaldehyde, washed in PBS, permeabilized with 0.5% Triton X-100/PBS 5 minutes RT, blocked in 5% normal goat serum/PBS 30 minutes RT, incubated in 1 °C antibody A2764 1:5000 in PBS overnight at 4 °C, washed in PBS, incubated in 2 °C antibody (Alexa 488 goat antirabbit; Molecular Probes) 1:400 + 4,6 diamino-2-phenylindole dihydrochloride 1:20,000 in PBS for 1 hour RT, and washed in PBS.
Fluorescence microscopy. Images were captured using a Nikon Eclipse E600 fluorescence microscope (Nikon USA, Melville, NY), a Cool Snap HQ digital camera (Photometrics, Tucson, AZ), and Metavue software (Molecular Devices, Sunnyvale, CA). Z-plane stacks were deconvolved using AutoQuant software (Media Cybernetics, Bethesda, MD). All images shown are maximum projections of deconvolved Z-plane stacks with the exception of FISH images using the HSA probe, which are maximum projections of nondeconvolved stacks. Image exposure times and deconvolution parameters were identical for PBS- and siCAG-treated samples. Objective: 100× Plan Apo 1.4 NA oil.
Electromyography. Electromyography analysis of myotonia was performed under general anesthesia by a blinded examiner as described previously.15 Myotonia was graded as follows: 0 indicates no myotonia; 1, occasional myotonic discharge in <50% of electrode insertions; 2, myotonic discharge in >50% of insertions; 3, myotonic discharge with nearly every insertion.
Statistical analysis. Group data are expressed as mean ± SD. Data comparison was performed by two-tailed Student's t-test, and P value of <0.0001 or <0.05 (depicted on figures as * or **, respectively) was considered significant.
SUPPLEMENTARY MATERIAL Figure S1. Semiquantitative RT-PCR for seven mouse transcripts that contain CUG or CAG repeats. Figure S2. FISH analysis of nuclear RNA foci in isolated FDB muscle fibers using a transgene-specific oligonucleotide probe. Figure S3. FISH showing nuclear foci of CUGexp transcripts in transverse sections of saline- or siCAG-treated TA muscle. Figure S4. Alternative splicing of DM1-dysregulated transcripts is not affected by electroporation of control siRNA in HSALR mice, or by siCAG in WT mice. Figure S5. CAG repeat strand is more active if introduced as siCAG duplex than as single-stranded oligoribonucleotide. Figure S6. AGO2 influence activity of siCAG. Table S1. Mouse Ref-Seq sequences of mRNAs containing at least (CTG)7 or (CAG)7 repeat tract. Table S2. Primers used for RT-PCR amplification of endogenous transcripts containing either CUG or CAG repeats.
Acknowledgments
This work comes from the University of Rochester Wellstone Muscular Dystrophy Cooperative Research Center (U54NS48843) with support from National Institutes of Health/National Institute of Arthritis and Musculoskeletal and Skin Diseases AR049077 and K24AR/NS48143 (to C.A.T.), National Institute of Neurological Disorders and Stroke K08NS064293 (to T.M.W.), and the Polish Ministry of Science and Higher Education grant N302 260938 (to K.S.) and the Foundation for Polish Science-TEAM program cofinanced by the European Union within the European Regional Development Fund (to K.S.). The authors declare no conflict of interest.
Supplementary Material
Semiquantitative RT-PCR for seven mouse transcripts that contain CUG or CAG repeats.
FISH analysis of nuclear RNA foci in isolated FDB muscle fibers using a transgene-specific oligonucleotide probe.
FISH showing nuclear foci of CUGexp transcripts in transverse sections of saline- or siCAG-treated TA muscle.
Alternative splicing of DM1-dysregulated transcripts is not affected by electroporation of control siRNA in HSALR mice, or by siCAG in WT mice.
CAG repeat strand is more active if introduced as siCAG duplex than as single-stranded oligoribonucleotide.
AGO2 influence activity of siCAG.
Mouse Ref-Seq sequences of mRNAs containing at least (CTG)7 or (CAG)7 repeat tract.
Primers used for RT-PCR amplification of endogenous transcripts containing either CUG or CAG repeats.
