

Abstract
Immunosuppression associated with ovarian cancer (OC) and resultant peritoneal carcinomatosis (PC) hampers the efficacy of many promising treatment options, including immunotherapies. It is hypothesized that oncolytic virus-based therapies can simultaneously kill OC and mitigate immunosuppression. Currently, reovirus-based anticancer therapy is undergoing phase I/II clinical trials for the treatment of OC. Hence, this study was focused on characterizing the effects of reovirus therapy on OC and associated immune microenvironment. Our data shows that reovirus efficiently killed OC cells and induced higher expression of the molecules involved in antigen presentation including major histocompatibility complex (MHC) class I, β2-microglobulin (β2M), TAP-1, and TAP-2. In addition, in the presence of reovirus, dendritic cells (DCs) overcame the OC-mediated phenotypic suppression and successfully stimulated tumor-specific CD8+ T cells. In animal studies, reovirus targeted local and distal OC, alleviated the severity of PC and significantly prolonged survival. These therapeutic effects were accompanied by decreased frequency of suppressive cells, e.g., Gr1.1+, CD11b+ myeloid derived suppressor cells (MDSCs), and CD4+, CD25+, FOXP3+ Tregs, tumor-infiltration of CD3+ cells and higher expression of Th1 cytokines. Finally, reovirus therapy during early stages of OC also resulted in the postponement of PC development. This report elucidates timely information on a therapeutic approach that can target OC through clinically desired multifaceted mechanisms to better the outcomes.
Introduction
Therapeutic options for ovarian cancer (OC) and associated peritoneal carcinomatosis (OC-PC) are limited. Unfortunately, OC is usually diagnosed at an advanced stage which correlates with high rates of recurrence and mortality. However, in recent years novel approaches based on immunological principles have shown promising results for the prevention, delay or management of OC-PC.1,2 These immunotherapies comprise modalities that either directly target tumor cells or further stimulate appropriate immune responses and indirectly attack tumors. Thus far, cellular (e.g., T cells or dendritic cells (DCs)), antibody-based (e.g., anti-CTLA4, anti-vascular endothelial growth factor (also called as bevacizumab or avastin)),3 cytokine-based (e.g., granulocyte-macrophage colony-stimulating factor, interferons) or immune-modulating agent-based (e.g., ONTAK— a interleukin (IL)-2 immunotoxin fusion protein denileukin difitox, ref. 4); therapies have been employed to target OC (reviewed in ref. 2,5).
Efficiency of these promising therapeutic options, however, is dampened due to an inherent suppressive milieu persisting in the OC-PC microenvironment. OC cells are known to deploy various evasion strategies that ultimately result in the escape from immune attack and subsequent elimination.6 For example, ovarian tumor cells produce factors such as vascular endothelial growth factor,7 CXCL12, and transforming growth factor-β8 that promote vascularization and metastasis, and additionally suppress antitumor T immunity. OC lesions display downregulated expression of molecules essential for antigen presentation (e.g., major histocompatibility complex (MHC) class I, ref. 9,10) or processing (e.g., transporter 1 (TAP-1) and TAP-2, ref. 9,11). Similarly, higher numbers of Gr1.1+, CD11b+ myeloid derived suppressor cells (MDSCs)12 or CD4+, CD25+, FOXP3+ T regulatory cells (Tregs)13 as well as dysfunctional dendritic cells (DCs) and impaired CD4+ or CD8+ T cells accompany advanced OC-PC. It is now well acknowledged that OC-PC associated immunosuppression correlates with poor prognosis.2,6,14
Reovirus, a naturally occurring benign human virus, preferentially kills cancer cells15,16 of many types and is currently under an international phase III clinical trial (NCT01166542) against head and neck cancers. Recently, reovirus-based phase I/II clinical trials (OSU-07022, GOG-0186H) were initiated for the treatment of OC. Although reovirus-based oncotherapy primarily targets cancer cells through direct killing (oncolysis), recent reports have documented additional immune-based mechanisms aiding tumor elimination.17,18,19 Along with others, our recent work has shown that, during its therapeutic administration, reovirus additionally alerts and educates the immune system against tumor antigens and facilitates the induction of beneficial antitumor immunity in melanoma,20 lung,20 and prostate17 cancer models. Thus, if appropriately managed reovirus-driven antitumor activities comprised of virus-mediated direct killing and antitumor immunity can target tumors simultaneously. Nonetheless, whether reovirus can target local or metastatic OC and affect immune microenvironment is unknown. In this context, this study was focused on characterizing the immunological consequences of intraperitoneal (i.p.) injections of reovirus in PC-bearing hosts and evaluating the applicability of reovirus therapy for the prevention of PC development or the treatment of already developed PC. We show that i.p. administered reovirus can target local as well as distally located OC cells. In addition, our data also demonstrates that reovirus resolves the PC-associated immune evasion mechanisms including those involved in antigen processing and presentation as well as T-cell suppression, and subsequently initiates clinically desired beneficial antitumor immune activities with a potential to establish long-term recurrence-free survival.
Results
OC cells express the molecules necessary for antigen presentation preceding oncolysis
To establish the applicability of reovirus therapy for the treatment of OC, we first evaluated the susceptibility of OC cells to reovirus-mediated oncolysis. For this purpose, mouse OC cells (ID8) were cultured in the presence of live reovirus (LRV) or UV-inactivated reovirus (UVRV) and then analyzed for cell death. As shown in a representative example (Figure 1a), ID8 cells were highly susceptible to oncolysis and most of these cells were killed by 72 hours post-infection with either 1, 10 or 100 multiplicity of infection (MOI) of LRV (Figure 1b), but not with UVRV (data not shown). Previously, we have established that LRV does not kill nontransformed, normal ovarian cells.21 Together, our data conclusively established the selective capacity of reovirus to efficiently kill OC cells.
Figure 1.

