

Abstract
Objectives
Pancreatic endocrine tumors (PETs) share numerous features with gastrointestinal neuroendocrine (carcinoid) tumors. Targets of novel therapeutic strategies previously assessed in carcinoid tumors were analyzed in PETs (44 cases).
Methods
Activating mutations in EGFR, KIT, and PDGFRA, and non-response mutations in KRAS, were evaluated. Copy number of EGFR and HER-2/neu was quantified by fluorescence in situ hybridization. Expression of EGFR, PDGFRA, VEGFR1, TGFBR1, Hsp90, SSTR2A, SSTR5, IGF1R, mTOR, and MGMT was measured immunohistochemically.
Results
Elevated EGFR copy number was found in 38% of cases, but no KRAS non-response mutations. VEGFR1, TGFBR1, PDGFRA, SSTR5, SSTR2A, and IGF1R exhibited the highest levels of expression in the largest percentages of PETs.
Anticancer drugs BMS-754807 (selective for IGF1R/IR), 17-(allylamino)-17-demethoxygeldanamycin (17-AAG, targeting Hsp90), and axitinib (directed toward VEGFR1–3/PDGFRA-B/KIT) induced growth inhibition of human QGP-1 PET cells with IC50 values (nM) of 273, 723, and 743, respectively. At growth-inhibiting concentrations, BMS-754807 inhibited IGF1R phosphorylation; 17-AAG induced loss of EGFR, IGF1R, and VEGFR2; and axitinib increased p21Waf1/Cip1(CDKN1A) expression without inhibiting VEGFR2 phosphorylation.
Conclusions
Results encourage further research into multi-drug strategies incorporating inhibitors targeting IGF1R or Hsp90 and into studies of axitinib combined with conventional chemotherapeutics toxic to tumor cells in persistent growth arrest.
Keywords: pancreatic endocrine tumors, molecular analysis
INTRODUCTION
Pancreatic endocrine tumors (PETs), including islet cell carcinomas, account for 1–3% of all pancreatic cancers [1]. An identifying characteristic of some PETs is their overproduction of polypeptide hormones (e.g., gastrin, glucagon, insulin, somatostatin, or vasoactive intestinal peptide); these tumors are classified as functional or nonfunctional based upon whether their excessive polypeptide synthesis induces clinical symptoms of hormonal syndromes. An analysis of 1483 cases of PETs in the Surveillance, Epidemiology, and End Results (SEER) database (1973–2000) indicates that, despite the categorization of this neoplasm as an indolent tumor, the median overall survival of patients with metastatic disease is only 17 months [2]. Patients seen at centers specializing in the treatment of neuroendocrine tumors seem to have superior survival when compared with patients in population-based databases like the SEER registry [3]. Complete surgical resection is the most successful treatment for patients with PETs; however, most patients present with advanced stages of disease [2], for which few current treatment options yield high tumor regression rates. Somatostatin therapy can ameliorate clinical symptoms and perhaps induce tumor growth stabilization [review 4], and, in recent phase III trials, mTOR inhibitor everolimus and tyrosine kinase inhibitor (TKI) sunitinib improved progression-free survival [5,6]; however, novel therapeutic strategies exhibiting increased antitumor activity would be beneficial.
Gastrointestinal neuroendocrine (carcinoid) tumors and pancreatic endocrine tumors share a number of characteristics, including neuroendocrine properties (originating from cells of the diffuse neuroendocrine system), certain ultrastructural features (such as cytoplasmic dense-core secretory granules), and similar biomarker expression (e.g., chromogranin, synaptophysin, and keratin). Both malignancies can be associated with clinical syndromes induced by tumor overproduction of bioactive peptides or amines which serve as a diagnostic marker for the functioning gastroenteropancreatic endocrine tumor, e.g., elevated levels in urine of serotonin catabolite 5-HIAA for carcinoid tumors, serum insulin for insulinoma PETs, serum gastrin for gastrinoma PETs, and serum glucagon for glucagonoma PETs. Patients with carcinoid tumors commonly present at advanced stages for which few treatment options are available; therefore, we recently evaluated a heterogeneous collection of 104 carcinoid tumors for growth factor receptors and downstream effectors and regulators targeted by protein kinase inhibitors and/or antibodies under development as anticancer therapeutics in other forms of cancer [7]. The results were supportive of further research into targeting Hsp90, IGF1R, and EGFR for development of new therapeutic strategies for some carcinoid tumors.
Due to the similarities shared by gastroenteropancreatic endocrine tumors, the present study investigated in 67 PETs the genetic abnormalities and protein expression of growth factor receptors and downstream regulators examined earlier in carcinoid tumors [7]. The genetic abnormalities analyzed were biomarkers of response to targeted therapeutics developed for other cancers, including activating mutations in EGFR exons 18, 19, and 21 and high EGFR copy number by fluorescence in situ hybridization (FISH), the former a marker for response to anti-EGFR TKI gefitinib in non—small-cell lung cancer (NSCLC) [8, 9] and the latter a biomarker predictive of sensitivity to gefitinib in NSCLC [10, 11] and to anti-EGFR monoclonal antibodies cetuximab and panitumumab in colorectal cancer [12 – 14]. Mutations in KRAS codons 12 and 13 were assessed as markers for non-response to anti-EGFR therapy, correlating with lack of sensitivity to cetuximab [15 – 17] and panitumumab [18] in colorectal cancer and to TKIs gefitinib and erlotinib in lung cancer [19 – 21]. High HER-2/neu copy number was measured by FISH as a biomarker predicting response to anti-HER2 monoclonal antibody trastuzumab in breast cancer [22]. Finally, mutations in KIT exons 9, 11, 13, and 17 and in PDGFRA exons 12, 14, and 18 were analyzed due to association with sensitivity to TKI imatinib in gastrointestinal stromal tumors [23, 24].
The protein expression analyzed was that of growth factor receptors and downstream effectors and regulators, as measured by immunohistochemistry (IHC). Immunohistochemical expression in PETs of the common therapeutic targets somatostatin receptors SSTR2A and SSTR5 was compared to that of EGFR, PDGFRA, VEGFR1, TGFBR1, Hsp90, IGF1R, and mTOR. Furthermore, the immunohistochemical absence of the de-methylating enzyme MGMT was measured as a marker for response of PETs to the DNA-methylating chemotherapeutic temozolomide [25].
Finally, follow-up in vitro studies were performed in QGP-1 cells, the sole well-established PET cell line, to measure the effect of therapeutics targeting four molecular markers that were strongly or moderately strongly expressed immunohistochemically in PETs (VEGFR1, PDGFRA, IGF1R, and Hsp90) and a biomarker with elevated gene copy number by FISH (EGFR). The effects on QGP-1 cells of the following anticancer drugs were compared: TKIs axitinib (selective for VEGFR1–3/PDGFRA-B/KIT), sunitinib (targeting ≥ nine receptor tyrosine kinases including VEGFR1–3/PDGFRA-B), BMS-754807 (specific for IGF1R/IR), and gefitinib (targeting EGFR), as well as Hsp90 inhibitor 17-(allylamino)-17-demethoxygeldanamycin (17-AAG).
We report here a multifaceted study based on mutational, gene copy number, immunohistochemical, and in vitro analyses, which assessed biomarkers for novel therapeutic strategies in a collection of 41 primary and 26 metastatic PETs, and compared the results to data obtained from neuroendocrine (carcinoid) tumors. This work was presented in preliminary form at the 100th Annual Meeting of the United States and Canadian Academy of Pathology in February 2011 [26].
