

Abstract
To continue our efforts toward the development of 99mTc PiB analogs, we have synthesized twenty-four neutral and lipophilic Re (as a surrogate of 99mTc) 2-arylbenzothiazoles, and explored their structure-activity relationship for binding to Aβ1–40fibrils. These Re complexes were designed and synthesized via the integrated approach, so their 99mTc analogs would have a greater chance of crossing the blood-brain barrier. While the lipophilicities (logPC18= 1.59–3.53) of these Re 2-arylbenzothiazoles were all within suitable range, their binding affinities (Ki= 30–617 nM) to Aβ1–40 fibrils varied widely depending on the selection and integration of the tetradentate chelator into the 2-phenylbenzothiazole pharmacophore. For potential clinical applications, further refinement to obtain Re 2-arylbenzothiazoles with better binding affinities (< 10 nM) will likely be needed. The integrated approach reported here to generate compact, neutral and lipophilic Re 2-arylbenzothiazoles could be applied to other potent pharmacophores as well to convert other current Aβ PET tracers to their 99mTc analogs for more widespread application via the use of SPECT scanners.
Keywords: Technetium-99m, Rhenium, SPECT, PiB, Flutemetamol, β-Amyloid plaques, Alzheimer’s disease
Alzheimer’s disease (AD) is a progressive and fatal neurodegenerative disorder characterized by irreversible memory impairment, continuous cognitive decline and behavioral disturbances. AD causes about two thirds of dementia in the elderly1. It is estimated that by the year of 2050, there will be 13.2 million cases of AD in the US2. At present, there is no medical treatment that cures or prevents AD. The production and accumulation of β-amyloid peptides (Aβ) is believed to be pivotal to the pathogenesis and progression of AD3, and therefore, research on the treatment of AD has focused on the anti-amyloid therapies4. It is well documented that the formation of Aβ plaques precedes the appearance of clinical symptoms5. In order to achieve the best therapeutic outcome, it may be necessary to identify potential subjects for therapy before neurons are damaged by Aβ aggregates. Therefore, the development of a noninvasive imaging method capable of quantifying the deposition of Aβ plaques could provide a useful tool for identifying preclinical cases of AD as candidates for early intervention and to follow the effectiveness of anti-amyloid therapy in individual patients6.
Toward this end, the development of Aβ plaque-targeting radiotracers for use with positron emission tomography (PET) and single photon emission tomography (SPECT) has been an active research topic in the past two decades7–8. PET and SPECT are effective nuclear imaging modalities to detect probes that bind saturable binding sites because their high sensitivity is suitable for extremely low tracer concentrations. Both modalities are now commonly coupled with computed tomography (CT) to generate hybrid images that provide the benefits of both structural and functional/molecular information. Currently, there are several 11C- and 18F-labeled Aβ PET tracers that have been successfully applied in clinical research studies of AD (Figure 1). Among these imaging agents, 2-(4-[11C]methylaminophenyl)-6-hydroxybenzothiazole (Pittsburgh Compound B, PiB)9 has a high signal-to-noise ratio and has been adopted to perform AD-related research studies worldwide. Unfortunately, due to its short half-life (20 min), the 11C label on PiB limits its use to major academic PET facilities with on-site cyclotrons and sophisticated radiochemistry laboratories. Promising radiotracers labeled with the longer half-life (110 min) radioisotope 18F have been developed. Among them, Florbetapir10 has recently been approved by the US Food and Drug Administration (FDA) for clinical use for ruling out AD. Manufacturers of other 18F-labeled tracers such as Florbetaben11, Flutemetamol12 and NAV469413 are expected to seek FDA approval in the next few years. These 18F-labeled PET tracers could increase the availability of Aβ imaging to all PET facilities, but this still represents a minority of modern hospitals with PET scanners. Many more hospitals have the capacity to perform SPECT imaging. Aβ imaging agents labeled with SPECT radionuclides, particularly inexpensive and readily available 99mTc will have more widespread clinical applicability especially in developing countries.
Figure 1.

PET Aβ imaging agents currently under clinical evaluation.
With the success in the development of the 2-arylbenzothiazole (2-ABT) based PET radiotracers, PiB and Flutemetamol, we were also interested in the development of 99mTc-labeled 2-ABTs for more widespread application using SPECT scanners. In contrast to most attempts by other investigators on the development of 99mTc-labeled 2-ABTs using the pendant approach14–16, we have previously demonstrated the feasibility of design and synthesis of three neutral and lipophilic Re (as a surrogate of 99mTc) 2-ABTs (compounds# 6, 12 and 21 in Table 1) with moderate Aβ binding affinity (30–87 nM) using the integrated approach to minimize their overall molecular weight (<550 daltons)17. Compound 6 was prepared using a thiol-triamine SN3 tetradentate chelator (Scheme 1A), while semi-rigid thiol-diamine-phenol (SN2O) and thiol-diamine-thiol (SN2S) chelators (Scheme 1B, X= O and S) were used for the preparation of 12 and 21, respectively. Besides the SN3, SN2O and SN2S chelators, the semi-rigid thiol-diamine-thioether (SN2Sether)18 chelator is also commonly used for the design and synthesis of neutral and lipophilic Tc/Re complexes. While SN3, SN2X, SN2Sether chelators all form stable Tc/Re complexes, the preparation of Tc/Re 2-ABTs by the use of different tetradentate chelators or the same chelator integrated at different positions of the pharmacophore might generate Tc/Re 2-ABTs with different binding affinity, lipophilicity and in vivo pharmacokinetics. The aim of this present work was to systematically explore the potential of utilizing SN3, SN2X, and SN2Sether chelators to generate compact, neutral, and lipophilic Re 2-ABTs, and to investigate the structure-activity relationship for their lipophilicity and binding affinity to aggregated Aβ.