Virus-mediated oncolysis and enhanced inherent antigen presentation molecule expression of OC cells. (a) ID8 cells infected in vitro with 1, 10 or 100 MOI of reovirus were harvested at 24, 48, and 72 hours, stained with annexin-V/7-AAD and then analyzed in flow cytometry for the detection of apoptotic cells. (b) Data shows representative profiles of ID8 cells at respective timepoints. (c) In addition, cells were stained with anti-MHC I (surface staining) and antireovirus antibodies (intracellular staining) at respective timepoints shown. (d) In addition, at 24 hours post-infection, cells were also analyzed in quantitative real-time PCR to quantitate the gene expression of selected genes (as shown) using the Livak and Schmittgen's 2-ΔΔCT method.36 Gene expression in LRV-treated cells was normalized against GAPDH gene expression, and then compared against that observed in PBS-treated cells to calculate fold change. In both b and d, LRV-treated samples were compared against PBS-treated samples using Student's t-test at 95% CI; ns = P > 0.05; *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001. Cumulative data from a single experiment is representative of atleast five other independent experiments. CI, confidence interval; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; IFN, interferon; LRV, live reovirus; MHC, major histocompatibility complex; MOI, multiplicity of infection; OC, ovarian cancer; ns, not significant; PBS, phosphate-buffered saline.
OC downmodulates the molecules that are either essential for antigen processing or presentation. Hence, we next investigated whether reovirus can overcome this immune evasion strategy. For this purpose, ID8 cells were cultured in the presence of LRV for 24 hours, and then analyzed for MHC class I protein and interferon (IFN)-β, MHC class I, β2-microglobulin (β2M), TAP-1, and TAP-2 mRNA expression. As shown in Figure 1c, untreated ID8 cells showed absence of MHC class I (0 hour) on the cell surface. Following incubation with LRV, however, most of ID8 cells displayed a gradually increasing expression of MHC class I. Interestingly, LRV also initiated a significantly increased expression of MHC class I, β2M, TAP-1, and TAP-2 along with IFN-β (Figure 1d). Thus, OC cells overcame MHC class I display inhibition and acquired higher expression of the molecules involved in antigen presentation pathway following reovirus infection.
Reovirus reverses ID8-mediated phenotypic and functional incapacitation of DCs
Since OC inhibits the expression of costimulatory molecules on DCs,6,22 the effect of reovirus on the ID8-induced inhibitory DC phenotype was investigated. For this purpose, ID8 cells and bone marrow-derived DCs (BMDCs)20 were co-cultured together in vitro for 24 or 72 hours, and then exposed to either 10 MOI of UVRV/LRV or phosphate-buffered saline (PBS) (control) for an additional 48 hours. The cultures were then monitored for the expression of MHC class II, CD40, CD80, CD86 molecules on the surface of CD11c+ cells. As shown in Figure 2a, ID8 cells significantly reduced expression of the abovementioned costimulatory molecules on DCs following a co-culture for 24 or 72 hours (“ID8 only” bars). Interestingly, the addition of both UVRV as well as LRV significantly enhanced the expression of these molecules on DCs. Thus, the data showed that both LRV and UVRV can reverse ID8-mediated suppression of costimulatory molecule expression on DCs.
Figure 2.
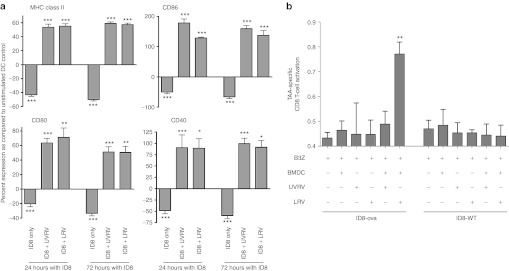
Overcoming OC-induced dysfunction of DCs. (a) ID8 cells and BMDCs were co-cultured together for 24 or 72 hours before infection with 10 MOI of reovirus for 48 hours. Cells were stained with antibodies against CD11c and either MHC II, CD86, CD80, and CD40 and analyzed using flow cytometry. Bars show respective percentages normalized against, and asterisks show statistical analysis as compared with, BMDCs cultured without ID8 or reovirus. The data is representative of at least five independent experiments. (b) 2 × 105 BMDCs were co-cultured with 2 × 105 ID8-ova cells and added with LRV/UVRV for 24 hours. Next, co-cultures were washed, added with 1 × 105 B3Z cells per well, incubated for additional 18–24 hours and then added with 0.15 mmol/l of CPRG for additional 4 hours. The breakdown of GPRG was read at 570 nm as a measure of CD8+ T-cell response. Statistical analysis was obtained by comparing readings from each experimental condition against that observed in only the B3Z wells; ns = P > 0.05; *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001. The data is cumulative from three independent experiments. BMDC, bone marrow-derived DCs; CPRG, chlorophenol red-β-D-galactopyranoside; DC, dendritic cell; MHC, major histocompatibility complex; MOI, multiplicity of infection; ns, not significant; LRV, live reovirus; TAA, tumor-associated antigen; UVRV, UV-inactivated reovirus.
Next, the B3Z antigen presentation assay17,23 was employed to analyze whether reovirus can endow DCs with a capacity to present tumor-associated antigen (TAA) to tumor-specific CD8+ T cells. Briefly, BMDCs incubated in the presence of wild-type (WT) or ova-expressing ID8 (ID8-ova) tumor cells were further exposed to UVRV/LRV, and then mixed with TAA-specific B3Z CD8+ T cells. ID8 WT cells do not express ova and hence served as a non-TAA–bearing negative control. As shown in Figure 2b, BMDCs co-cultured in the presence of ID8-ova alone or UVRV failed to activate B3Z cells. On the other hand, in the presence of LRV, B3Z cells underwent significantly greater activation as compared with that observed in the presence of PBS/UVRV. As expected, BMDCs cultured with ID8 WT cells did not activate B3Z T cells. These results showed the ability of reovirus to facilitate the presentation of OC-associated antigens to tumor-specific CD8+ T cells. Thus, although both LRV and UVRV can induce the phenotypic activation of DCs, only LRV facilitates the presentation of TAA to tumor-specific CD8+ T cells.