REFERENCES
- Orr HT., and, Zoghbi HY. Trinucleotide repeat disorders. Annu Rev Neurosci. 2007;30:575–621. doi: 10.1146/annurev.neuro.29.051605.113042. [DOI] [PubMed] [Google Scholar]
- Osborne RJ., and, Thornton CA. RNA-dominant diseases. Hum Mol Genet. 2006;15 Spec No 2:R162–R169. doi: 10.1093/hmg/ddl181. [DOI] [PubMed] [Google Scholar]
- Li LB., and, Bonini NM. Roles of trinucleotide-repeat RNA in neurological disease and degeneration. Trends Neurosci. 2010;33:292–298. doi: 10.1016/j.tins.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brook JD, McCurrach ME, Harley HG, Buckler AJ, Church D, Aburatani H.et al. (1992Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member Cell 68799–808. [DOI] [PubMed] [Google Scholar]
- Harper PS. W.B. Saunders: London, Philadelphia; 2001. Myotonic Dystrophy. [Google Scholar]
- Thornton CA, Johnson K., and, Moxley RT 3rd. Myotonic dystrophy patients have larger CTG expansions in skeletal muscle than in leukocytes. Ann Neurol. 1994;35:104–107. doi: 10.1002/ana.410350116. [DOI] [PubMed] [Google Scholar]
- Davis BM, McCurrach ME, Taneja KL, Singer RH., and, Housman DE. Expansion of a CUG trinucleotide repeat in the 3′ untranslated region of myotonic dystrophy protein kinase transcripts results in nuclear retention of transcripts. Proc Natl Acad Sci USA. 1997;94:7388–7393. doi: 10.1073/pnas.94.14.7388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JW, Urbinati CR, Teng-Umnuay P, Stenberg MG, Byrne BJ, Thornton CA.et al. (2000Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy EMBO J 194439–4448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dansithong W, Paul S, Comai L., and, Reddy S. MBNL1 is the primary determinant of focus formation and aberrant insulin receptor splicing in DM1. J Biol Chem. 2005;280:5773–5780. doi: 10.1074/jbc.M410781200. [DOI] [PubMed] [Google Scholar]
- Lin X, Miller JW, Mankodi A, Kanadia RN, Yuan Y, Moxley RT.et al. (2006Failure of MBNL1-dependent post-natal splicing transitions in myotonic dystrophy Hum Mol Genet 152087–2097. [DOI] [PubMed] [Google Scholar]
- Kuyumcu-Martinez NM, Wang GS., and, Cooper TA. Increased steady-state levels of CUGBP1 in myotonic dystrophy 1 are due to PKC-mediated hyperphosphorylation. Mol Cell. 2007;28:68–78. doi: 10.1016/j.molcel.2007.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne RJ, Lin X, Welle S, Sobczak K, O'Rourke JR, Swanson MS.et al. (2009Transcriptional and post-transcriptional impact of toxic RNA in myotonic dystrophy Hum Mol Genet 181471–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulders SA, van den Broek WJ, Wheeler TM, Croes HJ, van Kuik-Romeijn P, de Kimpe SJ.et al. (2009Triplet-repeat oligonucleotide-mediated reversal of RNA toxicity in myotonic dystrophy Proc Natl Acad Sci USA 10613915–13920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzyzosiak WJ, Sobczak K, Wojciechowska M, Fiszer A, Mykowska A., and, Kozlowski P. Triplet repeat RNA structure and its role as pathogenic agent and therapeutic target. Nucleic Acids Res. 2012;40:11–26. doi: 10.1093/nar/gkr729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler TM, Lueck JD, Swanson MS, Dirksen RT., and, Thornton CA. Correction of ClC-1 splicing eliminates chloride channelopathy and myotonia in mouse models of myotonic dystrophy. J Clin Invest. 2007;117:3952–3957. doi: 10.1172/JCI33355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler TM, Sobczak K, Lueck JD, Osborne RJ, Lin X, Dirksen RT.et al. (2009Reversal of RNA dominance by displacement of protein sequestered on triplet repeat RNA Science 325336–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- François V, Klein AF, Beley C, Jollet A, Lemercier C, Garcia L.et al. (2011Selective silencing of mutated mRNAs in DM1 by using modified hU7-snRNAs Nat Struct Mol Biol 1885–87. [DOI] [PubMed] [Google Scholar]
- Langlois MA, Boniface C, Wang G, Alluin J, Salvaterra PM, Puymirat J.et al. (2005Cytoplasmic and nuclear retained DMPK mRNAs are targets for RNA interference in myotonic dystrophy cells J Biol Chem 28016949–16954. [DOI] [PubMed] [Google Scholar]
- Mankodi A, Logigian E, Callahan L, McClain C, White R, Henderson D.et al. (2000Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat Science 2891769–1773. [DOI] [PubMed] [Google Scholar]