Materials and Methods
Patient samples
Forty-four patients were identified undergoing surgery at Mayo Clinic between 2001 and 2005 for PETs. All cases had accessible pathology slides as well as formalin-fixed, paraffin-embedded tumor blocks, and most had flash-frozen surgical specimens available for analysis. Prior to inclusion of a case in this study, an hematoxylin and eosin (H&E) stained slide from each tumor block associated with the case was reviewed (M.H.M. and R.V.L.) to confirm the PET diagnosis. Written research authorization was obtained from all patients for this study, as well as Mayo Clinic Institutional Review Board approval.
Tissue microarray construction
A tissue microarray (TMA) was constructed by the Tissue and Cell Molecular Analysis Shared Resource, Mayo Clinic, with a Beecher ATA-27 automated arrayer (Sun Prairie, WI). From 44 cases, 67 primary and metastatic PETs were selected. The most characteristic area from each tumor was circled on an H&E slide, and triplicate 0.6 mm cores were removed from the corresponding area in the associated formalin-fixed, paraffin-embedded tissue block and placed into a single recipient paraffin block. All of the tumor samples selected for constructing the TMA are listed, by case, in Supplementary Table S1.
Immunohistochemical analysis
Sections (5 µ) of the PET TMA were analyzed by IHC for EGFR, PDGFRA, VEGFR1, mTOR, IGF1R, Hsp90, TGFBR1, MGMT, SSTR2A, and SSTR5. Immunohistochemical staining was performed by the Tissue and Cell Molecular Analysis Shared Resource, Mayo Clinic. Positive controls for IHC stains were normal colon (for TGFBR1), normal pancreas (SSTR2A and SSTR5), breast cancer (EGFR, IGF1R, and PDGFRA), normal skin (VEGFR1), prostate cancer (mTOR and Hsp90), and colon cancer (MGMT). Negative controls for all stains were prepared by substituting diluent for primary antibodies. IHC of all biomarkers was scored based on intensity by two pathologists (R.V.L. and L.J.A.), with a score of 0 indicating absence of staining, and 1, 2, and 3 representing weak, moderate, and strong staining intensity, respectively. The immunohistochemical intensity score reported for the staining of tumor cells within each assessable PET was the average from the replicate TMA cores for that sample. All immunohistochemical antibodies and epitope retrieval methods are listed in Supplementary Table S2.
FISH analysis of gene copy number
PET TMA sections (5 µ) were analyzed by FISH for HER-2/neu and EGFR copy number by the Cytogenetics Shared Resource Laboratory, Mayo Clinic, by the methodology previously reported [27]. Briefly, thirty nuclei were scored per sample with quantitation of red signals (HER-2/neu or EGFR) and green signals [chromosome 17 centromere (CEP17) or chromosome 7 centromere (CEP7), respectively]. A ratio of HER-2/neu:CEP17 or EGFR:CEP7 of 0.8 – 1.30 was interpreted as normal, < 0.8 as gene deletion, 1.30 – 2.0 as gene duplication, and ≥ 2.0 as gene amplification. Aneusomy was defined to be a normal HER-2/neu:CEP17 or EGFR:CEP7 ratio with > 30% of cells having ≥ 3 CEP17 or CEP7 signals, respectively (i.e., a balanced gain in the number of gene copies and the number of chromosome copies). Aneuploid tumors were analyzed for mean EGFR copy number/cell to determine whether EGFR copy number was elevated (≥ 2.47/nucleus [13] or ≥ 2.92/cell [14]). HER-2/neu and EGFR FISH-positive samples were those demonstrating amplification or high aneusomy with ≥ 40% of cells having ≥ 4 copies of HER-2/neu or EGFR, respectively. Due to the inherent admixture of tumor and non-tumor cells within PETs, FISH-positivity for HER-2/neu and EGFR as well as FISH-determined elevation of EGFR copy number/cell was assigned the highest value score from the replicate TMA cores for each assessable neoplasm.
Mutational Analysis of Selected EGFR, KIT, PDGFRA, and KRAS exons
Thirty-six surgical specimens from the 44 cases in this study were flash-frozen following excision and stored at −70°C by the Tissue Core Resource of the Mayo Clinic Cancer Center SPORE in Pancreatic Cancer. DNA for mutational analyses was isolated from frozen tissue samples with the QIAamp DNA Mini Kit (Qiagen, Valencia, CA). Mutational analyses of EGFR (exons 18, 19, and 21) and KIT (exons 9, 11, 13, and 17) were performed as reported [28 and 27, respectively]; mutational analyses of KRAS (exon 2) and PDGFRA (exons 12, 14, and 18) were conducted as described [7]. Amplification and sequencing was repeated for all observed polymorphisms and mutations. All tissues assessed for EGFR, KIT, PDGFRA, and KRAS mutations are listed by case in Supplementary Table S1.
Cell culture
The QGP-1 (monolayer) PET cell line was purchased from the Japan Health Sciences Foundation’s Health Science Research Resources Bank (Osaka, Japan). The QGP-1 line was established from a human pancreatic islet cell carcinoma lesion in the late 1970s, exhibiting morphological and histological similarities to the original tumor [29]. The NCI-H727 (monolayer) human bronchopulmonary neuroendocrine tumor (carcinoid) cell line was purchased from American Type Culture Collection (Manasses, VA). Cell lines were cultured at 37°C in a humidified environment of 95%:5%, air:CO2 in RPMI 1640 media (Invitrogen; Carlsbad, CA) supplemented with 10% v/v heat-inactivated fetal bovine serum (PAA Labs; New Bedford, MA).
Cell proliferation assay
QGP-1 (4000/well) or NCI-H727 cells (4000/well) were seeded into 96-well plates (Corning; Corning, NY) in 100 µl aliquots of growth medium, and incubated (37°C) for 48 h. Each drug concentration was added in 100 µl of growth medium containing 0.125% v/v DMSO to six replicate wells; 100 µl of growth medium containing 0.125% v/v DMSO without drug was added to six replicate wells of control cells. Following incubation for 5 days (continuous drug exposure), cell growth was determined with the 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt (MTS) assay [30] upon addition to each well of 40 µl containing a 20:1 ratio of 2 mg/ml MTS (Promega; Madison, WI) and 0.92 mg/ml phenazine methosulfate (Sigma; St. Louis, MO). After incubation for 2 h, absorbance at 490 nm was measured on a SpectraMax Plus384 (Molecular Devices; Sunnyvale, CA) microplate reader. Dose response experiments for each drug were performed in triplicate, with growth of treated cells at each dosage compared to proliferation of control cells. IC50 values were estimated with Prism software (GraphPad; San Diego, CA) using nonlinear least squares regression analysis to fit the mean percent control value for each dosage to the best sigmoidal dose-response curve. Anticancer drugs axitinib, sunitinib, gefitinib, and BMS-754807 were purchased from ChemiTek (Indianapolis, IN); 17-AAG was provided by the National Cancer Institute (Bethesda, MD).