Therapeutic oncotherapy alleviates OC-PC disease severity and increases survival
To understand the therapeutic benefits of reovirus therapy during advanced stages of OC, animals with ID8-induced PC were treated with a reovirus regimen as per protocol in Figure 3a. As shown in Figure 3b, the animals injected with LRV displayed substantial alleviation of abdominal distension. This LRV-induced reduction in ascitic fluid volume was gradual and highly significant as compared to the animals injected with PBS/UVRV (Figure 3c). Most importantly, PC-bearing animals treated with LRV showed significantly higher survival as compared with PBS/UVRV-treated animals (Figure 3d). Collectively, our results demonstrated that reovirus therapy can alleviate disease pathology and increase survival of the hosts with OC-PC.
Figure 3.
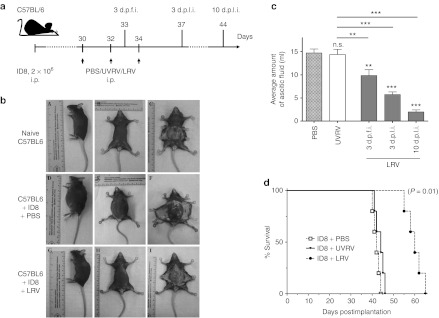
Virotherapy to target established advanced OC. (a) Female C57BL/6 mice were implanted i.p. with ID8 cells, injected with a regimen of PBS/UVRV/LRV as shown and then monitored for the (b) development of PC, (c) amount of ascitic fluid, and (d) survival. (b) Representative example of OC-PC–bearing animals treated with LRV against PBS-treated or non-OC–bearing animal. (c) Graphs represent volume of ascitic fluid collected from PBS/UVRV-treated animals on the day they were killed or LRV-treated animals on the days indicated. Data was analyzed with Student's t-tests at 95% CI; ns = P > 0.05; *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001. Statistics shown immediately on top of the bars was obtained by comparing the respective data against PBS control, while analysis above the horizontal lines were obtained through comparison of LRV-treated animals with that of UVRV-treated ones. (d) Survival in respective experimental groups (injected as per a) was calculated with the Kaplan–Meier survival method. The data is representative of three independent experiments. CI, confidence interval; d.p.f.i, days post first injection; d.p.l.i., days post last injection; i.p., intraperitoneally; LRV, live reovirus; ns, not significant; OC, ovarian cancer; PBS, phosphate-buffered saline; PC, peritoneal carcinomatosis; UVRV, UV-inactivated reovirus.
Reovirus i.p. injections target local and distal OC-PC immune microenvironment
To identify the capacity of i.p. injected reovirus to reach metastatic OC, we evaluated the presence of reovirus in various tissues collected from the animals treated as per protocol in Figure 3a. As shown in Figure 4a, high levels of reovirus transcripts were detectable in both local ascites and distal tumor cells, especially at 3 days post first injection (d.p.f.i.) and 3 days post last injection (d.p.l.i.). As anticipated, at 10 d.p.l.i. both ascites and tumor showed the lowest titers of reovirus, suggesting a possible immune-mediated elimination of reovirus. Of note, reovirus was also detected in the spleen at 3 d.p.f.i.; however, by 10 d.p.l.i. these levels were almost undetectable. Collectively, these results show that i.p. administered reovirus can successfully reach local and distal OC.
Figure 4.

Initiation of antitumor immunological events. C57BL/6 mice were injected as per protocol shown in Figure 3a, and then killed at the indicated timepoints to obtain respective tissues. These samples were processed, RNA was extracted, purified, and reverse transcribed using random hexamers. (a) Quantitative real-time PCR was conducted with the gene-specific primers for reovirus, (b) MHC class I, β2M, TAP-1, and TAP-2, (c) CD3, CD4, CD8, and IFN-γ, and (d) TNF-α, IL-4, IL-6, and IL-10 followed by analysis using the Livak and Schmittgen's 2-ΔΔCT method.36 Graphs illustrate the linear fold change of the indicated mRNA replicates normalized to GAPDH and PBS control. Statistical analysis was performed with two-tailed, Student's t-test as described for Figure 3; ns = P > 0.05; *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001. Data is representative of three independent experiments. d.p.f.i., days post first injection; d.p.l.i., days post last injection; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; IFN, interferon; IL, interleukin; MHC, major histocompatibility complex; ns, not significant; PBS, phosphate-buffered saline; TNF, tumor necrosis factor.
Suppressive microenvironment hampers the immune-mediated elimination and thus promotes the persistence of OC. Hence, we also analyzed whether reovirus can modulate the composition and phenotype of immune markers in and around the OC milieu. As shown in Figure 4b, reovirus enhances the expression of MHC class I and β2M in ascites as well as distal tumors at all the three timepoints analyzed as compared with the respective tissues collected from PBS-treated PC-bearing animals. In addition, the expression of TAP-1 and TAP-2, molecules involved in antigen processing, was also higher in ascites and tumor samples collected from LRV-treated animals at various timepoints. Thus, similar to our in vitro data, reovirus therapy enhances the expression of molecules involved in antigen processing and display in OC-PC.
Infiltration of OC microenvironment with immune cells, especially CD3+, CD4+ and CD3+, CD8+ T cells, is associated with a positive outcome. Hence, we also evaluated the CD3, CD4, and CD8 expression in the tumor microenvironment. As shown in Figure 4c, the expression of all these markers at 3 d.p.f.i. was unaffected in all three tissues tested, except for an elevated CD8 expression in ascites. At 3 d.p.l.i., however, significantly greater expression of CD3, CD4, and CD8 in ascites and of CD3 in tumor samples was evident following LRV injections as compared to those injected with PBS. Most importantly, at 10 d.p.l.i., highly significant expression of CD3, CD4, and CD8 was observed in distal tumors suggesting that the presence of intratumoral infiltration following a treatment with LRV.
