

Abstract
Hematopoietic stem cells (HSC) are found in several independent sites embryonically. Loss-of-function studies indicated that Notch1, but not Notch2 signaling was required for HSC emergence from the aortic-gonado-mesonephros (AGM) region. We previously showed that constitutive Notch1 activation impaired primitive erythroid differentiation, but its effects on HSC emergence from the AGM region were not studied. To further define specific roles of Notch receptors, we characterized HSC in mouse embryos expressing either Notch1 intracellular domain (ICD) or Notch4ICD in VE-cadherin or SM22α expressing populations. Although embryonic Notch1 activation in VE-cadherin populations led to lethality after E13.5, earlier defects in the fetal liver were observed. Embryos were analyzed at E12.5 to assess hematopoiesis and the phenotype of developing cells in the AGM region. We found that activation of Notch1 in the endothelial compartment in VE-cadherin expressing cells resulted in the absence of intra-aortic clusters and defects in fetal liver hematopoiesis. In contrast, although Notch4 expression is regulated during fetal hematopoiesis, activation of Notch4 in VE-cadherin expressing populations did not affect HSC phenotype, although later vascular remodeling was impaired. Likewise, activation of Notch1 in SM22α positive populations had no significant effect on hematopoiesis. Our results indicate a cell type-dependent activity and distinct features of Notch1 versus Notch4 signaling and their impact on HSC generation.
Keywords: Hematopoietic stem cell, Notch signaling, VE-cadherin, Embryonic hematopoiesis
Introduction
Hematopoietic stem cells (HSC) are found in several embryonic sites. Precisely how the first HSC are generated in the vertebrate embryo has been a matter of controversy. HSC originate within the aortic-gonado-mesonephros (AGM) region of the mouse embryo between E10.5 and E12.5 in clusters that can be morphologically detected in vivo (Dzierzak and Speck 2008; Medvinsky et al. 2011) and tracked in explant culture of AGM (Boisset et al. 2010). One source of HSC in the developing embryo is thought to be embryonic endothelium (Zovein et al. 2008; Chen et al. 2009; Bertrand et al. 2005; Kissa and Herbomel 2010), either directly, or via a transition from a hematopoietically committed pre-HSC into a mature HSC within the endothelial lining (Medvinsky et al. 2011). Recently, several studies highlighted the endothelial origins of HSC emergence (Boisset et al. 2010; Zovein et al. 2008; Chen et al. 2009; Kissa and Herbomel 2010; Eilken et al. 2009; Lancrin et al. 2009) and identified genes regulating this process. VE-cadherin-Cre transgenic mice were used to trace HSC emergence and to study the role of Runx1 in HSC emergence (Zovein et al. 2008; Chen et al. 2009). Fate tracing using SM22α-Cre transgenic mice indicated that mesenchyme can contribute to HSC emergence, but via an endothelial intermediate (Zovein et al. 2008). The endothelial originated HSC and progeny migrate to the fetal liver, and later to the bone marrow, and are capable of self-renewal and differentiation.
The Notch signaling pathway is a conserved intercellular signaling mechanism that regulates cell fate and tissue development (Artavanis-Tsakonas et al. 1999). There are four vertebrate members of this family, Notch1 through Notch4 (Artavanis-Tsakonas et al. 1999). By mouse embryonic day E8.5, transcript levels of Notch1 are highest in the presomitic mesoderm, mesenchyme, and endothelial cells, and lower levels are present in the neuroepithelium (Del Amo et al. 1992). Notch1 is expressed at E9.5–E10.5 in both endothelial cells and mesenchymal cells in the dorsal aorta (Del Amo et al. 1992; Reaume et al. 1992). Among the Notch family proteins, Notch1 and Notch2 are expressed in hematopoietic progenitor cells (Lardelli and Lendahl 1993). Multiple defects exhibited by Notch1-deficient embryos confound the determination of Notch1 function in HSC in vivo, however, ex vivo culture of Notch1 mutant embryos showed a requirement of Notch1 for HSC emergence. Notch1-deficient mouse embryonic stem cells are impaired in the ability to establish long-term, definitive HSC (Hadland et al. 2004). We previously demonstrated that the Notch1 activation in Tie2-expressing cell lineages resulted in early embryonic lethality due to cardiovascular and hematopoietic defects. To extend our studies and address HSC development, the VE-Cadherin Cre strain was used to activate our Notch1ICD transgenic allele. Notch1 and Notch4 have both been implicated in vascular morphogenesis, especially in angiogenesis (Krebs et al. 2000), and Notch4 is expressed in endothelial cells, but not in hematopoietic progenitor cells (Krebs et al. 2000). However, specific functions of Notch4 in HSC development are not understood. Therefore, we also utilized a Notch4ICD gain-of-function allele to compare with the phenotype of Notch1 activation. Our results show that imbalance of Notch1 signaling in endothelial/hematopoietic progenitors was sufficient to block HSC emergence or functional development. In contrast, activation of Notch4 in endothelial/hematopoietic progenitors, or activation of Notch1 in SM22α populations had no effect on HSC. Our data indicate that specific balance of Notch1 signaling in endothelial and hematopoietic progenitors is critical for HSC phenotype, and Notch activities are receptor dependent and cell type dependent.