Table 1.
Properties of Re 2-phenylbenzothiazoles including molecular weight (MW), binding affinity (Ki) to Aβ1–40, and lipophilicity (logPC18).
| Compd # | Structure | R | MW | Ki (nM) | LogPC18 |
|---|---|---|---|---|---|
| 1 |  |
H | 586 (498*) | 556 | 2.54 |
| 2 | F | 604 (516*) | 617 | 2.67 | |
| 3 | OMe | 616 (528*) | 85 | 2.37 | |
| 4 | 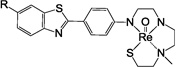 |
H | 586 (498*) | 378 | 2.65 |
| 5 | F | 604 (516*) | 118 | 2.84 | |
| 6 | OMe | 616 (528*) | 87 | 2.52 | |
| 7 |  |
H | 558 (470*) | 90 | 2.33 |
| 8 | F | 576 (488*) | 113 | 2.50 | |
| 9 | OMe | 588 (500*) | 61 | 2.21 | |
| 10 |  |
H | 545 (457*) | 109 | 1.70 |
| 11 | F | 563 (475*) | 64 | 1.90 | |
| 12 | OMe | 575 (487*) | 30 | 1.65 | |
| 13 |  |
H | 545 (457*) | 280 | 1.68 |
| 14 | F | 563 (475*) | 226 | 1.87 | |
| 15 | OMe | 575 (487*) | 140 | 1.59 | |
| 16 |  |
H | 561 (473*) | 264 | 2.49 |
| 17 | F | 579 (491*) | 93 | 2.65 | |
| 18 | OMe | 591 (503*) | 132 | 2.45 | |
| 19 |  |
H | 561 (473*) | 38 | 2.41 |
| 20 | F | 579 (491*) | 31 | 2.60 | |
| 21 | OMe | 591 (503*) | 43 | 2.30 | |
| 22 |  |
H | 575 (487*) | 200 | 3.35 |
| 23 | F | 593 (505*) | 148 | 3.53 | |
| 24 | OMe | 605 (517*) | 178 | 3.25 |
Molecular weight of their corresponding Tc analogs.
Scheme 1.

Design and synthesis of neutral Re 2-arylbenzothiazoles by modification of reported (A) SN3 chelating system, and (B) SN2X chelating system (X= O or S).
As shown in Table 1, besides 6, 12 and 21 that were reported earlier, we synthesized an additional twenty-one Re 2-ABTs with chelators integrated at different positions of 2-ABT pharmacophore for comparison. Our previous results19–20 indicated that substitution on the 2-ABT pharmacophore with an electron-donating group or a halogen significantly increases the binding affinity. For example, the Ki for an amino, N-methylamino, N,N-dimethylamino, fluoro, bromo, or iodo substitution at the 4’-position of 2-ABT were 37.0, 11.0, 4.0, 43.8, 8.8, and 2.6 nM, respectively. The definition of substitution positions is depicted on the structure of PiB shown in Fig. 1. The integration of a tetradentate chelator into the benzothiazole ring provides an electron-donating group at the 6-position (compounds 1–3), whereas the integration of a tetradentate chelator into the phenyl ring provides an electron-donating group at the 4’-position (compounds 4–6) or both the 3’- and 4’-position (compounds 7–24). To further enhance their binding affinity, we also synthesized analogs with a fluoro or methoxy substitution at the 6-position when a tetradentate chelator is integrated into the phenyl ring, or at the 4’-position when a tetradentate chelator is integrated into the benzothiazole ring. The choice of the small halogen fluorine and a methoxy group is to limit the overall size increase. In addition, substitution with an aromatic fluoro or methoxy group will not significantly change the overall lipophilicity.
The synthetic steps for the preparation of Re 2-ABT 1–24 are depicted in Schemes 2–9. The SN3 chelator used for the preparation of 1–6 (Schemes 2 and 3) was modified from the thiol-triamide21–22 chelator as shown in Scheme 1A. Complexation of the original thiol-triamide chelator with [Tc(V)O]3+ or [Re(V)O]3+ led to metal complexes with one negative charge due to the loss of four protons from three amide N-H groups and one thiol S-H group. It is well documented that charged Tc complexes do not cross the blood-brain barrier (BBB). In addition, Tc complexes derived from tetradentate chelators containing amide groups (such as monoamide-monoamine-dithiol, MAMA) showed less brain uptake when compared to those derived from their corresponding amino chelators (such as diaminedithiol, DADT)23–24. Therefore, we modified the original thiol-triamide chelator by replacing the three amide groups with three amino groups. In order to obtain neutral Tc/Re complexes, we also added one small methyl group to one of the aliphatic amino groups. After such modification (see Scheme 1A), all three protons were lost after complexation with [Re(V)O]3+, and the overall charge of 2-ABT 1–5 was balanced, which is in agreement with our previously reported results from the synthesis of 617.
Scheme 2.