In terms of cytokine response, ascites samples displayed a significantly higher expression of IFN-γ, tumor necrosis factor-α, IL-4, IL-6, IL-10 at all the indicated timepoints, except for IL-10 at 10 d.p.l.i. (Figure 4d). However, for tumor samples, significantly elevated expression was observed only for IFN-γ (Figure 4c) and tumor necrosis factor-α at 3 and 10 d.p.l.i. and for IL-10 at 10 d.p.l.i. Interestingly, spleen samples showed exclusive upregulation of IFN-γ expression at all the post-LRV timepoints as compared with that of PBS-treated sample. Taken together, our data shows the capacity of reovirus to drive beneficial Th1 cytokine response in the tumor microenvironment.
Reovirus modulates OC-PC–associated MDSCs and Tregs
Since a poor clinical outcome of OC often correlates with higher number of MDSCs12,24 and Tregs,13 we analyzed whether reovirus therapy can modulate the frequencies of these suppressor cells in the animals injected as per Figure 3a. As shown, PBS-treated OC-PC–bearing animals displayed significantly greater number of either Gr1.1+, CD11b+ (MDSCs) cells in spleen and ascetic fluid (Figure 5a) or CD4+, CD25+, FOXP3+ (Tregs) in the spleen and mesenteric lymph node (Figure 5b) as compared with naive, nontumor-bearing animals. Surprisingly, at 3 d.p.f.i., animals injected with LRV showed even higher numbers of both cell types which eventually decreased at 3 d.p.l.i. in respective organs. Finally, at 10 d.p.l.i., the numbers of both MDSCs and Tregs were significantly lower as compared with those observed in PBS-treated or in the LRV-treated OC-PC–bearing animals at 3 d.p.f.i.
Figure 5.
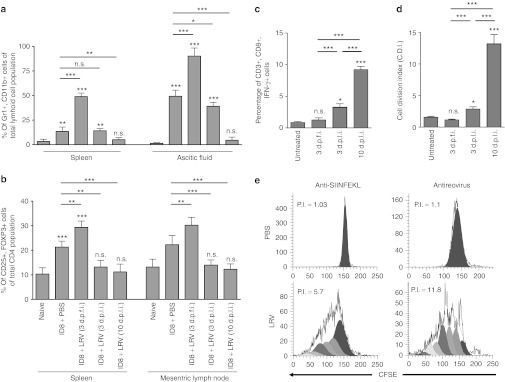
Modulation of Tregs and MDSCs during therapeutic targeting of OC-PC. Female C57BL/6 mice were injected with ID8 cells and a regimen of PBS/LRV as per protocol shown in Figure 3a. Animals were killed on the respective days as indicated to obtain single cell suspensions from spleen, MLN or ascetic fluid. These cells were directly stained with either (a) Gr1/CD11b or (b) CD4/CD25/FOXP3 and analyzed using flow cytometry. (c,d) Alternatively, ID8-ova cells were used to initiate OC-PC. These animals were treated with a regimen of PBS/LRV as per protocol shown in Figure 3a. Splenocytes obtained from these animals, on respective days as indicated, were stimulated with SIINFEKL peptide and (c) analyzed for the ability of CD3+,CD8+ cells to produce IFN-γ through intracellular staining or the (d) capacity of CD3+ cells to proliferate in CFSE assay. Statistical analysis was performed as explained for Figure 3d; ns = P > 0.05; *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001. (e) On 10 d.p.l.i., splenocytes were also stimulated with UVRV-pulsed BMDCs and assayed in CFSE-based flow cytometric assay to monitor for antireoviral T-cell proliferation. The representative histograms show profiles of antitumor and antireoviral T-cell response, with proliferative index (PI) as a measure of activation as calculated using ModFit LT computer algorithm. All the results are representative of atleast five independent experiments. BMDC, bone marrow-derived DCs; DC, dendritic cell; d.p.f.i., days post first injection; d.p.l.i., days post last injection; IFN, interferon; LRV, live reovirus; MDSC, myeloid derived suppressor cell; MLN, mesenteric lymph node; ns, not significant; OC, ovarian cancer; PBS, phosphate-buffered saline; PC, peritoneal carcinomatosis.
To further evaluate the effects of suppressor cell modulation on tumor-specific immunity, animals were injected with ID8-ova (as per Figure 3a) and analyzed for anti-ova immune response as a surrogate measure of antitumor immunity.20 Our data showed that lymphocytes collected from the PBS (control)-treated animals failed to produce IFN-γ (Figure 5c) or proliferate (Figure 5d) in response to SIINFEKL stimulation, suggesting the absence of antitumor immune response. The lymphocytes were still nonresponsive at 3 d.p.f.i. and only weakly responsive at 3 d.p.l.i. However at 10 d.p.l.i., splenocytes from LRV-treated animals displayed high proliferative index (also shown in a representative dot plot in Figure 5e) and IFN-γ production. Of note, at 10 d.p.l.i., splenocytes also showed a strong antireoviral T-cell proliferative response (Figure 5e). Taken together, our data showed that reovirus can modulate the frequencies of MDSCs and Tregs preceding the initiation of antitumor immunity in OC-PC–bearing hosts.
Reovirus oncotherapy during early stages of OC postpones the development of PC
Finally, we also evaluated the applicability of reovirus-based oncotherapy for the treatment of early stage OC. For this, C57BL/6 female mice were injected with ID8 cells, administered with PBS/UVRV/LRV therapeutic regimen starting at 7 d.p.i. as per Figure 6a and then monitored for ascites development and survival. As shown in Figure 6b, ID8-implated animals treated with LRV developed ascites at significantly later timepoints as compared to those treated with PBS or UVRV. This delayed PC development and was also associated with significantly greater survival (Figure 6c) in LRV-treated animals. Thus, our data suggests that reovirus-based treatment can be implemented during early stages of OC to delay PC development and achieve increased survival.