Materials and methods
Mouse transgenic strains
All experimental protocols involving mice were approved by the Institutional Animal Care and Use Committee at Maine Medical Center. The Notch1ICD-transgenic mice have been previously characterized (Venkatesh et al. 2008). VE-cadherin-Cre mice (B6.Cg-Tg(Cdh5-Cre)7Mlia/J, stock 006137) and Rosa26R-EYFP mice (B6.129X1-Gt(ROSA)26Sortm1(EYFP)Cos/J, stock 006148) were obtained from the Jackson Laboratory. Notch1ICD transgenic mice were bred with Rosa26R-EYFP mice to get double heterozygous transgenic Notch1ICD; Rosa26R-EYFP mice. Double heterozygous mice were bred with Rosa26R-EYFP mice to get female double transgenic mice with heterozygous Notch1ICD and homozygous Rosa26R-EYFP. These mice were bred with male VE-cadherin-Cre mice to get triple transgenic mice, and littermates were used for all comparisons. For each genotype, n = 8 or more embryos for the analysis. The average number of EYFP positive cells was 0.5 ± 0.21. The same breeding strategy was used for Notch4ICD transgenic mice.
Flow cytometric analysis
Flow cytometry was performed in a FACScalibur with the Cellquest program (Becton–Dickinson). For surface staining, cell suspensions from individual fetal liver were incubated on ice in the presence of various mixtures of labeled mAb. APC-conjugated Ter119 and PE-Cy7-conjugated CD45 were used to examine lineage contribution. All antibodies were purchased from eBioscience.
Results
Notch1 activation inhibits HSC generation from endothelial cells
The HSC of the AGM are incapable of in situ hematopoietic differentiation (Godin et al. 1999), and require migration to the fetal liver and later to bone marrow for differentiation and self-renewal (Delassus and Cumano 1996). VE-cadherin is a marker for AGM-derived hematopoietic cells (Nishikawa et al. 1998), and the VE-Cadherin-Cre transgenic strain is commonly used to target hematopoietic and endothelial cell populations (Zovein et al. 2008; Chen et al. 2009). We previously showed that Notch1 activation impaired primitive erythroid differentiation (Venkatesh et al. 2008). However, little is known about the role of Notch1 activation in HSC development from endothelial cells because of the early embryonic lethality when Notch1ICD is expressed in Tie2-expressing lineages. To study this, we intercrossed Rosa26R-EYFP with floxed Notch1ICD transgenic mice to get double transgenic Notch1ICD; Rosa26R-EYFP mice. These double transgenic mice were intercrossed with VE-Cadherin-Cre mice to activate the EYFP and NotchICD transgenes (Fig. 1a). Notably, VE-cadherin-Cre does label a sub-set of the population of hematopoietic cells in the fetal liver (Fig. 1b) and adult bone marrow (data not shown), which support the endothelial origins of HSC. To investigate the role of Notch activation in HSC generation from endothelial cells, we first studied adult bone marrow development since the endothelial-derived HSC and progeny migrate to the fetal liver, and later to the bone marrow, and are capable of self-renewal and differentiation. We were unable to derive triple transgenic mice when Notch1ICD; Rosa26R-EYFP double transgenic mice were intercrossed to VE-Cadherin-Cre mice, which suggested embryonic lethality following Notch1 activation in endothelial cells and/or hematopoietic populations.
Fig 1.