Synthesis of 1–3. Reagents: (a) 4-substituted benzoyl chloride, DMF, 140 °C, 18 h, 57–90 %; (b) SnCl2, EtOH, 3 h, reflux, 73–91 %; (c) 2-chloroacetyl chloride, K2CO3, THF, 18 h, rt, 94–99 %; (d) 2-(4-methoxybenzylthio)ethyl bromide, CH2Cl2, 18 h, reflux, 81 %; (e) NaOH, H2O, MeOH, 4 h, rt, 92 %; (f) KI, K2CO3, CH3CN, 14 h, reflux, 35–68 %; (g) LAH, THF, 18 h, rt, 38–80 %; (h) (i) TFA, anisole, rt, 1 h; (ii) Hg(OAc)2 0 °C, 1 h; (iii) H2S, EtOH; (iv) ReO(PPh3)2Cl3, NaOAc, MeOH, 18 h, 75 °C, 16–29 %.
Scheme 9.
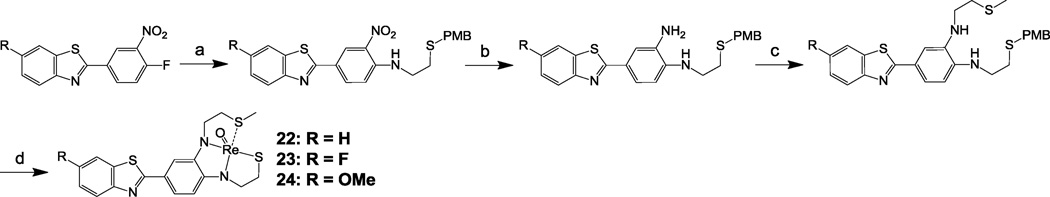
Synthesis of 22–24. Reagents: (a) 2-(4-methoxybenzylthio)ethylamine, K2CO3, DMF, 100 °C, 18 h, 41–96 %; (2) SnCl2, THF, EtOH, 3 h, reflux, 55–98 %; (c) 2-chloroethyl methyl sulfide, KI, K2CO3, CH3CN, 18 h, reflux, 40–64 %; (d) (i) TFA, anisole, rt, 1 h; (ii) Hg(OAc)2 0 °C, 1 h; (iii) H2S, EtOH; (iv) ReO(PPh3)2Cl3, NaOAc, MeOH, 18 h, 75 °C, 25–26 %.
Scheme 3.

Synthesis of 4–6. Reagents: (a) 4-nitrobenzoyl chloride, DMF, 140 °C, 18 h, 38–80 %; (b) SnCl2, EtOH, 3 h, reflux, 88–100 %; (c) 2-chloroacetyl chloride, K2CO3, THF, 18 h, rt, 92–96 %; (d) KI, K2CO3, CH3CN, 18 h, reflux, 63–88 %; (e) LAH, THF, 18 h, rt, 42–58 %; (f) (i) TFA, anisole, rt, 1 h; (ii) Hg(OAc)2 0° C, 1 h; (iii) H2S, EtOH; (iv) ReO(PPh3)2Cl3, NaOAc, MeOH, 18 h, 75 °C, 19–41 %.
Re 2-ABTs 7–9 (Scheme 4), 10–15 (Schemes 5 and 6), and 16–21 (Schemes 7 and 8) were synthesized using modified semi-rigid SN2X chelators (Scheme 1B, X= NH, O, and S, respectively) integrated into the 3’- and 4’-position of phenyl ring. This design was to further reduce their overall molecular weight to 500 daltons or less as suggested by the Rule of Five25, and in hopes that their corresponding 99mTc analogs would show rapid and high brain entry. These semi-rigid SN2X chelating systems were modified from previously reported thiol-diamide-X systems (X= NX, O and S)26. As shown in Scheme 1B, the complexation of [Re(V)O]3+ with thiol-diamide-X chelating systems led to Re complexes with one negative charge. Similar to our modification to the thiol-triamide chelator shown in Scheme 1A, we also replaced the two amide groups with two amino groups in order to produce neutral Re complexes and increase brain uptake of their 99mTc analogs. As expected, after complexation with [Re(V)O]3+, only three protons were lost. The aliphatic amino N-H group that has relatively higher pKa value was not deprotonated after Re complexation reaction, and therefore, the overall charge was balanced. These results were consistent with our previously reported results for the synthesis of 12 and 2117.
Scheme 4.

Synthesis of 7–9. Reagents: (a) 4-fluoro-3-nitrobenzoyl chloride, DMF, 140 °C, 18 h, 58–92 %; (b) K2CO3, DMF, 100°C, 18 h, 27–81 %; (c) SnCl2, EtOH, 3 h, reflux, 10–81 %; (d) (i) TFA, anisole, rt, 1 h; (ii) Hg(OAc)2 0 °C, 1 h; (iii) H2S, EtOH; (iv) ReO(PPh3)2Cl3, NaOAc, MeOH, 18 h, 75 °C, 10–39 %.
Scheme 5.

Synthesis of 10–12. Reagents: (a) 3-hydroxy-4-nitro benzaldehyde, DMSO, 125 °C, 18 h, 15–65 %; (b) MOMCl, DIPEA, THF, 18 h, reflux, 51–71 % (c) NaBH4, Cu(OAc)2, EtOH, rt, 18 h, 96–100 %; (d) 2-chloroacetyl chloride, K2CO3, THF, 18 h, rt, 64–82 %; (e) 2-(4-methoxybenzylthio)ethylamine, KI, K2CO3, CH3CN, 18 h, reflux, 80–98 %; (f) LAH, THF, 18 h, rt, 43–45 %; (g) (i) TFA, anisole, rt, 1 h; (ii) Hg(OAc)2 0 °C, 1 h; (iii) H2S, EtOH; (iv) ReO(PPh3)2Cl3, NaOAc, MeOH, 18 h, 75 °C, 17–49 %.