Figure 6.
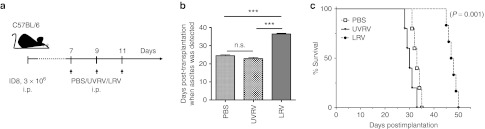
Use of reovirus-based therapy for the prevention of PC development. (a) Female C57BL/6 mice were implanted with 3 × 106 ID8 cells on day 0 and then injected with a regimen of PBS/UVRV/LRV injections as shown in the schematic. The animals were monitored for the development of (b) ascites and (c) survival. The graphs show the cumulative data on the average days post-tumor implantation when ascites was first detected in respective groups of animals. Statistical analysis was performed through intergroup comparison using Student's t-tests at 95% CI; ns = P > 0.05; *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001. CI, confidence interval; i.p., intraperitoneally; LRV, live reovirus; ns, not significant; PBS, phosphate-buffered saline; PC, peritoneal carcinomatosis; UVRV, UV-inactivated reovirus.
Discussion
Immunosuppressive OC microenvironment hinders therapeutic targeting of OC as it fosters metastasis and protects against immunity-mediated elimination. In this report, we demonstrate that reovirus-based anti-OC therapy mitigates the detrimental effects of OC microenvironment and successfully targets OC. We show that reovirus (i) efficiently targets local as well as metastatic OC cells; (ii) overturns immune evasion mechanisms and enhance immune recognition of cancer cells; (iii) modulates the frequencies of suppressive immune cells and aids in the development of antitumor immunity; and (iv) prevents the development of PC or targets already developed peritoneal disease to promote better outcomes from OC.
Antitumor T-cell immunity dictates the outcome of OC.2,14 The higher numbers of tumor-infiltrating CD3+ T cells and IFN-γ25 expression are associated with improved survival in OC patients26 and represent independent predictors of better prognosis in multivariate analysis.27,28 We observed that reovirus initiated highly significant expression of CD3 at 3 and 10 d.p.l.i. in both ascites and distal tumor. Most importantly, at 10 d.p.l.i, spleen as well as tumor samples from LRV-treated animals displayed elevated expression of CD8, suggesting that both intratumoral and peripheral responses bore similar kinetics and that the tumors were infiltrated with CD8+ T cells. Interestingly, significantly higher IFN-γ expression was observed at 3 and 10 d.p.l.i., suggesting the capacity of LRV to drive beneficial Th1 cytokine response.29 On the other hand, these findings also suggest the role of non-CD4+, non-CD8+ immune cells (e.g., natural killer and natural killer T cells) in mounting an IFN-γ immune response. Together, it can be surmised that reovirus therapy initiates a clinically desired T cell and IFN-γ immune response.
Regulatory cells such as Gr1.1+, CD11b+ MDSCs or CD4+,CD25+,FOXP3+ Tregs foster the suppressive milieu and inhibit antitumor immunity. Patients with late stages of OC display higher frequencies of Tregs in both tumor mass and ascites.13 Similarly, greater numbers of Gr1.1+, CD11b+ cells are detected in humans24 and mice12 with OC. Our results concur with these reports as we also observed the higher numbers of Tregs in the spleen and mesenteric lymph nodes as well as MDSCs in the spleen and ascites of OC-PC–bearing animals. Presence of both these major suppressive cells correlates with poor prognosis and higher mortality rates.13 Not surprisingly, many scientific initiatives in recent times are focused on the options that can alleviate the burden of these suppressive cells. Interestingly, we observed a spike in the numbers of both MDSCs and Tregs immediately following reovirus injection. We believe that this mechanism is essential to minimize the possible “collateral damage” for the host arising from antiviral immune response. Nonetheless, by 10 d.p.l.i. the frequency of these suppressive cells significantly decreased, and almost returned to the levels as those observed in naive animals. These findings suggest that reovirus therapy can alleviate the burden of MDSCs and Tregs. It should be noted that the downregulation of these cells was also accompanied by the development of antitumor T-cell response in spleen, as well as significantly high expression of CD4, CD8, and IFN-γ in tumor. Collectively, results show that reovirus successfully ablates the numbers, and possibly inhibitory functions, of Tregs and MDSCs preceding the development of antitumor immune response.
Evasion from antigen presentation is the central mechanism through which OC avoids immune recognition. In OC patients, higher levels of MHC class I are associated with improved treatment responsiveness and survival, while low level of HLA class I expression is an independent indicator of poor prognosis.10 In the absence of appropriate antigen processing and presentation, tumor-specific T cells fail to identify and subsequently kill OC cells.11 To achieve maximum benefits of immunological interventions, it is essential to aim for strategies that can improve the recognition of tumor cells. Our data shows that reovirus therapy increases the expression of MHC class I, β2M, TAP-1, and TAP-2 in ID8 cells in vitro as well as in tumor and ascites in vivo and inadvertently achieves such a function. It should be noted that LRV also promotes DC-mediated tumor antigen presentation to CD8+ T cells. These results support our hypothesis that reovirus overcomes tumor-associated antigen presentation dysfunction.