Notch1 activation blocks hematopoietic stem cell emergence from endothelial cells. a Gross appearance of surviving N1ICD;VE-cadherin-Cre; EYFP transgenic embryos and a littermate control (VE-cadherin-Cre;EYFP) at E12.5. The fetal liver is indicated by arrow. b Analysis of EYFP+ cells by flow cytometry in the fetal liver obtained from E12.5 EYFP single positive mice (SP), VE-cadherin-Cre; EYFP double positive (DP), and N1ICD; VE-cadherin-Cre; EYFP triple positive transgenic embryos (TP). c Quantitative analysis of EYFP+ cells from fetal liver at E12.5 of each genotype. Bars represent the mean ± standard deviation. d Representative flow cytometric analysis of single cells from fetal liver to detect EYFP and CD45 (top) or EYFP and Ter119 (bottom). e Quantitative analysis of the percentage of double positive EYFP/CD45 or EYFP/Ter119 for each genotype. Bars represent mean ± standard deviation. f–g Representative histological sections through aortae from E10.5 VE-cadherin-Cre; EYFP embryos (f) compared to N1ICD; VE-cadherin-Cre; EYFP (g) embryos. Sections were stained with hematoxylin/eosin. Arrow in f shows a hematopoietic cluster on the ventral wall of the dorsal aorta
To determine when the Notch1ICD gain-of-function embryos were dying, we isolated them from timed matings. When isolated at E12.5, the Notch1ICD gain-of-function embryos could be identified easily due to an abnormal fetal liver (Fig. 1a). When isolated at E13.5 or later, the Notch1ICD transgenic embryos had severe defects or had already died. Thus, the fetal liver at E12.5 was collected to analyze the result of Notch activation in fetal liver hematopoiesis. FACS analysis of fetal liver at E12.5 indicated that about 10 % (10.1 ± 2.4, n = 8) were EYFP+ in VE-cadherin; EYFP double transgenic embryos, and about 0.4 % (0.4 ± 0.3, n = 10) were EYFP+ in single transgenic embryos, which could be explained by the cellular auto-fluorescence. Surprisingly, we did not observe a significant increase in the EYFP+ population in triple transgenic embryos compared with single transgenic embryos (Fig. 1b). Staining of the fetal liver with CD45 or Ter119 showed that 2 % of the population expressed both CD45 and EYFP, and 8 % expressed both Ter119 and EYFP in the double transgenic embryos (Fig. 1d, e). There was no significant change in the proportion of EYFP positive cells expressing CD45 or TER119 in the triple positive (TP) embryos compared to the single positive embryos (SP). These data suggest that Notch1 activation in endothelial cells significantly inhibited HSC emergence or subsequent differentiation and proliferation during fetal liver hematopoiesis.
We next examined the presence and number of aortic clusters in E10.5 embryos of each genotype. Aortic clusters are morphologically defined cellular structures found intraluminally attached to the ventral floor of the dorsal aorta in the AGM region. These clusters contain definitive hematopoietic progenitor activity. We did not observe any aortic clusters in Notch1ICD conditional transgenic embryos at E10.5, while 5–6 aortic clusters per embryo were identified in littermate control embryos (Fig. 1f, g). The lack of aortic clusters in embryos with Notch1 activation further supports an inhibition of HSC emergence when Notch is activated in the endothelial population.
Notch4 activation in endothelial cells does not affect HSC
Notch4 is specifically expressed in endothelial cells, yet the Notch4 null mice have normal developmental angiogenesis (Krebs et al. 2000). However, the compound Notch1 and Notch4 null has a more severe phenotype compared with Notch1 null mice alone (Krebs et al. 2000), suggesting that Notch1 and Notch4 have non-redundant roles in angiogenesis. Indeed, the Notch4ICD conditional transgenic has defects in vasculogenesis and angiogenesis (Carlson et al. 2005; Uyttendaele et al. 2001). Notch4 is expressed in endothelial cells, but not in hematopoietic progenitor cells (Krebs et al. 2000), suggesting that Notch4 is silenced during endothelial cell differentiation into hematopoietic cells. However, the role of Notch4 during this process is unknown. To study this, we conditionally expressed Notch4ICD in endothelial cells using the same Cre-loxP strategy. We first generated floxed Notch4ICD transgenic mice. The human Notch4ICD construct (Havrda et al. 2006; amino acids 1,406–1,963) with an HA epitope tag was cloned downstream of a floxed GFP sequence driven by the cytomegalovirus (CMV) enhancer/chicken β-actin promoter (N4ICD) (Fig. 2a). The transgenic allele expresses GFP constitutively. Upon Cre recombination, the GFP sequence is excised, and N4ICD is conditionally expressed. We developed two independent founder lines (FVB/N) that have an identical phenotype. To test transgene regulation, we examined N4ICD expression in 293T cells. GFP, but not N4ICD, was produced in 293T cells transfected with CAG-N4ICD. When CMV-Cre was cotransfected with CAG-N4ICD, we observed robust N4ICD expression by immunoblot with loss of GFP by fluorescence microscopy (Fig. 2b, c). The activity of Notch4ICD in activating canonical CBF1-mediated transcriptional activation was validated using a CBF1 luciferase reporter assay (Fig. 2d).