Scheme 6.

Synthesis of 13–15. Reagents: (a) 4-hydroxy-3-nitro benzaldehyde, DMSO, 125 °C, 18 h, 28–35 %; (b) MOMCl, DIPEA, THF, reflux, 18 h, 98–99 %; (c) NaBH4, Cu(OAc)2, EtOH, rt, 18 h, 92–97 %; (d) 2-chloroacetyl chloride, K2CO3, THF, 18 h, rt, 79–91 %; (e) 2-(4-methoxybenzylthio)ethylamine, KI, K2CO3, CH3CN, 18 h, reflux, 40–96 %; (f) LAH, THF, 17 h, rt, 43–59 %; (g) (i) TFA, anisole, rt, 1 h; (ii) Hg(OAc)2 0 °C, 1h; (iii) H2S, EtOH; (iv) ReO(PPh3)2Cl3, NaOAc, MeOH, 18 h, 75 °C, 20–41 %.
Scheme 7.

Synthesis of 16–18. Reagents: (a) 4-methoxy-α-toluenethiol, K2CO3, DMF, 18 h, 100 °C, 35–77 %; (b) SnCl2, THF, EtOH, 3 h, reflux, 57–95 %; (c) 2-chloroacetyl chloride, K2CO3, THF, 18 h, rt, 90–93 %; (d) 2-(4-methoxybenzylthio)ethylamine, KI, K2CO3, CH3CN, 18 h, reflux, 43–84 %; (e) LAH, THF, 18 h, rt, 24–53 %; (f) (i) TFA, anisole, rt, 1 h; (ii) Hg(OAc)2 0 °C, 1 h; (iii) H2S, EtOH; (iv) ReO(PPh3)2Cl3, NaOAc, MeOH, 18 h, 75 °C, 25–36 %.
Scheme 8.

Synthesis of 19–21. Reagents: (a) 3-fluoro-4-nitrobenzoyl chloride, DMF, 140°C, 18 h, 73–99 %; (b) 4-methoxy-α-toluenethiol, K2CO3, DMF, 18 h, 95 °C, 55–96 %; (c) SnCl2, THF, EtOH, 3 h, reflux, 67–79 %; (d) 2-chloroacetyl chloride, K2CO3, THF, 18 h, rt, 81–98 %; (e) 2-(4-methoxybenzylthio)ethylamine, KI, K2CO3, CH3CN, 18 h, reflux, 75–93 %; (f) LAH, THF, 18 h, rt, 56–81 %; (g) (i) TFA, anisole, rt, 1 h; (ii) Hg(OAc)2 0 °C, 1 h; (iii) H2S, EtOH; (iv) ReO(PPh3)2Cl3, NaOAc, MeOH, 18 h, 75 °C, 12–23 %.
Syntheses of 22–24 are shown in Scheme 9 using the previously reported semi-rigid SN2Sether chelator18. The methyl thioether group was chosen to keep the overall molecular weight at minimum. After complexation with [Re(V)O]3+, as expected, neutral Re 2-ABT 22–24 were obtained resulting from the loss of three protons (two aromatic N-H and one thiol S-H groups). The final step in the preparation of Re 2-ABT 1–24 involved a two-stage reaction, deprotection of p-methoxybenzyl (PMB)/methoxymethyl (MOM) protecting groups to restore the chelating core followed by Re complexation reaction using Re(V)O(PPh3)2Cl327. In spite of potential existence of cis- and anti-isomers, similar to our previous results for the syntheses of 6, 12 and 2117, only one single isomer was isolated for each of these additional twenty-one Re 2-ABTs reported here, and their identities were confirmed by NMR Spectroscopy28.
Re 2-ABT 1–24 are moderately lipophilic with logPC18 (PC18: estimation of Poct by a reverse-phase HPLC method19) in the range of 1.59–3.53 (Table 1). Among them, 10–15 derived from the SN2O chelator displayed the lowest lipophilicity (logPC18= 1.59–1.90), whereas 22–24 derived from the SN2Sether chelator had the highest lipophilicity (logPC18= 3.25–3.53). Replacing the phenol of the SN2O chelator in 10–15 with a thiol resulted in Re SN2S derivatives 16–21 with an average increase of 0.75 in their logPC18 values (10–12 vs 19–21; 13–15 vs 16–18). The Re 2-ABTs derived from the same tetradentate chelator (SN2O or SN2S) but with different integration patterns into the phenyl ring (the amino group substituted at the 3’- or 4’-position) had similar lipophilicity (10–12 vs 13–15; 16–18 vs 19–21). Compared with 1–6 derived from the SN3 chelating system with only one lateral amino group of the chelator integrated into the 2-ABT backbone, 7–9 derived from the semi-rigid SN3 chelator with two amino groups integrated into the phenyl ring had lower lipophilicity due to the presence of an amino N-H proton at the 3’-position and one less ethylene moiety in the overall structure. If comparing the Re 2-ABTs with the same tetradentate chelator but with different substitution (H, F and OMe) at the 6- or 4’-position, the methoxy-substituted 2-ABTs had the lowest logPC18 values with an average of 0.10 and 0.25 lower than those of their respective un-substituted and fluoro-substituted Re 2-ABTs.