Peritoneally administered reovirus was able to target metastatic tumor sites as we detected reovirus in both distal tumors and ascites. As anticipated however, the levels of reovirus decreased over time, and were accompanied by a strong antireoviral T-cell response in the periphery. It is now acknowledged that antiviral immunity adversely affects the efficacy of oncolysis by prematurely eliminating reovirus from the host. Paradoxically however, this antiviral response also performs antitumor activities. Reovirus preferentially infects tumors, and hence antiviral immune response aimed at virally infected cells, indirectly and inadvertently targets tumor cells. Thus, even though antiviral immune response works against reovirus replication and spread, it performs beneficial antitumor activities. Indeed, the host immune responses are shown to be essential for the efficient implementation of reovirus therapy.30
In conclusion, our study demonstrates the applicability of reovirus therapy for the treatment of OC and associated PC. Most importantly, we demonstrate a therapeutic option that initiates multi-prong attack on OC which can be implemented under clinical situations to achieve better outcomes for patients suffering with OC. It should be noted that, apart from the direct anticancer activities, therapy-modulated tumor microenvironment can also encourage simultaneous implementation of the other complementary immunotherapies to further potentiate better outcomes from OC. Thus, an appropriately implemented reovirus anti-OC therapy represents a powerful treatment option for the management of early as well as advanced stages of OC.
Materials and Methods:
Reovirus, cell lines, and reagents. Reovirus (serotype 3, Dearing strain) was grown and purified as per established protocol.15 Mouse epithelial OC ID8 cell line31 was obtained from Edith Lord (University of Rochester, Rochester, NY)32 and cultured in RPMI with 5% (vol/vol) Glutamax, 10 % fetal bovine serum, 1X sodium pyruvate, 1X nonessential amino acids, and 1X Anti-Anti (Invitrogen, Carlsbad, CA). B3Z23 cell line was kindly provided by Nilabh Shastri, University of California, Berkely, CA and cultured as per published protocol. Antibodies and peptides were purchased from respective vendors as follows: eBioscience (San Diego, CA): APC-anti-Mouse MHC Class I molecule Kb (AF6-88.5.5.3), unconjugated anti-Mouse CD3e (145-2C11), unconjugated anti-Mouse CD28 (37.51), Alexa 488 anti-CD11c (N418), PE-Cy5-anti-Mouse MHC Class II (I-A/I-E) (M5/114.15.2), PE-anti-Mouse CD86 (B7-2) (GL1), APC-anti-Mouse CD80 (B7-1) (16-10A1), APC-anti-Mouse CD40 (1C10), APC-anti-Mouse Ly-6G (Gr-1) (RB6-8C5), PerCP-Cy5.5-anti-Mouse CD11b (M1/70), FITC-anti-Mouse CD4 (RM4-5), PerCP-Cy5.5-anti-Mouse CD25 (PC61.5), PE-anti-Mouse/Rat FoxP3 (FJK-16S), PE-anti-Mouse CD3e (eBio500A2), APC-anti-Mouse IFNγ (XMG1.2); Invitrogen: Alexa 488-Annexin V, 5- (and -6)-carboxyfluorescein diacetate (CFSE); BD Biosciences (Mississauga, Ontario, Canada): 7-Amino-Actinomycin D, PerCP-Rat anti-Mouse CD8a (53-6.7); Jackson ImmunoResearch LaboRatories (West Grove, PA): Cy2-goat anti-rabbit. GenScript (Piscataway, NJ): Ovalbumin peptide- SIINFEKL (ova257-264) and Control LCMV peptide-KAVYNFATM (LCMV gp33–41).
In vivo experimental manipulations. All experimental procedures performed during this study were governed by the approval of Ethics Committee at Dalhousie University, Halifax, Nova Scotia, Canada. Six to eight weeks old female WT C57BL/6 mice were obtained from Charles River Laboratory (Montreal, Quebec, Canada). Mice were injected according to protocols shown in respective figures.
Lymphocyte isolation and BMDC generation. Lymphocytes and BMDCs were generated as described previously.17,33 For lymphocytes, single cell suspension of splenocytes was treated with RBC-lysing ammonium chloride (ACK) buffer, washed and verified for cell viability. For BMDCs, BM hematopoietic progenitor cells obtained from tibia and femur bones were treated with ACK buffer, washed and cultured in the presence of complete RPMI 1640 containing with 10% vol/vol FCS, 2 mmol/l glutamine, 0.1 mmol/l nonessential amino acids, 50 U/ml penicillin/streptomycin, 0.1 mmol/l 2–ME (all obtained from Invitrogen) and supplemented with 20 ng/ml granulocyte-macrophage colony-stimulating factor (Shenandoah Biotech, Warwick, PA) and 10 ng/ml IL-4 (Shenandoah Biotech) for 6–8 days.
Tumor antigen presentation assay. B3Z assay was used to evaluate the ability of BMDCs to present surrogate TAA ovalbumin (ova) to TAA-specific CD8+ T cells as previously described.20,23,34 Ova–specific CD8+ T–cell hybridoma B3Z expresses β–galactosidase following recognition of ova immunodominant peptide (SIINFEKL) in the context of H2–Kb and catalyses the breakdown of β-gal substrate, e.g., chlorophenol red-β-D-galactopyranoside (CPRG). For this assay, 2 × 105 BMDCs were co-cultured with 2 x 105 ID8-ova cells and added with LRV 24 hr. Next, co-cultures were washed, added with 1 × 105 B3Z cells per well, incubated for an additional 18–24 hours and then added with 0.15 mmol/l of CPRG for an additional 4 hours. The breakdown of CPRG was read at 570 nm and used as a measure of CD8+ T-cell activation.
T-cell functional assay. T cell activation was visualized and quantified with 5–(and–6)carboxyfluorescein diacetate succinimidyl ester (CFSE)-based cell proliferation assay as previously described.20,35 Briefly, CFSE-labeled splenocytes were cultured in the presence of SIINFEKL or control peptide and then monitored in flow cytometry for cell division through halving of CFSE fluorescence. The CFSE fluorescence halving was deconvoluted using CellQuest Pro software (Becton Dickinson, Franklin Lakes, NJ), FCS express or ModFit LT softwares as described previously.35 Cell division index was defined by dividing the percentage of cells with halved CFSE fluorescence after stimulation with an antigen by percentage of cells with halved CFSE-fluorescence cultured in medium only. Proliferation index was obtained through computerized algorithm in ModFit LT software (Verity Software House, Topsham, ME) as per manufacturer's instructions.