Fig 2.
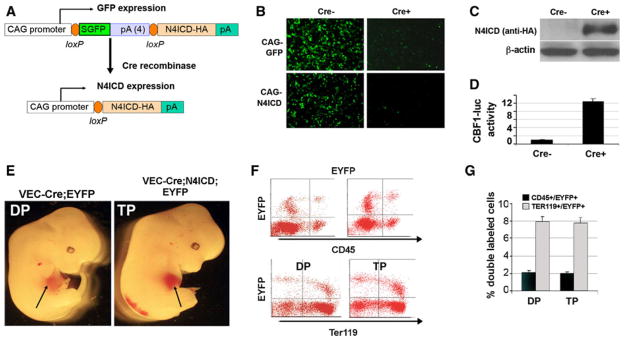
Expression of Notch4ICD in VE-cadherin-expressing populations does not impede hematopoietic stem cell development. a The conditional Notch4ICD (N4ICD) transgenic construct drives constitutive expression of GFP in the absence of Cre recombinase. Following Cre recombination, the GFP coding sequence is excised, and N4ICD is expressed. b–c The CAG-GFP and CAG-N4ICD constructs were validated in 293T cells without Cre recombinase (Cre−) or using co-transfection of a CMV-Cre expression plasmid (Cre+). GFP expression was monitored by immunofluorescence microscopy. c Cell lysates from the 293T experiment were collected, and CAG-N4ICD lysates were immunoblotted with anti-HA to detect N4ICD or β-actin as a control. d A CBF1–luciferase reporter construct was cotransfected with CAG-N4ICD in the absence (Cre−) or presence (Cre+) of CMV-Cre plasmid into 293T cells. Normalized luciferase activities are presented as mean ± standard deviation. e Gross appearance of control VE-cadherin-Cre;EYFP double positive transgenics (DP) compared to VE-cadherin-Cre; Notch4ICD; EYFP triple positive transgenic embryos (TP) at E12.5. The fetal liver is indicated by arrow. f Analysis of EYFP+ cells by flow cytometry in the fetal liver obtained from E12.5 VE-cadherin; EYFP double transgenic embryos (DP) or VE-cadherin; Notch4ICD; EYFP triple transgenic embryos (TP). Shown are representative flow cytometric analyses of fetal liver cells stained to detect CD45 or Ter119. g Quantitative analysis of the percentage of double positive CD45 and EYFP or Ter119 and EYFP in each genotype. Bars represent mean ± standard deviation
To determine the consequence of endothelial Notch4 activation on HSC phenotype, we used a similar breeding strategy as described above with the Notch1ICD transgenic strain. Notch4ICD; Rosa26R-EYFP double transgenic mice were intercrossed with VE-Cadherin-Cre mice. To compare with the phenotypes of Notch1ICD conditional transgenic mice, embryos were collected at E12.5, and the fetal liver was utilized for FACS analysis. When isolated at E12.5, we did not observe any significant morphological or gross developmental defects in Notch4ICD transgenic embryos (Fig. 2e). The analysis of the fetal liver at E12.5 indicated there was no significant difference in the EYFP+ population of triple positive (TP) embryos compared with double positive (DP) embryos (Fig. 2f). Furthermore, staining to detect CD45 and Ter119 yielded similar percentages of double positive CD45/EYFP or Ter119/EYFP in both groups (Fig. 2f, g). These data show that unlike Notch1 activation, Notch4 activation in endothelial cells does not impede HSC emergence and fetal liver hematopoiesis.