Re 2-ABTs 1–24 bind Aβ1–40 fibrils with moderate to poor affinity (Ki= 30–617 nM, Table 1) as determined by previously published in vitro competition binding assays19,29 using [3H]BTA-1 as the radioactive control compound. In general, compared to the un-substituted Re 2-ABTs, substitution with a fluoro or a methoxy group at the 6- or 4’-position enhanced their binding affinity to Aβ1–40 fibrils. The integration of an SN3 chelator into the phenyl ring (in 4–6) rather than the benzothiazole ring (in 1–3) resulted in Re 2-ABTs with better binding affinities. Replacing the free rotating Re-SN3 complex in 4–6 with a semi-rigid Re-SN3 complex in 7–9 further enhance the binding affinity. As discussed above, different integration patterns of the same tetradentate chelator (SN2O or SN2S) had little effects on the overall lipophilicity of the resulted Re 2-ABTs (10–12 vs 13–15; 16–18 vs 19–21). However, the binding affinities of these 2-ABTs were strongly influenced by the integration patterns of the tetradentate chelator. When comparing the 2-ABTs 10–15 with the semi-rigid SN2O chelator, 10–12 (Ki= 30–109 nM) with an amino group of the chelator substituted at the 4’-position had 2.6- to 4.7-fold better binding affinity than their corresponding analogs 13–15 (Ki= 140–280 nM) with the amino group substituted at the 3’-position. Similar results were obtained when comparing the binding affinity of Re 2-ABTs 16–21 with the semi-rigid SN2S chelator. The binding affinities of 19–21 (Ki= 31–43 nM) with an amino group of the SN2S chelator substituted at the 4’-position were 3.1- to 6.9-fold better than those of their corresponding analogs 16–18 (Ki= 93–264 nM) with the amino group substituted at the 3’-position. These results are in agreement with the fact that most of the promising PET tracers derived from the 2-ABT pharmacophore including [11C]PiB, [11C]AZD2184, and [18F]Flutemetamol (see Fig. 1) have an amino group substituted at the 4’-position.
In summary, we have synthesized twenty-four neutral and compact Re 2-ABTs, and measured their lipophilicity and binding affinity to aggregated Aβ. These Re 2-ABTs were designed and prepared via the integrated approach, so their 99mTc analogs would have a greater chance of crossing the BBB, and bind to Aβ plaques deposited in the brain parenchyma. While the lipophilicities of these 2-ABTs were within suitable range (logPC18 = 1–4), their binding affinities (Ki= 30–617 nM) to Aβ1–40 fibrils varied widely depending on the selection of the chelators, and the ways the chelators were integrated into the 2-ABT pharmacophore. Based on the binding affinity data, we have identified two promising semi-rigid chelators, SN2O and SN2S. The Re 2-ABTs 12 and 20 derived from the SN2O and SN2S chelators, respectively, had fairly good binding affinity (Ki ~ 30 nM) to Aβ1–40 fibrils. However, before translation into their 99mTc analogs and for potential clinical application, further modification to obtain Re 2-ABTs with even better binding affinity will likely be needed since most of the clinical Aβ imaging agents have binding affinities less than 10 nM. This might be achievable by optimizing the substitution at the 6-position of the 2-ABT pharmacophore with other potent electron-donating groups, and/or by the 3D-QSAR analysis as recently reported by Kim30 and Yang31. The integrated approach reported here to generate compact, neutral and lipophilic Re 2-arylbenzothiazoles could be applied to generate Re complexes of other potent pharmacophores, including stilbene and benzoxazole. Once potent Re complexes are obtained, their 99mTc analogs will have great potential to extend current Aβ imaging practices from PET to SPECT.
Acknowledgements
This work was supported by the US National Institutes of Health (R21EB009497).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Nussbaum RL, Ellis CE. N. Engl. J. Med. 2003;348:1356. doi: 10.1056/NEJM2003ra020003. [DOI] [PubMed] [Google Scholar]
- 2.Hebert LE, Scherr PA, Bienias JL, Bennett DA, Evans DA. Arch. Neurol. 2003;60:1119. doi: 10.1001/archneur.60.8.1119. [DOI] [PubMed] [Google Scholar]
- 3.Hardy J, Selkoe DJ. Science. 2002;297:353. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 4.Pangalos MN, Jacobsen SJ, Reinhart PH. Biochem. Soc. Trans. 2005;33:553. doi: 10.1042/BST0330553. [DOI] [PubMed] [Google Scholar]
- 5.Pike KE, Savage G, Villemagne VL, Ng S, Moss SA, Maruff P, Mathis CA, Klunk WE, Masters CL, Rowe CC. Brain. 2007;130:2837. doi: 10.1093/brain/awm238. [DOI] [PubMed] [Google Scholar]
- 6.Mathis CA, Lopresti BJ, Klunk WE. Nucl. Med. Biol. 2007;34:809. doi: 10.1016/j.nucmedbio.2007.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mathis CA, Wang Y, Klunk WE. Curr. Pharm. Des. 2004;10:1469. doi: 10.2174/1381612043384772. [DOI] [PubMed] [Google Scholar]