Quantitative real-time PCR. The RNA extractions were conducted via Trizol methodology and cDNA was synthesized using enzyme Supercript II. Each sample of cDNA was quantitated and diluted to a similar concentration of 10 µg/ml. The Stratagene MX3000P PCR machine was used for the quantitative real-time PCR, using GoTaq qPCR Master mix (Promega, Madison, WI) for amplification and quantification. All primers, as described in Supplementary Table S1 used were purchased from Invitrogen. GAPDH was used for normalization of the genes of interest. The results were collected and analyzed using Livak and Schmittgen's 2-ΔΔCT method.36 To calculate fold change, signals were first normalized against GAPDH and then compared against respective PBS-treated controls.
Flow cytometry and statistical analysis. Data was acquired with FACSCalibur flow cytometer (BD Biosciences) and analyzed using CellQuest Pro (BD Biosciences, San Jose, CA) and FCS Express V3 (De Novo Software, Los Angeles, CA) softwares. ModFit LT (Verity Software House) was also used for deconvolution of CFSE florescence and estimation of proliferation index. For statistical analysis, two-tailed, Student's t-test with 95% confidence interval was used and P values of <0.05 were considered as significant. Asterisks were used to denote P values as: not significant = P > 0.05; *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001. Survival rates were measured with Kaplan–Meier survival analysis with 95% confidence interval coupled with log-rank test, and the difference between survival curves of different populations was considered when P values of ≤0.05 were observed.
SUPPLEMENTARY MATERIAL Table S1. List of gene-specific primer sequences used for quantitative real-time PCR.
Acknowledgments
This work was supported by an operating grant from the Canadian Institutes for Health Research to P.W.K.L. D.C. and E.H. received the Norah Stephen Summer Studentship, while D.-G.A. holds the postdoctoral fellowship from the Beatrice Hunter Cancer Research Institute. S.G. is funded by the Canadian Institutes for Health Research postdoctoral fellowship. The authors declared no conflict of interest.
Supplementary Material
List of gene-specific primer sequences used for quantitative real-time PCR.
REFERENCES
- Cannon MJ., and, O'Brien TJ. Cellular immunotherapy for ovarian cancer. Expert Opin Biol Ther. 2009;9:677–688. doi: 10.1517/14712590902932897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson BH. The impact of T-cell immunity on ovarian cancer outcomes. Immunol Rev. 2008;222:101–116. doi: 10.1111/j.1600-065X.2008.00614.x. [DOI] [PubMed] [Google Scholar]
- Perren TJ, Swart AM, Pfisterer J, Ledermann JA, Pujade-Lauraine E, Kristensen G.et al. (2011A phase 3 trial of bevacizumab in ovarian cancer N Engl J Med 3652484–2496. [DOI] [PubMed] [Google Scholar]
- Barnett B, Kryczek I, Cheng P, Zou W., and, Curiel TJ. Regulatory T cells in ovarian cancer: biology and therapeutic potential. Am J Reprod Immunol. 2005;54:369–377. doi: 10.1111/j.1600-0897.2005.00330.x. [DOI] [PubMed] [Google Scholar]
- Thibodeaux SR., and, Curiel TJ. Immune therapy for ovarian cancer: promise and pitfalls. Int Rev Immunol. 2011;30:102–119. doi: 10.3109/08830185.2011.567361. [DOI] [PubMed] [Google Scholar]
- Yigit R, Massuger LF, Figdor CG., and, Torensma R. Ovarian cancer creates a suppressive microenvironment to escape immune elimination. Gynecol Oncol. 2010;117:366–372. doi: 10.1016/j.ygyno.2010.01.019. [DOI] [PubMed] [Google Scholar]
- Shen GH, Ghazizadeh M, Kawanami O, Shimizu H, Jin E, Araki T.et al. (2000Prognostic significance of vascular endothelial growth factor expression in human ovarian carcinoma Br J Cancer 83196–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou W, Machelon V, Coulomb-L'Hermin A, Borvak J, Nome F, Isaeva T.et al. (2001Stromal-derived factor-1 in human tumors recruits and alters the function of plasmacytoid precursor dendritic cells Nat Med 71339–1346. [DOI] [PubMed] [Google Scholar]
- Vitale M, Pelusi G, Taroni B, Gobbi G, Micheloni C, Rezzani R.et al. (2005HLA class I antigen down-regulation in primary ovary carcinoma lesions: association with disease stage Clin Cancer Res 1167–72. [PubMed] [Google Scholar]
- Shehata M, Mukherjee A, Deen S, Al-Attar A, Durrant LG., and, Chan S. Human leukocyte antigen class I expression is an independent prognostic factor in advanced ovarian cancer resistant to first-line platinum chemotherapy. Br J Cancer. 2009;101:1321–1328. doi: 10.1038/sj.bjc.6605315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han LY, Fletcher MS, Urbauer DL, Mueller P, Landen CN, Kamat AA.et al. (2008HLA class I antigen processing machinery component expression and intratumoral T-Cell infiltrate as independent prognostic markers in ovarian carcinoma Clin Cancer Res 143372–3379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang R, Cai Z, Zhang Y, Yutzy WH, 4th, Roby KF., and, Roden RB. CD80 in immune suppression by mouse ovarian carcinoma-associated Gr-1+CD11b+ myeloid cells. Cancer Res. 2006;66:6807–6815. doi: 10.1158/0008-5472.CAN-05-3755. [DOI] [PubMed] [Google Scholar]
- Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P.et al. (2004Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival Nat Med 10942–949. [DOI] [PubMed] [Google Scholar]