Western immunoblot analysis
Whole cell lysates were prepared from cultured cells with RIPA lysis buffer (Santa Cruz Biotechnology; Santa Cruz, CA) according to manufacturer’s instructions; protein was quantitated using the DC Protein Assay (Bio-Rad; Hercules, CA). Western blot analysis was performed following electrophoresis of samples that were loaded onto 10% or 12% w/v separating gels (Criterion XT, Bio-Rad) on the basis of protein concentration; after transfer to PVDF membranes (Bio-Rad), immunoreactive proteins were detected using SuperSignal enhanced chemiluminescent substrates (Pierce; Rockford, IL) and Hyblot CL film (Denville Scientific; Metuchen, NJ). Primary antibodies employed were anti-EGFR (Santa Cruz sc-03), anti-IGF1R beta (Cell Signaling 3027; Danvers, MA), anti-phospho-IGF1R beta (Tyr1131)/Insulin Receptor beta (Tyr1146) (Cell Signaling 3021), anti-VEGFR2 (Cell Signaling 2479), anti-phospho-VEGFR2 (Tyr1059) (Cell Signaling 3817), anti-p21Waf1/Cip1(CDKN1A) (Cell Signaling 2947), anti-Hsp90 (Cell Signaling 4874), and anti-α/β-tubulin (Cell Signaling 2148); the secondary antibody was goat anti-rabbit IgG-HRP (Santa Cruz sc-2004).
Results
Patient cases
Of 44 patients in this study, 21 were female and 23 were male (Table 1). The median age at the time of surgery was 52 years (range 19–84).
Table 1.
Characteristics of cases of pancreatic endocrine tumors (44)
| Sex | |
| Male | 23 |
| Female | 21 |
| Median age (range) at surgery | 52 y (19 – 84) |
| Subtype | |
| Non-functional | 38 |
| Functional | |
| Insulinoma | 4 |
| Gastrinoma | 1 |
| Glucagonoma | 1 |
| Previous therapy | 5 |
| History of other cancer | 9 |
| Tumor sites in TMA* | |
| Pancreas | 41 |
| Liver | 16 |
| Lymph node | 7 |
| Duodenum | 2 |
| Diaphragm | 1 |
TMA constructed with 41 PET primaries and 26 metastatic PETs.
The majority of patients (38) had nonfunctional PETs, and five patients had received treatment for pancreatic endocrine tumors prior to surgery (cases identified in Table 2). The classification of all cases according to the WHO 2004 clinicopathological criteria [31] is provided in Table 2.
Table 2.
Pancreatic endocrine tumors exhibiting strong biomarker expression
| Case | WHO 2004 classification |
Tumor site | VEGFR1 | TGFBR1 | PDGFRA | SSTR5 | SSTR2A | IGF1R | Hsp90 | EGFR | mTOR | MGMT | Mean EGFR ≥2.47/cell |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2a | Pancreas | +b | −b | − | − | − | − | − | − | − | − | − |
| Liver | + | + | + | + | + | + | − | − | − | — | − | ||
| 2c | 2 | Lymph node | + | + | + | + | + | − | − | − | − | − | + |
| Liver | + | + | + | + | + | + | − | − | − | − | + | ||
| 3 | 1.2 | Pancreas | + | + | − | + | + | − | + | − | − | − | − |
| 4 | 2 | Pancreas | + | + | + | + | + | + | + | − | − | − | − |
| Lymph node | + | + | + | − | + | − | − | − | − | − | − | ||
| Liver | − | + | − | + | + | + | − | − | − | − | − | ||
| 5d | 2 | Pancreas | + | + | + | + | + | − | − | − | − | − | + |
| Diaphragm | − | ||||||||||||
| 6 | 1.2 | Pancreas | − | − | − | − | − | − | − | − | − | − | − |
| 7 | 2 | Pancreas | + | + | − | + | + | − | − | − | − | − | − |
| Liver | + | + | + | + | + | − | − | − | + | − | − | ||
| 8e,f | 2 | Pancreas | + | + | + | − | − | + | − | − | − | − | − |
| Liver | − | + | − | + | − | + | − | − | − | − | − | ||
| 9 | 2 | Pancreas | + | − | + | + | + | − | − | − | − | − | − |
| Lymph node | + | − | + | + | + | − | − | − | − | − | − | ||
| 10 | 2 | Pancreas | + | − | + | − | − | + | − | − | − | − | − |
| Liver | − | − | − | − | − | − | − | − | − | — | − | ||
| 11 | 2 | Pancreas | + | + | + | + | + | − | − | − | − | — | + |
| 12 | 2 | Pancreas | − | − | − | − | − | + | − | − | − | − | − |
| Liver | − | − | + | − | − | − | − | − | − | − | − | ||
| 13h1 | 2 | Pancreas | + | + | + | − | + | + | + | − | − | — | + |
| Lymph node | + | + | + | + | + | − | − | − | − | − | + | ||
| Liver | + | + | + | + | + | − | − | − | − | — | + | ||
| 14 | 1.2 | Pancreas | + | − | − | + | − | − | − | ||||
| 15 | 2 | Pancreas | + | + | + | + | + | − | − | − | − | — | + |
| Liver | + | + | + | − | + | + | − | − | − | − | + | ||
| 16 | 1.2 | Pancreas | − | + | + | + | − | + | − | − | − | − | − |
| 17 | 1.2 | Pancreas | − | − | − | − | − | − | − | − | − | − | − |
| Lymph node | + | + | + | + | + | + | + | − | − | − | − | ||
| 18h1 | 1.2 | Pancreas | + | + | + | + | − | + | − | − | − | − | |
| 19 | 1.2 | Pancreas | + | − | − | − | − | + | − | − | − | − | |
| 20c | 2 | Pancreas | + | + | − | − | − | − | − | − | − | − | + |
| Lymph node | + | + | + | − | + | − | − | − | − | — | + | ||
| 21 | 1.2 | Pancreas | + | + | + | − | + | + | − | − | − | − | + |
| 22g,h1 | 1.1 | Pancreas | + | + | + | + | + | + | − | − | − | − | − |
| 23 | 1.2 | Pancreas | + | + | − | + | + | + | − | − | − | − | − |
| 24h1 | 1.2 | Pancreas | + | − | + | + | − | + | − | − | − | − | + |
| 25 | 1.2 | Pancreas | + | − | + | + | + | + | − | − | − | − | + |
| 26 | 2 | Pancreas | − | − | − | + | − | + | − | − | − | − | − |
| Liver | + | + | − | + | + | − | − | − | − | — | − | ||
| 27 | 2 | Pancreas | + | − | + | − | + | + | − | − | − | — | − |
| Duodenum | + | + | + | − | + | + | − | − | − | — | + | ||
| 28c | 2 | Liver | + | + | − | + | − | − | + | − | − | — | + |
| 29 | 2 | Pancreas | − | + | + | − | − | − | + | − | − | − | − |
| Liver | − | + | − | − | − | + | − | − | − | − | − | ||
| 30 | 1.2 | Pancreas | − | − | − | + | − | − | − | − | − | − | − |
| 31 | 2 | Pancreas | + | + | + | + | + | − | + | − | − | − | − |
| Liver | + | + | + | + | + | − | − | − | − | — | + | ||
| 32 | 2 | Pancreas | + | + | − | − | − | − | − | − | − | − | − |
| Liver | − | − | − | − | − | + | − | − | − | − | − | ||
| 33g,h2 | 2 | Pancreas | + | + | + | − | + | + | − | − | − | − | − |
| Duodenum | + | + | + | + | − | − | − | − | − | − | − | ||
| 34 | 2 | Pancreas | + | + | + | + | − | + | + | − | − | — | − |
| 35 | 2 | Pancreas | + | − | + | − | − | + | − | − | − | − | − |
| 36e,h3 | 2 | Pancreas | + | + | + | − | − | − | − | − | − | − | − |
| Lymph node | + | + | + | + | + | − | − | − | − | − | − | ||
| Liver | + | + | − | + | − | − | − | + | − | − | − | ||
| 37 | 1.2 | Pancreas | + | + | − | + | + | − | − | − | − | — | |
| 38 | 1.2 | Pancreas | + | + | + | − | − | − | + | − | − | − | − |
| 39 | 1.1 | Pancreas | + | + | + | + | + | + | − | − | − | − | − |
| 40 | 1.2 | Pancreas | + | − | + | − | − | + | − | − | − | − | − |
| 41 | 1.2 | Pancreas | + | + | + | − | + | + | − | − | − | − | + |
| 42 | 1.2 | Pancreas | + | + | + | − | + | − | − | − | − | — | − |
| 43 | 2 | Pancreas | + | − | − | − | − | − | − | − | − | − | + |
| 44 | 2 | Liver | + | − | + | + | + | − | − | − | − | — | + |
1.1: Well differentiated endocrine tumor with 'benign behavior'; 1.2: Well differentiated endocrine tumor with uncertain behavior; 2: Well differentiated endocrine carcinoma
For VEGFR1, TGFBR1, PDGFRA, SSTR5, SSTR2A, IGF1R, Hsp90, EGFR, mTOR, and MGMT “+” indicates an IHC intensity score of 3, and “−“ an IHC score < 3; for MGMT “—“ indicates a score of 0
Tumor(s) FISH-positive for EGFR and HER-2/neu
PET therapy prior to this instance of surgery:
radiation therapy,
somatostatin analog therapy,
chemotherapy, and
surgery
Functioning PET:
insulinoma,
gastrinoma, and
glucagonoma
Sixty-seven PETs (Table 1) from the 44 cases were accessible in formalin-fixed, paraffin-embedded blocks and were included in the TMA construction, with liver the most frequent metastatic site represented. Supplemental Table S1 lists all PETs, by case, included in the TMA.