- 8.Mori T, Maeda J, Shimada H, Higuchi M, Shinotoh H, Ueno S, Suhara T. Psychogeriatrics. 2012;12:106. doi: 10.1111/j.1479-8301.2012.00409.x. [DOI] [PubMed] [Google Scholar]
- 9.Klunk WE, Engler H, Nordberg A, Wang YM, Blomqvist G, Holt DP, Bergstrom M, Savitcheva I, Huang GF, Estrada S, Ausen B, Debnath ML, Barletta J, Price JC, Sandell J, Lopresti BJ, Wall A, Koivisto P, Antoni G, Mathis CA, Langstrom B. Ann. Neurol. 2004;55:306. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- 10.Clark CM, Schneider JA, Bedell BJ, Beach TG, Bilker WB, Mintun MA, Pontecorvo MJ, Hefti F, Carpenter AP, Flitter ML, Krautkramer MJ, Kung HF, Coleman RE, Doraiswamy PM, Fleisher AS, Sabbagh MN, Sadowsky CH, Reiman PEM, Zehntner SP, Skovronsky DM. JAMA J. Am. Med. Assoc. 2011;305:275. doi: 10.1001/jama.2010.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rowe CC, Ackerman U, Browne W, Mulligan R, Pike KL, O'Keefe G, Tochon-Danguy H, Chan G, Berlangieri SU, Jones G, Dickinson-Rowe KL, Kung HF, Zhang W, Kung MP, Skovronsky D, Dyrks T, Hall G, Krause S, Friebe M, Lehman L, Lindemann S, Dinkelborg LM, Masters CL, Villemagne VL. Lancet Neurol. 2008;7:129. doi: 10.1016/S1474-4422(08)70001-2. [DOI] [PubMed] [Google Scholar]
- 12.Nelissen N, Van Laere K, Thurfjell L, Owenius R, Vandenbulcke M, Koole M, Bormans G, Brooks DJ, Vandenberghe R. J. Nucl. Med. 2009;50:1251. doi: 10.2967/jnumed.109.063305. [DOI] [PubMed] [Google Scholar]
- 13.Cselenyi Z, Jonhagen ME, Forsberg A, Halldin C, Julin P, Schou M, Johnstrom P, Varnas K, Svensson S, Farde L. J. Nucl. Med. 2012;53:415. doi: 10.2967/jnumed.111.094029. [DOI] [PubMed] [Google Scholar]
- 14.Serdons K, Vanderghinste D, Van Eeckhoudt M, Cleynhens J, de Groot T, Bormans G, Verbruggena A. J. Label. Compd. Radiopharm. 2009;52:227. [Google Scholar]
- 15.Chen X, Yu P, Zhang L, Liu B. Bioorg. Med. Chem. Lett. 2008;18:1442. doi: 10.1016/j.bmcl.2007.12.071. [DOI] [PubMed] [Google Scholar]
- 16.Serdons K, Verduyckt T, Cleynhens J, Terwinghe C, Mortelmans L, Bormansa G, Verbruggen A. Bioorg. Med. Chem. Lett. 2007;17:6086. doi: 10.1016/j.bmcl.2007.09.055. [DOI] [PubMed] [Google Scholar]
- 17.Lin KS, Debnath ML, Mathis CA, Klunk WE. Bioorg. Med. Chem. Lett. 2009;19:2258. doi: 10.1016/j.bmcl.2009.02.096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McBride BJ, Baldwin RM, Kerr JM, Wu JL, Schultze LM, Salazar NE, Chinitz JM, Byrne EF. J. Med. Chem. 1993;36:81. doi: 10.1021/jm00053a010. [DOI] [PubMed] [Google Scholar]
- 19.Mathis CA, Wang YM, Holt DP, Huang GF, Debnath ML, Klunk WE. J. Med. Chem. 2003;46:2740. doi: 10.1021/jm030026b. [DOI] [PubMed] [Google Scholar]
- 20.KlunK WE, Mathis CA. 0142269A1. U.S. Patent. 2009
- 21.Eisenhut M, Mohammed A, Mier W, Schoensiegel F, Friebe M, Mahmood A, Jones AG, Haberkorn U. J. Med. Chem. 2002;45:5802. doi: 10.1021/jm021026z. [DOI] [PubMed] [Google Scholar]
- 22.Hansen L, Cini R, Taylor A, Jr, Marzilli LG. Inorg. Chem. 1992;31:2801. [Google Scholar]
- 23.Zhuang ZP, Kung MP, Hou C, Ploessl K, Kung HF. Nucl. Med. Biol. 2005;32:171. doi: 10.1016/j.nucmedbio.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 24.Oya S, Plossl K, Kung MP, Stevenson DA, Kung HF. Nucl. Med. Biol. 1998;25:135. doi: 10.1016/s0969-8051(97)00153-4. [DOI] [PubMed] [Google Scholar]
- 25.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Adv. Drug. Deliver. Rev. 1995;23:3. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 26.Le Gal J, Tisato F, Bandoli G, Gressier M, Jaud J, Michaud S, Dartiguenave M, Benoist E. Dalton Trans. 2005;23:3800. doi: 10.1039/b508661b. [DOI] [PubMed] [Google Scholar]
- 27.General procedures for the synthesis of Re complexes 1–24: A solution of the respective PMB/MOM-protected precursor (0.2 mmol) in trifluoroacetic acid (5 mL) and anisole (0.2 mL) was stirred at room temperature for 1 h, then cooled in ice/water bath. Hg(OAc)2 (0.48 mmol for the preparation of 16–21, and 0.24 mmol for the preparation of other Re complexes) was added, and the resulting solution was stirred at 0 °C for another 1 h. After removing volatile trifluoroacetic acid under reduced pressure, the residue was washed with diethyl ether (20 mL), and then dissolved in ethanol (15 mL). To this solution was bubbled in hydrogen sulfide for 10 min, the resulting black precipitate was removed by filtration through celite. The filtrate was concentrated under reduced pressure. ReO(PPh3)2Cl3 (208 mg, 0.25 mmol) and NaOAc methanolic solution (1.0 M, 20 mL) were added to the residue, and the resulting solution was heated at 75 °C for 18 h. After cooling to room temperature, the solution was diluted with water (50 mL), and extracted with chloroform (50 mL). The organic phase was dried over anhydrous magnesium sulfate and concentrated under reduced pressure. The crude product was purified by chromatography on silica gel eluting with 10:90 methanol/ethyl acetate to give the expected product in 10–49 % yields.