- Gavalas NG, Karadimou A, Dimopoulos MA., and, Bamias A. Immune response in ovarian cancer: how is the immune system involved in prognosis and therapy: potential for treatment utilization. Clin Dev Immunol. 2010;2010:791603. doi: 10.1155/2010/791603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffey MC, Strong JE, Forsyth PA., and, Lee PW. Reovirus therapy of tumors with activated Ras pathway. Science. 1998;282:1332–1334. doi: 10.1126/science.282.5392.1332. [DOI] [PubMed] [Google Scholar]
- Norman KL., and, Lee PW. Reovirus as a novel oncolytic agent. J Clin Invest. 2000;105:1035–1038. doi: 10.1172/JCI9871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gujar SA, Pan DA, Marcato P, Garant KA., and, Lee PW. Oncolytic virus-initiated protective immunity against prostate cancer. Mol Ther. 2011;19:797–804. doi: 10.1038/mt.2010.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prestwich RJ, Ilett EJ, Errington F, Diaz RM, Steele LP, Kottke T.et al. (2009Immune-mediated antitumor activity of reovirus is required for therapy and is independent of direct viral oncolysis and replication Clin Cancer Res 154374–4381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Errington F, Steele L, Prestwich R, Harrington KJ, Pandha HS, Vidal L.et al. (2008Reovirus activates human dendritic cells to promote innate antitumor immunity J Immunol 1806018–6026. [DOI] [PubMed] [Google Scholar]
- Gujar SA, Marcato P, Pan D., and, Lee PW. Reovirus virotherapy overrides tumor antigen presentation evasion and promotes protective antitumor immunity. Mol Cancer Ther. 2010;9:2924–2933. doi: 10.1158/1535-7163.MCT-10-0590. [DOI] [PubMed] [Google Scholar]
- Hirasawa K, Nishikawa SG, Norman KL, Alain T, Kossakowska A., and, Lee PW. Oncolytic reovirus against ovarian and colon cancer. Cancer Res. 2002;62:1696–1701. [PubMed] [Google Scholar]
- Scarlett UK, Cubillos-Ruiz JR, Nesbeth YC, Martinez DG, Engle X, Gewirtz AT.et al. (2009In situ stimulation of CD40 and Toll-like receptor 3 transforms ovarian cancer-infiltrating dendritic cells from immunosuppressive to immunostimulatory cells Cancer Res 697329–7337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson S., and, Shastri N. LacZ inducible, antigen/MHC-specific T cell hybrids. Int Immunol. 1994;6:369–376. doi: 10.1093/intimm/6.3.369. [DOI] [PubMed] [Google Scholar]
- Tomihara K, Guo M, Shin T, Sun X, Ludwig SM, Brumlik MJ.et al. (2010Antigen-specific immunity and cross-priming by epithelial ovarian carcinoma-induced CD11b(+)Gr-1(+) cells J Immunol 1846151–6160. [DOI] [PubMed] [Google Scholar]
- Marth C, Fiegl H, Zeimet AG, Müller-Holzner E, Deibl M, Doppler W.et al. (2004Interferon-gamma expression is an independent prognostic factor in ovarian cancer Am J Obstet Gynecol 1911598–1605. [DOI] [PubMed] [Google Scholar]
- Zhang L, Conejo-Garcia JR, Katsaros D, Gimotty PA, Massobrio M, Regnani G.et al. (2003Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer N Engl J Med 348203–213. [DOI] [PubMed] [Google Scholar]
- Leffers N, Gooden MJ, de Jong RA, Hoogeboom BN, ten Hoor KA, Hollema H.et al. (2009Prognostic significance of tumor-infiltrating T-lymphocytes in primary and metastatic lesions of advanced stage ovarian cancer Cancer Immunol Immunother 58449–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomsová M, Melichar B, Sedláková I., and, Steiner I. Prognostic significance of CD3+ tumor-infiltrating lymphocytes in ovarian carcinoma. Gynecol Oncol. 2008;108:415–420. doi: 10.1016/j.ygyno.2007.10.016. [DOI] [PubMed] [Google Scholar]
- Kusuda T, Shigemasa K, Arihiro K, Fujii T, Nagai N., and, Ohama K. Relative expression levels of Th1 and Th2 cytokine mRNA are independent prognostic factors in patients with ovarian cancer. Oncol Rep. 2005;13:1153–1158. [PubMed] [Google Scholar]
- Prestwich RJ, Ilett EJ, Errington F, Diaz RM, Steele LP, Kottke T.et al. (2009Immune-mediated antitumor activity of reovirus is required for therapy and is independent of direct viral oncolysis and replication Clin Cancer Res 154374–4381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roby KF, Taylor CC, Sweetwood JP, Cheng Y, Pace JL, Tawfik O.et al. (2000Development of a syngeneic mouse model for events related to ovarian cancer Carcinogenesis 21585–591. [DOI] [PubMed] [Google Scholar]
- Sorensen EW, Gerber SA, Sedlacek AL, Rybalko VY, Chan WM., and, Lord EM. Omental immune aggregates and tumor metastasis within the peritoneal cavity. Immunol Res. 2009;45:185–194. doi: 10.1007/s12026-009-8100-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gujar SA, Jenkins AK, Macparland SA., and, Michalak TI. Pre-acute hepadnaviral infection is associated with activation-induced apoptotic death of lymphocytes in the woodchuck (Marmota monax) model of hepatitis B. Dev Comp Immunol. 2010;34:999–1008. doi: 10.1016/j.dci.2010.05.001. [DOI] [PubMed] [Google Scholar]
- Sanderson S, Campbell DJ., and, Shastri N. Identification of a CD4+ T cell-stimulating antigen of pathogenic bacteria by expression cloning. J Exp Med. 1995;182:1751–1757. doi: 10.1084/jem.182.6.1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gujar SA., and, Michalak TI. Flow cytometric quantification of T cell proliferation and division kinetics in woodchuck model of hepatitis B. Immunol Invest. 2005;34:215–236. [PubMed] [Google Scholar]
- Livak KJ., and, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
List of gene-specific primer sequences used for quantitative real-time PCR.