Notch4 activation in VE-cadherin populations leads to vascular remodeling defects
Because the Notch4 gain-of-function embryos did not present with apparent HSC deficiencies in mid-gestation, we evaluated later stages of development to validate activity of the transgene. Previous data have shown that knock-in of a Notch4ICD allele in early endothelial cell populations expressing VEGFR (flk1) leads to mid-gestation embryonic lethality with vascular patterning defects (Uyttendaele et al. 2001). Because VE-cadherin is expressed later than VEGFR, we predicted a vascular remodeling defect that occurred later in embryogenesis. We analyzed E13.5 (Fig. 3a), E 14.5 (Fig. 3b), and E16.5 (Fig. 3c), and found that expression of Notch4ICD in VE-cadherin expressing populations indeed led to vascular remodeling defects including hemorrhaging and loss of vascular integrity. These data are consistent with known activities of Notch4 in vascular patterning and maturation. These results also confirm that Notch4 activation, unlike Notch1 activation, does not alter earlier HSC phenotype and fetal liver hematopoiesis when expressed in the same endothelial/hematopoietic compartment.
Fig 3.
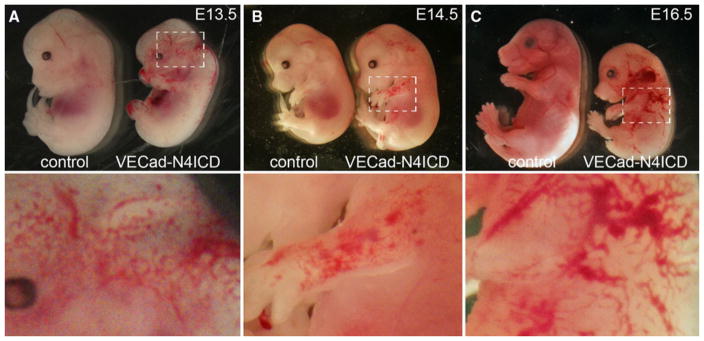
Notch4 activation in VE-cadherin cells leads to embryonic vascular remodeling defects. The Notch4ICD transgenic strain was crossed to VE-cadherin-Cre to generate VE-cadherin-Cre; Notch4ICD embryos. Litters were analyzed to compare controls (no N4ICD transgene) to embryos with N4ICD expression at E13.5 (a), E14.5 (b), and E16.5 (c). The embryos with activated Notch4 signaling showed vascular hemorrhaging and defective vascular structure. The dotted areas in the upper panels are shown below at higher magnification
Notch1 activation in the mesenchyme does not inhibit HSC emergence
In addition to an endothelial cell source of HSC, there is evidence that mesenchyme in the AGM may also give rise to HSC (Bertrand et al. 2005). To perturb Notch1 signaling in the mesenchyme, we activated the Notch1-ICD transgene using the SM22α Cre strain (Zovein et al. 2008). SM22α is expressed in adult vascular smooth muscle, and transiently in embryonic cardiac and somitic mesoderm (Li et al. 1996; Zhang et al. 2001). Recently, an SM22α expressing population was shown to contribute to developing hematopoietic cells (Zovein et al. 2008). Notch1ICD; Rosa26R-EYFP double transgenic mice were intercrossed with SM22a-Cre mice, and embryos collected at E13.5. Fetal livers were used for FACS analysis. We did not observe significant anatomical defects at E13.5 (Fig. 4a), although these embryos die postnatally. Notch1 activation in SM22α positive populations did not alter HSC emergence from the mesenchyme, and there were no changes in the proportion of EYFP positive cells (Fig. 4b, c). CD45 staining further confirmed that Notch activation in the SM22α positive population did not impede HSC emergence and fetal liver hematopoiesis (Fig. 4d). These findings show that a tightly regulated balance of Notch1 in endothelial cells is required for normal HSC emergence, but altered Notch1 signaling in mesenchymal cells does not have a major impact.
Fig 4.