Mutational Analysis of EGFR, KIT, PDGFRA, and KRAS
Thirty-six PETs (from 35 of the 44 cases) were available as frozen specimens, providing DNA for analysis of mutations in selected exons of EGFR, KIT, PDGFRA, and KRAS. Supplemental Table S1 lists, by case, the tissues (all primary tumors except one) examined by mutational analysis.
No PET analyzed displayed KRAS exon 2 mutations that are associated with non-response to anti-EGFR monoclonal antibodies [15 – 18] and TKIs [19 – 21]. No PET exhibited EGFR mutations predictive of gefitinib sensitivity nor KIT or PDGFRA mutations associated with clinical response to imatinib.
Polymorphisms in PDGFRA and EGFR were detected in some PETs during mutational analyses. All tumors analyzed were homozygous for the synonymous single nucleotide polymorphism (SNP) dbSNP rs1873778 in PDGFRA exon 12 (A1701G, where number “1” is assigned to the “A” in the translation initiation codon of the cDNA; amino acid position 567). PETs from 16% of the cases examined were heterozygous for the synonymous SNP dbSNP rs2228230 in PDGFRA exon 18 (C2472T; amino acid 824). In addition, tumor in two cases exhibited heterozygosity for the synonymous SNP dbSNP rs2229066 in EGFR exon 21 (C2508T; amino acid 836).
FISH analysis of EGFR and HER-2/neu copy number
EGFR
FISH analysis determined that some of the assessable cases had PETs displaying EGFR aneusomy with EGFR copy number at elevated levels associated with the response of colorectal cancer to panitumumab and cetuximab: 38% exhibited ≥ 2.47/nucleus, 28% ≥ 2.92/cell. Of the 19 cases in which multiple tumor tissues from the same patient were assessable for elevated EGFR copy number, all sites were positive in 4 cases and all negative in 13. Table 2 presents the PETs exhibiting elevated EGFR copy number, on a case-by-case basis.
Three cases with PETs displaying elevated EGFR copy number were also determined to be “FISH-positive,” with EGFR copy number at high levels associated with gefitinib response (aneusomy with ≥ 40% of cells having ≥ 4 copies of EGFR [10, 11]). EGFR FISH-positive tumors, all from cases with non-functional PETs and all tumors from each case exhibiting FISH-positivity, are indicated in Table 2.
HER-2/neu
HER-2/neu aneusomy was detected in 33% of the assessable cases, although PET HER-2/neu copy number was below levels predictive of anti-HER2 response (defined as > 6 HER-2/neu copies/cell by FISH analysis [32]). However, three cases with HER-2/neu aneusomy displayed high copy number for both EGFR and HER-2/neu (aneusomy with ≥ 40% of cells having ≥ 4 gene copies), a combination predictive of gefitinib sensitivity greater than that for patients with high EGFR copy number alone [33]. Table 2 provides the cases with tumors that were FISH-positive for both HER-2/neu and EGFR, all well differentiated endocrine carcinomas.
Immunohistochemical analysis
The immunohistochemical staining of pancreatic endocrine tumors for SSTR2A, SSTR5, EGFR, PDGFRA, VEGFR1, TGFBR1, Hsp90, IGF1R, mTOR, and MGMT is illustrated in Figure 1. All of the immunohistochemical stains showed a diffuse cytoplasmic pattern with the exception of MGMT which was a nuclear stain and SSTR2A which was predominantly membranous. IGF1R, mTOR, and SSTR5 in most cases stained in a diffuse pattern, however, there were a few cases that had more of a nuclear staining pattern with weak cytoplasmic staining; the significance of these findings was uncertain. Predominantly focal staining was not noted with any of the antibodies. A PET sample was assigned a score of 1 if the tissue had weak staining diffusely or only a few cells with weak staining.
Figure 1.
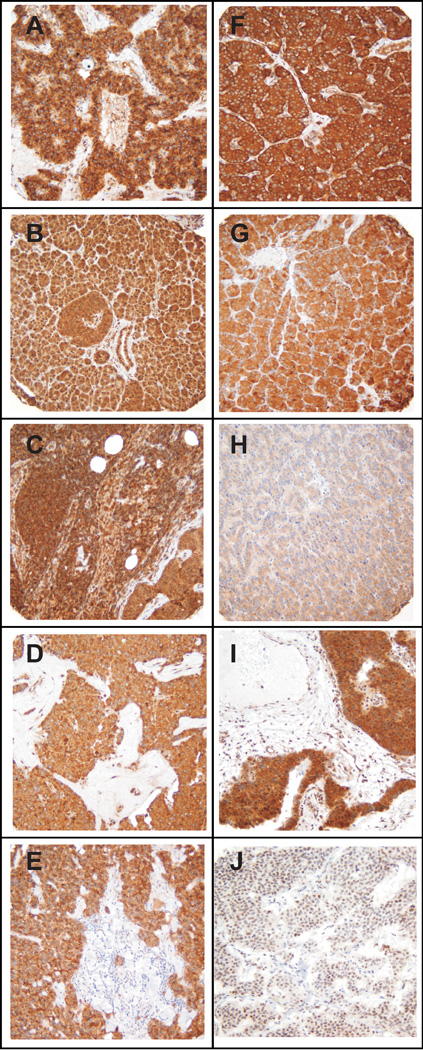
Immunohistochemical analysis of pancreatic endocrine tumors. Panels illustrate individual PET specimens in the TMA (original magnification × 200) exhibiting strong immunostaining for (A) VEGFR1, (B) TGFBR1, (C) PDGFRA, (D) SSTR5, (E) SSTR2A, (F) IGF1R, (G) Hsp90, (H) EGFR, (I) mTOR, and (J) MGMT.