- 28.NMR spectra were recorded using a Bruker Avance 400inv NMR spectrometer. Chemical shifts (δ) are reported in parts per million (ppm). Compound 1 (DMSO-d6): 2.04–2.16 (m, 1H), 2.71–2.79 (m, 1H), 3.19 (s, 3H), 3.21–3.27 (m, 2H), 3.39–3.45 (m, 1H), 3.49–3.57 (m, 1H), 3.61–3.70 (m, 1H), 3.83–3.90 (m, 1H), 3.84 (s, 3H), 3.91–3.40 (m, 2H), 3.66–4.73 (m, 1H), 7.36 (dd, J = 8.8, 2.4 Hz, 1H), 7.52–7.64 (m, 3H), 7.70 (d, J = 2.4 Hz, 1H), 7.81 (d, J = 8.8 Hz, 1H), 8.02 (dd, J = 7.8, 2.0 Hz, 2H); Compound 2 (DMSO-d6): 2.06–2.16 (m, 1H), 2.71–2.79 (m, 1H), 3.19 (s, 3H), 3.21–3.27 (m, 2H), 3.39–3.46 (m, 1H), 3.49–3.58 (m, 1H), 3.62–3.71 (m, 1H), 3.83–3.90 (m, 1H), 3.91–3.40 (m, 2H), 3.66–4.73 (m, 1H), 7.33–7.42 (m, 3H), 7.73 (d, J = 2.0 Hz, 1H), 7.81 (d, J = 8.8 Hz, 1H), 8.04–8.09 (m, 2H); Compound 3 (DMSO-d6): 2.06–2.16 (m, 1H), 2.71–2.79 (m, 1H), 3.19 (s, 3H), 3.21–3.29 (m, 2H), 3.39–3.46 (m, 1H), 3.50–3.59 (m, 1H), 3.62–3.70 (m, 1H), 3.83–3.90 (m, 1H), 3.84 (s, 3H), 3.91–3.40 (m, 2H), 3.66–4.73 (m, 1H), 7.10 (d, J = 8.8 Hz, 2H), 7.32 (dd, J = 8.8, 2.4 Hz, 1H), 7.67 (d, J = 2.4 Hz, 1H), 7.76 (d, J = 8.8 Hz, 1H), 7.96 (d, J = 8.8 Hz, 2H); Compound 4 (DMSO-d6): 2.13–2.20 (m, 1H), 2.53–2.58 (m, 1H), 2.75–2.79 (m, 1H), 3.20 (s, 3H), 3.22–3.40 (m, 2H), 3.43–3.50 (m, 2H), 3.64–3.71 (m, 1H), 3.84–3.90 (m, 2H), 3.94–3.99 (m, 1H), 4.59–4.64 (m, 1H), 7.24 (d, J = 8.8 Hz, 2H), 7.36–7.40 (m, 1H), 7.47–7.51 (m, 1H), 7.79 (d, J = 8.8 Hz, 2H), 7.96 (d, J = 8.1 Hz, 1H), 8.70 (d, J = 7.9 Hz, 1H); Compound 5 (DMSO-d6): 2.13–2.20 (m, 1H), 2.53–2.58 (m, 1H), 2.75–2.79 (m, 1H), 3.20 (s, 3H), 3.22–3.40 (m, 2H), 3.43–3.50 (m, 2H), 3.64–3.70 (m, 1H), 3.80–3.91 (m, 2H), 3.93–3.99 (m, 1H), 4.60–4.64 (m, 1H), 7.24 (d, J = 8.9 Hz, 2H), 7.32–7.38 (m, 1H), 7.87 (d, J = 8.8 Hz, 2H), 7.95–8.01 (m, 2H); Compound 7 (DMSO-d6): 2.17–2.25 (m, 1H), 3.07–3.23 (m, 3H), 3.30 (s, 3H), 3.74–3.79 (m, 1H), 4.06–4.11 (m, 1H), 4.62–4.69 (m, 1H), 6.88 (d, J= 8.1 Hz, 1H), 7.34–7.38 (m, 2H), 7.46–7.50 (m, 1H), 7.58 (d, J = 1.8 Hz, 1H), 7.95 (d, J = 7.9 Hz, 1H), 8.05 (d, J = 7.5 Hz, 1H), 11.3 (s, 1H); Compound 8 (DMSO-d6): 2.16–2.25 (m, 1H), 3.05–3.23 (m, 3H), 3.29 (s, 3H), 3.73–3.80 (m, 1H), 4.04–4.11 (m, 1H), 4.60–4.69 (m, 1H), 6.87 (d, J = 8.0 Hz, 1H), 7.30–7.36 (m, 2H), 7.54 (d, J = 1.6 Hz), 7.93–7.98 (m, 2H), 11.30 (s, 1H); Compound 9 (DMSO-d6): 2.15–2.25 (m, 1H), 3.05–3.23 (m, 3H), 3.27 (s, 3H), 3.73–3.80 (m, 1H), 3.83 (s, 3H), 4.02–4.10 (m, 1H), 4.60–4.69 (m, 1H), 