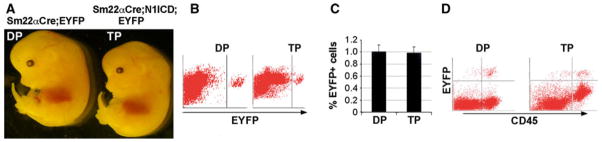
Notch1 activation in SM22α expressing populations does not impede hematopoietic stem cell emergence. a Gross appearance of SM22α-Cre; Notch1ICD double transgenic embryos and littermate controls at E13.5. b Analysis of EYFP positive cells by flow cytometry in the fetal liver obtained from E13.5 SM22α; EYFP double transgenic embryos (DP) or SM22α; Notch1ICD; EYFP triple transgenic embryos (TP). c Quantitative analysis of EYFP positive cells from E13.5 fetal liver of each genotype. Bars represent mean ± standard deviation. d Representative flow cytometric analysis of single fetal liver cells using CD45
Discussion
HSC originate within the AGM region of the midgestation embryo, and it has been proposed that both endothelial cells and mesenchyme cells contribute to this population. In the present study, we compared genetic activation of the Notch1 or Notch4 pathways, and found a selective requirement for tightly regulated endothelial Notch1 signaling for normal HSC development. In contrast, alterations in endothelial Notch4 signaling did not affect HSC emergence and fetal liver hematopoiesis. In addition, HSC development from VE-cadherin-expressing endothelial cells is highly sensitive to changes in Notch1 signaling activity, whereas perturbation of Notch1 signaling in mesenchymal cells expressing SM22α does not change HSC development. Our results show a clear cell type-dependent role of Notch1 activation in this context.
HSC of the AGM are incapable of in situ hematopoietic differentiation (Godin et al. 1999). Therefore, the progression of AGM-derived HSC to the fetal liver and later to the bone marrow provides the niche for self-renewal and differentiation. Notch signaling is specifically required for hematopoietic cell emergence from the AGM region, but not for either primitive or definitive yolk sac hematopoiesis (Hadland et al. 2004; Burns et al. 2005; Kumano et al. 2003; Robert-Moreno et al. 2005, 2008). Both endothelial cells and mesenchyme apparently contribute to HSC emergence, however, the cell specific role of Notch in this process is only starting to be understood. For example, analysis of Mib1 (Yoon et al. 2008), a ubiquitin E3 ligase required for Notch ligand processing, was performed using both global knockout and Tie2-Cre-mediated knockout. Mib1 deficient fetuses had no hematopoietic progenitors in the AGM region, but loss of Mib1 in Tie2-expressing populations had a milder hematopoietic defect. These differences suggest that cell populations other than endothelial cells require proper Notch ligand-receptor interaction for HSC development from the AGM. In addition, analysis of endothelial cells derived from Notch1 mutant embryos show that Notch1 is required for HSC emergence from endothelial cells (Kumano et al. 2003). To complement these studies, we tested the dosage dependence of Notch1 by constitutively activating signaling in VE-cadherin-expressing endothelial cells. Our results support the idea that lack of Notch1 signaling or mutations that may increase Notch1 signaling would both result in defective HSC development in the AGM.
In addition to Notch1, signaling via Notch4 regulates embryonic angiogenesis. Although the Notch4 null mice appear overtly normal, the combination of Notch1 and Notch4 mutation yields embryonic vascular defects that are more severe than Notch1 deficiency alone (Krebs et al. 2000). In addition, Notch4 gain-of-function alleles have defects in both vasculogenesis and angiogenesis (Carlson et al. 2005; Uyttendaele et al. 2001). To test imbalanced Notch4 signaling in HSC development, we generated floxed Notch4ICD transgenic mice. When the Notch4ICD transgene is activated in Tie2-expressing populations, embryonic lethality occurs before E9.5 (unpublished data), which limited our study to activation at later times. When Notch4 was activated in the VE-cadherin expressing population, there were no defects of HSC emergence. This was also true when Notch4 was activated in SM22α expressing embryonic populations. Our findings support the notion that the balance of Notch4 signaling is not critical in the process of HSC emergence. Thus, Notch1 in VE-cadherin populations has a sensitive dosage requirement that is cell type and Notch receptor-specific.