The immunohistochemical staining intensity score for each assessable PET in the TMA is presented for each biomarker in Figure 2with scores grouped by location of malignancy (primary or metastasis). With the exception of MGMT, all biomarkers were expressed in all tumors except for one liver metastasis with no detectable VEGFR1, EGFR, or mTOR staining. The biomarkers for which the largest number of PETs exhibited the strongest IHC staining were VEGFR1, TGFBR1, PDGFRA, SSTR5, SSTR2A, and IGF1R, with intensity scores of 3 for 80%, 69%, 65%, 55%, 55%, and 47% of all tumors, respectively. The biomarker with the lowest immunohistochemical expression level was MGMT, with complete absence of staining in 24% of PETs (32% of cases); in eight of these cases with available primary as well as metastatic tumor tissues, only one exhibited MGMT deficiency across all sites. For each target protein analyzed immunohistochemically, the staining intensity score averaged over all primaries was not significantly different from the score averaged over all metastases (data not shown) except for VEGFR1, with a mean ± SEM of 2.94 ± 0.02 for primaries and 2.72 ± 0.13 for metastases (P < 0.05).
Figure 2.

Immunohistochemical staining intensity of ten biomarkers in PETs. Individual IHC staining intensity scores for all PETs assessable in the TMA (n = 67) were grouped for comparison by site of malignancy (primary or metastasis).
Table 2 summarizes, by case, the combination of molecular markers that were strongly expressed (immunohistochemical staining intensity score of 3) in each assessable PET. The percentage of cases with assessable primary as well as metastatic tumors in which all sites exhibited the same biomarker level, i.e., an IHC intensity score of either 3 or < 3 (for MGMT, either < 3 or 0), were as follows: For biomarkers EGFR and mTOR, 95%; TGFBR1 and Hsp90, 74%; VEGFR1 and SSTR2A, 68%; MGMT, 63%; SSTR5, 58%; PDGFRA, 47%; and IGF1R, 37%. Of cases in which multiple tumor tissues from the same patient were analyzed, a majority exhibited strong immunohistochemical expression across all sites for biomarkers VEGFR1 and TGFBR1 (58% and 58%, respectively). Most PETs that exhibited strong expression of VEGFR1 also displayed strong staining for PDGFRA (75%), a similar occurrence for SSTR5 and SSTR2A (72%). Of note, strong EGFR immunohistochemical staining was not exhibited by any PET that displayed elevated EGFR copy number.
In vitro effects of anticancer drugs in PET cells
A dearth of well-established human PET cell lines currently exists [4]. Accordingly, our in vitro PET studies were constrained to one PET line, QGP-1 [29], for measuring the effects on cellular proliferation and downstream functionality of anticancer drugs targeting biomarkers identified in a collection of PETs. MTS assays performed with QGP-1 cells determined that growth inhibition was produced by increasing concentrations of the following therapeutic agents: axitinib and sunitinib (TKIs targeting VEGFR1–3 and PDGFRA-B, among others), BMS-754807 (a TKI specific for IGF1R and IR), gefitinib (a TKI selective for EGFR), as well as 17-AAG (an Hsp90 inhibitor active in bronchopulmonary NET cell lines [7]). Table 3 presents IC50 values for the inhibition of QGP-1 proliferation induced by each of these compounds.
Table 3.
Anticancer drugs that inhibited QGP-1 human PET cell proliferation
| Inhibitor | Specificity | IC50 (nM)* |
|---|---|---|
| BMS-754807 | IGF1R/IR | 273 |
| 17-AAG | Hsp90 | 723 |
| Axitinib | VEGFR1–3, PDGFRA-B, KIT | 743 |
| Sunitinib | VEGFR1–3, PDGFRA-B, KIT, FLT3, CSF1R, RET | 2707 |
| Gefitinib | EGFR | 24080 |
Cell proliferation was determined by the MTS assay; data were the average of ≥ three experiments.
The three most potent growth inhibitors, BMS-754807, 17-AAG, and axitinib, were assessed in follow-up experiments with QGP-1 cells to measure functional effects downstream of their targeted proteins, with results from QGP-1 cells compared, in turn, to those obtained in the human NCI-H727 bronchopulmonary neuroendocrine tumor (carcinoid) cell line. Western immunoblot analysis indicated that exposure of QGP-1 cells (24 h) to BMS-754807 concentrations increasing from 0.1 µM to 10 µM resulted in increasing inhibition of the constitutive phosphorylation of IGF1R (n = 3). Figure 3A illustrates the inhibition induced by increasing BMS-754807 concentrations on QGP-1 cell growth as well as on constitutive IGF1R phosphorylation. In comparison, BMS-754807 was less potent at inhibiting NCI-H727 carcinoid cell growth (IC50 of 428 nM) but reduced constitutive phosphorylation of IGF1R in the NCI-H727 line in a similar fashion [see 7].
Figure 3.



Effects of anticancer drugs in QGP-1 PET cell line. (A) Left: QGP-1 cells (4000/well) were incubated (continuous exposure, 5 days) in 96-well plates at 37°C with increasing concentrations of anticancer drug BMS-754807 (targeting IGF1R/IR) in serum-containing medium, with cell proliferation measured by the MTS assay; right: QGP-1 cells (500,000/well) were incubated (continuous exposure, 24 h) in 6-well plates at 37°C with increasing concentrations of BMS-754807 in serum-containing medium, with effect on constitutive IGF1R phosphorylation determined by western immunoblotting of equal quantities of protein from whole cell lysates. (B) Effect of increasing concentrations of anticancer drug 17-AAG (targeting Hsp90) on QGP-1 cell growth and levels of indicated biomarkers, with experiments performed as described in (A). (C) Effect of increasing concentrations of anticancer therapeutic axitinib (targeting VEGFR1–3, PDGFRA-B, and KIT) on QGP-1 cell growth and levels of indicated biomarkers, with experiments performed as described in (A). MTS data presented were the average ± SE of three experiments; western immunoblotting results were representative blots from one of three experiments.
Incubation of QGP-1 cells (24 h) with 17-AAG concentrations ranging from 0.3 µM to 10 µM resulted in dose-dependent decreases in the levels of the client proteins EGFR, IGF1R, and VEGFR2 (the principal mediator of VEGF signaling [review 34]) as measured by western analysis (n = 3). Figure 3B illustrates in QGP-1 cells the 17-AAG-induced dose-dependent decrease in cell proliferation and in cellular levels of EGFR, IGF1R, and VEGFR2. In comparison, 17-AAG, with ten-fold greater antiproliferative activity in the NCI-H727 carcinoid line (IC50 of 70.4 nM), decreased in a dose-related fashion the levels in NCI-H727 cells of EGFR and IGF1R [see 7] but not VEGFR2 (data not shown).
Western blot analysis determined that QGP-1 cells exposed (24h) to axitinib concentrations from 0.1 µM to 10 µM exhibited increasing levels of p21Waf1/Cip1(CDKN1A) (p21) expression with no change in constitutive VEGFR2 phosphorylation (n = 3). Figure 3C presents data on the growth inhibition, increased p21 expression, and constant level of constitutive VEGFR2 phosphorylation seen in QGP-1 cells exposed to axitinib in a dose-related fashion. In contrast, while sunitinib was moderately less potent in NCI-H727 cells (IC50 of 3.24 µM; n = 4), axitinib had little effect on the proliferation of the NCI-H727 carcinoid line. An axitinib concentration of 10 µM inhibited NCI-H727 growth less than 50% (n = 3), the solubility limit in RPMI preventing an accurate IC50 determination.