- Mankodi A, Takahashi MP, Jiang H, Beck CL, Bowers WJ, Moxley RT.et al. (2002Expanded CUG repeats trigger aberrant splicing of ClC-1 chloride channel pre-mRNA and hyperexcitability of skeletal muscle in myotonic dystrophy Mol Cell 1035–44. [DOI] [PubMed] [Google Scholar]
- Krol J, Sobczak K, Wilczynska U, Drath M, Jasinska A, Kaczynska D.et al. (2004Structural features of microRNA (miRNA) precursors and their relevance to miRNA biogenesis and small interfering RNA/short hairpin RNA design J Biol Chem 27942230–42239. [DOI] [PubMed] [Google Scholar]
- Hwang HW, Wentzel EA., and, Mendell JT. A hexanucleotide element directs microRNA nuclear import. Science. 2007;315:97–100. doi: 10.1126/science.1136235. [DOI] [PubMed] [Google Scholar]
- Krol J, Fiszer A, Mykowska A, Sobczak K, de Mezer M., and, Krzyzosiak WJ. Ribonuclease dicer cleaves triplet repeat hairpins into shorter repeats that silence specific targets. Mol Cell. 2007;25:575–586. doi: 10.1016/j.molcel.2007.01.031. [DOI] [PubMed] [Google Scholar]
- Nakamori M, Gourdon G., and, Thornton CA. Stabilization of expanded (CTG)·(CAG) repeats by antisense oligonucleotides. Mol Ther. 2011;19:2222–2227. doi: 10.1038/mt.2011.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlet-B N, Savkur RS, Singh G, Philips AV, Grice EA., and, Cooper TA. Loss of the muscle-specific chloride channel in type 1 myotonic dystrophy due to misregulated alternative splicing. Mol Cell. 2002;10:45–53. doi: 10.1016/s1097-2765(02)00572-5. [DOI] [PubMed] [Google Scholar]
- Liquori CL, Ricker K, Moseley ML, Jacobsen JF, Kress W, Naylor SL.et al. (2001Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9 Science 293864–867. [DOI] [PubMed] [Google Scholar]
- Sato N, Amino T, Kobayashi K, Asakawa S, Ishiguro T, Tsunemi T.et al. (2009Spinocerebellar ataxia type 31 is associated with “inserted” penta-nucleotide repeats containing (TGGAA)n Am J Hum Genet 85544–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White MC, Gao R, Xu W, Mandal SM, Lim JG, Hazra TK.et al. (2010Inactivation of hnRNP K by expanded intronic AUUCU repeat induces apoptosis via translocation of PKCΔ to mitochondria in spinocerebellar ataxia 10 PLoS Genet 6e1000984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ.et al. (2011Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS Neuron 72245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi H, Abe K, Matsuura T, Ikeda Y, Hitomi T, Akechi Y.et al. (2011Expansion of intronic GGCCTG hexanucleotide repeat in NOP56 causes SCA36, a type of spinocerebellar ataxia accompanied by motor neuron involvement Am J Hum Genet 89121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Matsui M, Gagnon KT, Schwartz JC, Gabillet S, Arar K.et al. (2009Allele-specific silencing of mutant huntingtin and ataxin-3 genes by targeting expanded CAG repeats in mRNAs Nat Biotechnol 27478–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Mezer M, Wojciechowska M, Napierala M, Sobczak K., and, Krzyzosiak WJ. Mutant CAG repeats of Huntingtin transcript fold into hairpins, form nuclear foci and are targets for RNA interference. Nucleic Acids Res. 2011;39:3852–3863. doi: 10.1093/nar/gkq1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiszer A, Mykowska A., and, Krzyzosiak WJ. Inhibition of mutant huntingtin expression by RNA duplex targeting expanded CAG repeats. Nucleic Acids Res. 2011;39:5578–5585. doi: 10.1093/nar/gkr156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lueck JD, Mankodi A, Swanson MS, Thornton CA., and, Dirksen RT. Muscle chloride channel dysfunction in two mouse models of myotonic dystrophy. J Gen Physiol. 2007;129:79–94. doi: 10.1085/jgp.200609635. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Semiquantitative RT-PCR for seven mouse transcripts that contain CUG or CAG repeats.
FISH analysis of nuclear RNA foci in isolated FDB muscle fibers using a transgene-specific oligonucleotide probe.
FISH showing nuclear foci of CUGexp transcripts in transverse sections of saline- or siCAG-treated TA muscle.
Alternative splicing of DM1-dysregulated transcripts is not affected by electroporation of control siRNA in HSALR mice, or by siCAG in WT mice.
CAG repeat strand is more active if introduced as siCAG duplex than as single-stranded oligoribonucleotide.
AGO2 influence activity of siCAG.
Mouse Ref-Seq sequences of mRNAs containing at least (CTG)7 or (CAG)7 repeat tract.
Primers used for RT-PCR amplification of endogenous transcripts containing either CUG or CAG repeats.