References
- Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: cell fate control and signal integration in development. Science. 1999;284(5415):770–776. doi: 10.1126/science.284.5415.770. [DOI] [PubMed] [Google Scholar]
- Bertrand JY, Giroux S, Golub R, Klaine M, Jalil A, Boucontet L, Godin I, Cumano A. Characterization of purified intraembryonic hematopoietic stem cells as a tool to define their site of origin. Proc Natl Acad Sci USA. 2005;102(1):134–139. doi: 10.1073/pnas.0402270102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boisset JC, van Cappellen W, Andrieu-Soler C, Galjart N, Dzierzak E, Robin C. In vivo imaging of haematopoietic cells emerging from the mouse aortic endothelium. Nature. 2010;464(7285):116–120. doi: 10.1038/nature08764. [DOI] [PubMed] [Google Scholar]
- Burns CE, Traver D, Mayhall E, Shepard JL, Zon LI. Hematopoietic stem cell fate is established by the Notch-Runx pathway. Gene Dev. 2005;19(19):2331–2342. doi: 10.1101/gad.1337005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson TR, Yan Y, Wu X, Lam MT, Tang GL, Beverly LJ, Messina LM, Capobianco AJ, Werb Z, Wang R. Endothelial expression of constitutively active Notch4 elicits reversible arteriovenous malformations in adult mice. Proc Natl Acad Sci USA. 2005;102(28):9884–9889. doi: 10.1073/pnas.0504391102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen MJ, Yokomizo T, Zeigler BM, Dzierzak E, Speck NA. Runx1 is required for the endothelial to haematopoietic cell transition but not thereafter. Nature. 2009;457(7231):887–891. doi: 10.1038/nature07619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Amo FF, Smith DE, Swiatek PJ, Gendron-Maguire M, Greenspan RJ, McMahon AP, Gridley T. Expression pattern of Motch, a mouse homolog of Drosophila Notch, suggests an important role in early postimplantation mouse development. Development (Cambridge, England) 1992;115(3):737–744. doi: 10.1242/dev.115.3.737. [DOI] [PubMed] [Google Scholar]
- Delassus S, Cumano A. Circulation of hematopoietic progenitors in the mouse embryo. Immunity. 1996;4(1):97–106. doi: 10.1016/s1074-7613(00)80302-7. [DOI] [PubMed] [Google Scholar]
- Dzierzak E, Speck NA. Of lineage and legacy: the development of mammalian hematopoietic stem cells. Nat Immunol. 2008;9(2):129–136. doi: 10.1038/ni1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eilken HM, Nishikawa S, Schroeder T. Continuous single-cell imaging of blood generation from haemogenic endothelium. Nature. 2009;457(7231):896–900. doi: 10.1038/nature07760. [DOI] [PubMed] [Google Scholar]
- Godin I, Garcia-Porrero JA, Dieterlen-Lievre F, Cumano A. Stem cell emergence and hemopoietic activity are incompatible in mouse intraembryonic sites. J Exp Med. 1999;190(1):43–52. doi: 10.1084/jem.190.1.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadland BK, Huppert SS, Kanungo J, Xue Y, Jiang R, Gridley T, Conlon RA, Cheng AM, Kopan R, Longmore GD. A requirement for Notch1 distinguishes 2 phases of definitive hematopoiesis during development. Blood. 2004;104(10):3097–3105. doi: 10.1182/blood-2004-03-1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havrda MC, Johnson MJ, O’Neill CF, Liaw L. A novel mechanism of transcriptional repression of p27kip1 through Notch/HRT2 signaling in vascular smooth muscle cells. Thromb Haemost. 2006;96(3):361–370. doi: 10.1160/TH06-04-0224. [DOI] [PubMed] [Google Scholar]
- Kissa K, Herbomel P. Blood stem cells emerge from aortic endothelium by a novel type of cell transition. Nature. 2010;464(7285):112–115. doi: 10.1038/nature08761. [DOI] [PubMed] [Google Scholar]
- Krebs LT, Xue Y, Norton CR, Shutter JR, Maguire M, Sundberg JP, Gallahan D, Closson V, Kitajewski J, Callahan R, Smith GH, Stark KL, Gridley T. Notch signaling is essential for vascular morphogenesis in mice. Gene Dev. 2000;14(11):1343–1352. [PMC free article] [PubMed] [Google Scholar]