In analyzing the PET cells for genetic abnormalities, EGFR aneusomy was detected by FISH in the QGP-1 line, with elevated EGFR copy number predictive of sensitivity to panitumumab and cetuximab; these results were found in NCI-H727 carcinoid cells as well [see 7]. QGP-1 cells were also EGFR FISH-positive, with high aneusomy associated with gefitinib response, as were NCI-H727 cells [see 7]. Mutational analyses detected no EGFR, KIT, or PDGFRA activating mutations in either the QGP-1 or NCI-H727 line. However, QGP-1 (as well as NCI-H727) cells were homozygous for the dbSNP rs1873778 in PDGFRA exon 12; the NCI-H727 line was heterozygous for the dbSNP rs2228230 in PDGFRA exon 18. Furthermore, in contradistinction to the reported absence in QGP-1 cells of mutations in KRAS codons 12 and 13 associated with non-response to anti-EGFR therapy [35], our analysis indicated that QGP-1 cells harbored the KRAS non-response codon 12 mutation G35T (encoding G12V); NCI-H727 cells also exhibited this mutation, which was heterozygous in both lines. Consistent with the KRAS mutational status of QGP-1 cells, gefitinib exhibited the lowest antiproliferative activity of the therapeutic agents analyzed in the QGP-1 line, with a 9- to 90-fold higher IC50 value (Table 3). In NCI-H727 cells, increasing concentrations of gefitinib inhibited proliferation in a dose-related, biphasic-like fashion, with gefitinib concentrations ≥ 5 µM inhibiting growth more than 50% (n = 6). Of note, the non-response KRAS mutation detected in QGP-1 PET cells was absent from all PET tissues analyzed.
Finally, immunohistochemical analysis of QGP-1 cells for the ten molecular markers evaluated in PET TMA sections determined that strong immunohistochemical staining (IHC staining intensity score of 3) was exhibited by all but three biomarkers: MGMT and Hsp90 (intensity score of 2) and SSTR2A (score of 1). In the NCI-H727 line, all molecular markers were strongly expressed except MGMT (score of 2). Interestingly, the biomarkers that exhibited the strongest IHC staining in the largest number of PETs (VEGFR1, TGFBR1, PDGFRA, SSTR5, SSTR2A, and IGF1R) were all strongly expressed in QGP-1 PET cells as well, with the exception of SSTR2A.
Discussion
Of 1483 cases of PETs diagnosed and recorded in the SEER registry from 1973–2000, 80.9% of the patients presented with metastatic or regionally advanced tumors [2]. Whereas somatostatin therapy is effective for clinical symptoms and may induce tumor growth stabilization [review 4], PETs generally have a short-lived duration of response to conventional chemotherapeutic agents [review 36]; while recent advances have been made in significantly prolonging progression-free survival with everolimus and sunitinib [5, 6], new therapeutic strategies, especially strategies inducing higher radiographic tumor response rates, are needed. We analyzed a collection of PETs for genetic abnormalities and protein expression of a variety of growth factor receptors and downstream effectors and regulators targeted by therapeutics developed for other forms of cancer and assessed previously in a heterogeneous collection of 104 neuroendocrine (carcinoid) tumors [7].
From analyses for genetic abnormalities in PETs, the EGFR FISH assay provided results of note. Elevated EGFR copy number was detected in 38% of all assessable cases, whereas moderate EGFR expression was displayed immunohistochemically in the majority of PETs. In comparison, the first immunohistochemical study of both EGFR and activated-EGFR in a heterogeneous collection of metastatic PETs (48 cases) reported EGFR expression in only 25% of PET primaries and 18% of metastatic PETs; these values were 23% and 32%, respectively, for phosphorylated-EGFR expression [37]. Interestingly, 73% of the cases with elevated EGFR copy number in this report were well differentiated endocrine carcinomas; these results, together with the absence of KRAS non-response mutations in PETs, suggested further research into the role of anti-EGFR monoclonal antibodies for selected PETs.
Protein expression of ten biomarkers was compared immunohistochemically, including that for SSTR2A and SSTR5. Of the five human somatostatin receptor subtypes, SSTR2 and SSTR5 have the highest binding affinity for somatostatin analogs employed clinically for gastroenteropancreatic neuroendocrine tumors (primarily octreotide and lanreotide) [review 38]. The high expression levels of SSTR2A (the long form of SSTR2) and SSTR5 in the majority of PETs were consistent with the therapeutic importance of somatostatin analogs in PET disease for improving clinical symptoms and potentially stabilizing tumor growth. Although somatostatin analogs have been shown to improve time to progression in carcinoid tumors [39], no similar studies in patients with PETs have been performed. Despite the lack of clinical data, somatostatin analogs are frequently employed in patients with PETs.
In contrast, MGMT displayed the lowest immunohistochemical expression level of ten biomarkers analyzed in this collection of 67 PETs, with 24% exhibiting no detectable staining. MGMT deficiency, a predictive marker for PET sensitivity to temozolomide [25], was less common in this study than in an earlier report of 37 archival PETs in which 51% were MGMT deficient [25].
Our results indicated that, aside from the prevalence of somatostatin receptors in both malignancies, PETs exhibited a different immunohistochemical expression profile for growth factor receptor and downstream regulators than did a heterogeneous collection of neuroendocrine (carcinoid) tumors (104 samples consisting predominantly of bronchopulmonary (21%) and small bowel (17%) primaries as well as liver (29%) and lymph node (10%) metastases [7]). The biomarkers for which the largest number of PETs exhibited the strongest IHC staining were, in decreasing order, VEGFR1, TGFBR1, PDGFRA, and IGF1R as compared to the sequence Hsp90, TGFBR1, and IGF1R in carcinoid tumors (Hsp90 and IGF1R in small bowel primaries alone) [7]. The expression of high levels of numerous growth factor receptors and regulators in large percentages of PETs encouraged follow-up in vitro studies of the effects on PET cells of anticancer drugs targeting these biomarkers.
QGP-1 cell growth was inhibited by anticancer drug BMS-754807 (an anti-IGF1R/IR TKI currently in phase I clinical trials for treatment of solid tumors) at nM concentrations that inhibited downstream constitutive IGF1R phosphorylation. In addition, QGP-1 proliferation was sensitive to chemotherapeutic 17-AAG (an Hsp90 small molecule inhibitor in phase II clinical trials for treatment of advanced malignancies) at nM concentrations that induced loss of client proteins EGFR, IGF1R, and VEGFR2. Finally, the therapeutic agent axitinib (a TKI targeting VEGFR1–3/PDGFRA-B/KIT in phase II clinical trials for treatment of solid tumors) inhibited QGP-1 growth at nM concentrations that increased expression of cyclin-dependent kinase inhibitor p21 without inhibiting constitutive VEGFR2 phosphorylation, a pattern suggestive of treatment-induced senescence in cancer [review 40].
While this report is the first immunohistochemical study of IGF1R in a heterogeneous collection of PETs, IGF1R was detected by IHC in 31 (of 32) gastrinomas assessed previously [41]. No clinical trials of anti-IGF1R small molecule TKI monotherapy for PETs have been undertaken, for comparison of treatment response to data presented here on in vitro BMS-754807 antiproliferative activity and strong PET IGF1R expression. However, anti-IGF1R monoclonal antibody monotherapy for PETs has been investigated in clinical studies. Results from a recent phase II clinical trial with dalotuzumab (MK-0646) indicated an absence of antitumor activity in ten patients with metastatic PETs; IGF1R tumor expression data were unavailable [42]. Of note, it is well established that functional reciprocal cross-talk between EGFR and IGF1R occurs in other forms of cancer [43], in which adaptive activation of HER family members occurs upon inhibition of IGF1R (and vice versa). Thus, further research into developing anti-IGF1R TKIs for potential PET anticancer treatment might benefit from investigating these therapeutics as part of multi-drug, rather than single-agent, strategies targeting the HER family of receptors as well.
Similarly, no clinical studies in patients with PETs have been performed with single-drug axitinib, the more selective and potent of the two anti-VEGFR1–3/PDGFRA-B TKIs analyzed in QGP-1 cells, for comparison of treatment response to data here on in vitro antiproliferative activity and strong tumor expression of VEGFR1 and PDGFRA. However, sunitinib monotherapy has been investigated in phase II and III clinical trials in patients with PETs, although no VEGFR1–3 and PDGFRA-B tumor expression data are available [44, 6]. Characteristics of the PET growth inhibition induced by axitinib and sunitinib in vitro and in vivo, respectively, are consistent with a cytostatic mechanism. Axitinib induced evidence of senescence in PET cells, a response possibly exploitable in follow-up combination studies with conventional chemotherapeutics cytotoxic to growth arrested cells. Sunitinib monotherapy in a phase II trial in patients with PETs induced a level of tumor shrinkage classified as stable disease by Response Evaluation Criteria in Solid Tumors in a majority (62.1%) of patients, while the objective response rate was 16.7% [44]. Furthermore, in a recent phase III study of sunitinib monotherapy in patients with PETs, the median progression-free survival in the sunitinib-treated group was more than double that of the placebo-treated group, whereas the objective response rate was 9.3% [6]. Taken together, these results are encouraging of combination, rather than monotherapy, studies of axitinib to fully explore its potential for development as antitumor therapy in PETs, i.e., by combining axitinib with conventional chemotherapeutics toxic to tumor cells in growth arrest.
In contrast to the in vitro effects of TKIs BMS-754807 and axitinib, antiproliferative concentrations of inhibitor 17-AAG, targeting the ubiquitous molecular chaperone Hsp90, depleted multiple growth factor receptors in QGP-1 cells in a concurrent fashion (EGFR, IGF1R, and VEGFR2). No clinical trials of 17-AAG monotherapy for PETs have been undertaken; however, Hsp90 was moderately strongly expressed in the majority of these tumors, suggesting that further investigation into the potential for targeting Hsp90 for development of new anticancer drugs for this malignancy might benefit from multi-drug strategies that include a cytotoxic chemotherapeutic with a different mode of action.
Pancreatic endocrine tumors are commonly diagnosed at advanced stages of disease for which few current therapies induce high tumor regression rates. Ongoing investigations in our laboratory are based on this multifaceted study, which encouraged further research into the role in some PETs of multi-drug strategies incorporating anti-EGFR monoclonal antibody, anti-IGF1R TKI BMS-754807, or Hsp90 inhibitor 17-AAG as well as combination studies with axitinib and conventional chemotherapeutics maximally toxic to growth-arrested cancer cells.
Supplementary Material
Acknowledgments
Sources of Funding: This study received support from the Mayo Comprehensive Cancer Center grant (CA15083). The Mayo Clinic SPORE in Pancreatic Cancer grant (P50 CA102701) provided funding for construction of the PET TMA, including salary support (M.H.M.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of Interest No conflicts of interest were declared.
References
- 1.Halfdanarson TR, Rubin J, Farnell MB, et al. Pancreatic endocrine neoplasms: epidemiology and prognosis of pancreatic endocrine tumors. Endocr Relat Cancer. 2008;15:409–427. doi: 10.1677/ERC-07-0221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Halfdanarson TR, Rabe KG, Rubin J, et al. Pancreatic neuroendocrine tumors (PNETs): incidence, prognosis and recent trend toward improved survival. Ann Oncol. 2008;19:1727–1733. doi: 10.1093/annonc/mdn351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Strosberg JR, Halfdanarson TR. Survival analyses of pancreatic neuroendocrine tumors: Contrasting institutional databases with population-based studies. J Clin Oncol. 2011;29(suppl4):186. [Google Scholar]
- 4.Modlin IM, Oberg K, Chung DC, et al. Gastroenteropancreatic neuroendocrine tumours. Lancet Oncol. 2008;9:61–72. doi: 10.1016/S1470-2045(07)70410-2. [DOI] [PubMed] [Google Scholar]
- 5.Yao JC, Shah MH, Ito T, et al. Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med. 2011;364:514–523. doi: 10.1056/NEJMoa1009290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Raymond E, Dahan L, Raoul J-L, et al. Sunitinib malate for the treatment of pancreatic neuroendocrine tumors. N Engl J Med. 2011;364:501–513. doi: 10.1056/NEJMoa1003825. [DOI] [PubMed] [Google Scholar]
- 7.Gilbert JA, Adhikari LJ, Lloyd RV, Rubin J, et al. Molecular markers for novel therapies in neuroendocrine (carcinoid) tumors. Endocr Relat Cancer. 2010;17:623–636. doi: 10.1677/ERC-09-0318. [DOI] [PubMed] [Google Scholar]
- 8.Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non—small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 9.Paez JG, Janne PA, Lee JC, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 10.Cappuzzo F, Hirsch FR, Rossi E, et al. Epidermal growth factor receptor gene and protein and gefitinib sensitivity in non-small-cell lung cancer. J Natl Cancer Inst. 2005;97:643–655. doi: 10.1093/jnci/dji112. [DOI] [PubMed] [Google Scholar]
- 11.Hirsch FR, Varella-Garcia M, McCoy J, et al. Increased epidermal growth factor receptor gene copy number detected by fluorescence in situ hybridization associates with increased sensitiviy to gefitinib in patients with bronchioloalveolar carcinoma subtypes: a southwest oncology group study. J Clin Oncol. 2005;23:6838–6845. doi: 10.1200/JCO.2005.01.2823. [DOI] [PubMed] [Google Scholar]
- 12.Moroni M, Veronese S, Benvenuti S, et al. Gene copy number for epidermal growth factor receptor (EGFR) and clinical response to antiEGFR treatment in colorectal cancer: a cohort study. Lancet Oncol. 2005;6:279–286. doi: 10.1016/S1470-2045(05)70102-9. [DOI] [PubMed] [Google Scholar]
- 13.Sartore-Bianchi A, Moroni M, Veronese S, et al. Epidermal growth factor receptor gene copy number and clinical outcome of metastatic colorectal cancer treated with panitumumab. J Clin Oncol. 2007;25:3238–3245. doi: 10.1200/JCO.2007.11.5956. [DOI] [PubMed] [Google Scholar]
- 14.Cappuzzo F, Finocchiaro G, Rossi E, et al. EGFR FISH assay predicts for response to cetuximab in chemotherapy refractory colorectal cancer patients. Ann Oncol. 2008;19:717–723. doi: 10.1093/annonc/mdm492. [DOI] [PubMed] [Google Scholar]
- 15.Lievre A, Bachet J-B, Le Corre D, et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 2006;66:3992–3995. doi: 10.1158/0008-5472.CAN-06-0191. [DOI] [PubMed] [Google Scholar]
- 16.De Roock W, Piessevaux H, De Schutter J, et al. KRAS wild-type state predicts survival and is associated to early radiological response in metastatic colorectal cancer treated with cetuximab. Ann Oncol. 2008;19:508–515. doi: 10.1093/annonc/mdm496. [DOI] [PubMed] [Google Scholar]