- Kumano K, Chiba S, Kunisato A, Sata M, Saito T, Nakagami-Yamaguchi E, Yamaguchi T, Masuda S, Shimizu K, Takahashi T, Ogawa S, Hamada Y, Hirai H. Notch1 but not Notch2 is essential for generating hematopoietic stem cells from endothelial cells. Immunity. 2003;18(5):699–711. doi: 10.1016/s1074-7613(03)00117-1. [DOI] [PubMed] [Google Scholar]
- Lancrin C, Sroczynska P, Stephenson C, Allen T, Kouskoff V, Lacaud G. The haemangioblast generates haematopoietic cells through a haemogenic endothelium stage. Nature. 2009;457(7231):892–895. doi: 10.1038/nature07679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lardelli M, Lendahl U. Motch A and motch B—two mouse Notch homologues coexpressed in a wide variety of tissues. Exp Cell Res. 1993;204(2):364–372. doi: 10.1006/excr.1993.1044. [DOI] [PubMed] [Google Scholar]
- Li L, Miano JM, Mercer B, Olson EN. Expression of the SM22alpha promoter in transgenic mice provides evidence for distinct transcriptional regulatory programs in vascular and visceral smooth muscle cells. J Cell Biol. 1996;132(5):849–859. doi: 10.1083/jcb.132.5.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medvinsky A, Rybtsov S, Taoudi S. Embryonic origin of the adult hematopoietic system: advances and questions. Development. 2011;138(6):1017–1031. doi: 10.1242/dev.040998. [DOI] [PubMed] [Google Scholar]
- Nishikawa SI, Nishikawa S, Kawamoto H, Yoshida H, Kizumoto M, Kataoka H, Katsura Y. In vitro generation of lymphohematopoietic cells from endothelial cells purified from murine embryos. Immunity. 1998;8(6):761–769. doi: 10.1016/s1074-7613(00)80581-6. [DOI] [PubMed] [Google Scholar]
- Reaume AG, Conlon RA, Zirngibl R, Yamaguchi TP, Rossant J. Expression analysis of a Notch homologue in the mouse embryo. Dev Biol. 1992;154(2):377–387. doi: 10.1016/0012-1606(92)90076-s. [DOI] [PubMed] [Google Scholar]
- Robert-Moreno A, Espinosa L, de la Pompa JL, Bigas A. RBPjkappa-dependent Notch function regulates Gata2 and is essential for the formation of intra-embryonic hematopoietic cells. Development (Cambridge, England) 2005;132(5):1117–1126. doi: 10.1242/dev.01660. [DOI] [PubMed] [Google Scholar]
- Robert-Moreno A, Guiu J, Ruiz-Herguido C, Lopez ME, Ingles-Esteve J, Riera L, Tipping A, Enver T, Dzierzak E, Gridley T, Espinosa L, Bigas A. Impaired embryonic haematopoiesis yet normal arterial development in the absence of the Notch ligand Jagged1. EMBO J. 2008;27(13):1886–1895. doi: 10.1038/emboj.2008.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uyttendaele H, Ho J, Rossant J, Kitajewski J. Vascular patterning defects associated with expression of activated Notch4 in embryonic endothelium. Proc Natl Acad Sci USA. 2001;98(10):5643–5648. doi: 10.1073/pnas.091584598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesh DA, Park KS, Harrington A, Miceli-Libby L, Yoon JK, Liaw L. Cardiovascular and hematopoietic defects associated with Notch1 activation in embryonic Tie2-expressing populations. Circ Res. 2008;103(4):423–431. doi: 10.1161/CIRCRESAHA.108.177808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon MJ, Koo BK, Song R, Jeong HW, Shin J, Kim YW, Kong YY, Suh PG. Mind bomb-1 is essential for intra-embryonic hematopoiesis in the aortic endothelium and the subaortic patches. Mol Cell Biol. 2008;28(15):4794–4804. doi: 10.1128/MCB.00436-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JC, Kim S, Helmke BP, Yu WW, Du KL, Lu MM, Strobeck M, Yu Q, Parmacek MS. Analysis of SM22alpha-deficient mice reveals unanticipated insights into smooth muscle cell differentiation and function. Mol Cell Biol. 2001;21(4):1336–1344. doi: 10.1128/MCB.2001.21.4.1336-1344.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zovein AC, Hofmann JJ, Lynch M, French WJ, Turlo KA, Yang Y, Becker MS, Zanetta L, Dejana E, Gasson JC, Tallquist MD, Iruela-Arispe ML. Fate tracing reveals the endothelial origin of hematopoietic stem cells. Cell Stem Cell. 2008;3(6):625–636. doi: 10.1016/j.stem.2008.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]