- 17.Karapetis CS, Khambata-Ford S, Jonker DJ, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med. 2008;359:1757–1765. doi: 10.1056/NEJMoa0804385. [DOI] [PubMed] [Google Scholar]
- 18.Amado RG, Wolf M, Peeters M, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26:1626–1634. doi: 10.1200/JCO.2007.14.7116. [DOI] [PubMed] [Google Scholar]
- 19.Pao W, Wang TY, Riely GJ, et al. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med. 2005;2:57–61. doi: 10.1371/journal.pmed.0020017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eberhard DA, Johnson BE, Amler LC, et al. Mutations in the epidermal growth factor receptor and in KRAS are predictive and prognostic indicators in patients with non—small-cell lung cancer treated with chemotherapy alone and in combination with erlotinib. J Clin Oncol. 2005;23:5900–5909. doi: 10.1200/JCO.2005.02.857. [DOI] [PubMed] [Google Scholar]
- 21.Massarelli E, Varella-Garcia M, Tang X, et al. KRAS mutation is an important predictor of resistance to therapy with epidermal growth factor receptor tyrosine kinase inhibitors in non—small cell lung cancer. Clin Cancer Res. 2007;13:2890–2896. doi: 10.1158/1078-0432.CCR-06-3043. [DOI] [PubMed] [Google Scholar]
- 22.Perez EA, Roche PC, Jenkins RB, et al. HER2 testing in patients with breast cancer: Poor correlation between weak positivity by immunohistochemistry and gene amplification by fluorescence in situ hybridization. Mayo Clin Proc. 2002;77:148–154. doi: 10.4065/77.2.148. [DOI] [PubMed] [Google Scholar]
- 23.Heinrich MC, Corless CL, Demetri GD, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol. 2003;21:4342–4349. doi: 10.1200/JCO.2003.04.190. [DOI] [PubMed] [Google Scholar]
- 24.Corless CL, Schroeder A, Griffith D, et al. PDGFRA mutations in gastrointestinal stromal tumors: Frequency, spectrum and in vitro sensitivity to imatinib. J Clin Oncol. 2005;23:5357–5364. doi: 10.1200/JCO.2005.14.068. [DOI] [PubMed] [Google Scholar]
- 25.Kulke MH, Hornick JL, Frauenhoffer C, et al. O6-Methylguanine DNA methyltransferase deficiency and response to temozolomide-based therapy in patients with neuroendocrine tumors. Clin Cancer Res. 2009;15:338–345. doi: 10.1158/1078-0432.CCR-08-1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Adhikari LJ, Gilbert JA, Lloyd RV, et al. Biomarker expression in pancreatic endocrine tumors. Mod Pathol. 2011;24(Suppl1s):133A. [Google Scholar]
- 27.Gilbert JA, Goetz MP, Reynolds CA, et al. Molecular analysis of metaplastic breast carcinoma: high EGFR copy number via aneusomy. Mol Cancer Ther. 2008;7:944–951. doi: 10.1158/1535-7163.MCT-07-0570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gilbert JA, Lloyd RV, Ames MM. Lack of mutations in EGFR in gastroenteropancreatic neuroendocrine tumors. N Engl J Med. 2005;353:209–210. doi: 10.1056/NEJM200507143530219. [DOI] [PubMed] [Google Scholar]
- 29.Kaku M, Nishiyama T, Yagawa K, et al. Establishment of a carcinoembryonic antigen-producing cell line from human pancreatic carcinoma. Gann. 1980;71:596–601. [PubMed] [Google Scholar]
- 30.Cory AH, Owen TC, Barltrop JA, et al. Use of an aqueous soluble tetrazolium/formazan assay for cell growth assays in culture. Cancer Commun. 1991;3:207–212. doi: 10.3727/095535491820873191. [DOI] [PubMed] [Google Scholar]
- 31.Heitz PU, Komminoth P, Perren A, et al. Pancreatic endocrine tumours: Introduction. In: DeLellis RA, Lloyd RV, Heitz PU, Eng C, editors. Pathology and Genetics of Tumours of Endocrine Organs. World Health Organization Classification of Tumours. Lyon, France: IARC Press; 2004. pp. 177–182. [Google Scholar]
- 32.Wolff AC, Hammond MEH, Schwartz JN, et al. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for human epidermal growth factor receptor 2 testing in breast cancer. J Clin Oncol. 2007;25:118–145. doi: 10.1200/JCO.2006.09.2775. [DOI] [PubMed] [Google Scholar]
- 33.Cappuzzo F, Varella-Garcia M, Shigematsu H, et al. Increased HER2 gene copy number is associated with response to gefitinib therapy in epidermal growth factor receptor-positive non—small-cell lung cancer patients. J Clin Oncol. 2005;23:5007–5018. doi: 10.1200/JCO.2005.09.111. [DOI] [PubMed] [Google Scholar]
- 34.Roskoski R., Jr VEGF receptor protein-tyrosine kinases: Structure and regulation. Biochem Biophys Res Commun. 2008;375:287–291. doi: 10.1016/j.bbrc.2008.07.121. [DOI] [PubMed] [Google Scholar]
- 35.Kalthoff H, Schmiegel W, Roeder C, et al. p53 and K-RAS alterations in pancreatic epithelial cell lesions. Oncogene. 1993;8:289–298. [PubMed] [Google Scholar]
- 36.Metz DC, Jensen RT. Gastrointestinal neuroendocrine tumors: Pancreatic endocrine tumors. Gastroenterology. 2008;135:1469–1492. doi: 10.1053/j.gastro.2008.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Papouchado B, Erickson LA, Rohlinger AL, et al. Epidermal growth factor receptor and activated epidermal growth factor receptor expression in gastrointestinal carcinoids and pancreatic endocrine carcinomas. Mod Pathol. 2005;18:1329–1335. doi: 10.1038/modpathol.3800427. [DOI] [PubMed] [Google Scholar]
- 38.Patel YC. Somatostatin and its receptor family. Front Neuroendocrinol. 1999;20:157–198. doi: 10.1006/frne.1999.0183. [DOI] [PubMed] [Google Scholar]
- 39.Rinke A, Muller H-H, Schade-Brittinger C, et al. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: a report from the PROMID Study Group. J Clin Oncol. 2009 Oct 1;27(28):4656–4663. doi: 10.1200/JCO.2009.22.8510. [DOI] [PubMed] [Google Scholar]
- 40.Ewald JA, Desotelle JA, Wilding G, et al. Therapy-induced senescence in cancer. J Natl Cancer Inst. 2010;102:1536–1546. doi: 10.1093/jnci/djq364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Furukawa M, Raffeld M, Mateo C, et al. Increased expression of insulin-like growth factor I and/or its receptor in gastrinomas is associated with low curability, increased growth, and development of metastases. Clin Cancer Res. 2005;11:3233–3242. doi: 10.1158/1078-0432.CCR-04-1915. [DOI] [PubMed] [Google Scholar]
- 42.Reidy DL, Hollywood E, Segal M, et al. A phase II clinical trial of MK-0646, an insulin-like growth factor-1 receptor inhibitor (IGF-1R), in patients with metastatic well-differentiated neuroendocrine tumors (NETs) J Clin Oncol. 2010;28(15Suppl):4163. [Google Scholar]
- 43.Haluska P, Carboni JM, TenEyck C, et al. HER receptor signaling confers resistance to the insulin-like growth factor-I receptor inhibitor, BMS-536924. Mol Cancer Ther. 2008;7:2589–2598. doi: 10.1158/1535-7163.MCT-08-0493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kulke MH, Lenz H-J, Meropol NJ, et al. Activity of sunitinib in patients with advanced neuroendocrine tumors. J Clin Oncol. 2008;26:3403–3410. doi: 10.1200/JCO.2007.15.9020. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.