6.85 (d, J = 8.0 Hz, 1H), 7.06 (dd, J = 8.8, 2.4 Hz, 1H), 7.29 (dd, J = 8.0, 2.0 Hz, 1H), 7.51 (d, J = 1.6 Hz, 1H), 7.62 (d, J = 2.4 Hz, 1H), 7.83 (d, J = 8.8 Hz, 1H), 11.30 (s, 1H); Compound 10 (DMSO-d6): 2.14–2.28 (m, 1H), 2.84–2.95 (m, 1H), 2.98–3.05 (m, 1H), 3.17–3.27 (m, 1H), 3.61–3.71 (m, 1H), 3.86–4.00 (m, 2H), 4.38–4.46 (m, 1H), 7.00 (d, J = 8.0 Hz, 1H), 7.36–7.41 (m, 1H), 7.47–7.53 (m, 2H), 7.64 (d, J = 1.6 Hz, 1H), 7.98 (d, J = 8.0 Hz, 1H), 8.07 (d, J = 7.2 Hz, 1H), 9.46 (s, 1H); Compound 11 (DMSO-d6): 2.15–2.23 (m, 1H), 2.83–2.94 (m, 1H), 2.97–3.05 (m, 1H), 3.18–3.28 (m, 1H), 3.60–3.71 (m, 1H), 3.86–4.01 (m, 2H), 4.36–4.46 (m, 1H), 6.99 (d, J = 8.0 Hz, 1H), 7.35 (td, J = 8.8, 2.4 Hz, 1H), 7.48 (dd, J = 8.0, 2.0 Hz, 1H), 7.57–7.64 (m, 1H), 7.96–8.02 (m, 2H), 9.46 (s, 1H); Compound 13 (DMSO-d6): 2.15–2.27 (m, 1H), 2.85–2.95 (m, 1H), 2.96–3.04 (m, 1H), 3.18–3.27 (m, 1H), 3.64–3.74 (m, 1H), 3.91–4.00 (m, 2H), 4.47–4.57 (m, 1H), 7.12 (d, J = 8.0 Hz, 1H), 7.36–7.42 (m, 1H), 7.44–7.52 (m, 2H), 7.55 (d, J = 1.6 Hz, 1H), 7.98 (d, J = 7.6 Hz, 1H), 8.07 (d, J = 7.6 Hz, 1H), 9.47 (s, 1H); Compound 14 (DMSO-d6): 2.15–2.27 (m, 1H), 2.85–2.95 (m, 1H), 2.96–3.04 (m, 1H), 3.18–3.29 (m, 1H), 3.63–3.74 (m, 1H), 3.91–4.00 (m, 2H), 4.46–4.55 (m, 1H), 7.12 (d, J = 8.4 Hz, 1H), 7.36 (td, J = 8.8, 2.4 Hz, 1H), 7.44 (dd, J = 8.0, 2.0 Hz, 1H), 7.53 (d, J = 2.0 Hz, 1H), 7.96–8.03 (m, 2H), 9.47 (s, 1H); Compound 15 (DMSO-d6): 2.13–2.28 (m, 1H), 2.80–3.02 (m, 2H), 3.18–3.33 (m, 1H), 3.60–3.73 (m, 1H), 3.83 (s, 3H), 3.87–4.02 (m, 2H), 4.43–5.02 (m, 1H), 7.02–7.11 (m, 2H), 7.38 (dd, J = 8.1, 1.8 Hz, 1H), 7.49 (d, J = 1.8 Hz, 1H), 7.64 (d, J = 2.4 Hz, 1H), 7.85 (d, J = 8.7 Hz, 1H), 9.46 (s, 1H); Compound 16 (DMSO-d6): 2.06–2.20 (m, 1H), 2.90–3.05 (m, 2H), 3.34–3.42 (m, 2H), 3.92–4.02 (m, 1H), 4.31–4.40 (m, 1H), 4.45–4.54 (m, 1H), 7.42–7.48 (m, 1H), 7.51–7.57 (m, 2H), 7.66–7.72 (m, 2H), 8.04 (d, J = 7.6 Hz, 1H), 8.12 (d, J =7.6 Hz, 1H), 9.67 (s, 1H); Compound 17 (DMSO-d6): 2.06–2.16 (m, 1H), 2.91–3.04 (m, 2H), 3.27–4.43 (m, 2H), 3.92–4.01 (m, 1H), 4.30–4.39 (m, 1H), 4.45–4.55 (m, 1H), 7.40 (td, J = 8.8, 2.4 Hz, 1H), 7.52 (dd, J = 8.0, 1.6 Hz, 1H), 7.65–7.70 (m, 2H), 8.02–8.08 (m, 2H), 9.69 (s, 1H); Compound 18 (DMSO-d6): 2.02–2.18 (m, 1H), 2.90–3.05 (m, 2H), 3.85 (s, 3H), 3.88–4.03 (m, 2H), 4.28–4.53 (m, 2H), 7.12 (dd, J = 7.5, 2.1 Hz, 1H), 7.47 (d, J = 7.8 Hz, 1H), 7.55–7.75 (m, 3H), 7.92 (d, J = 8.7 Hz, 1H), 9.67 (s, 1H); Compound 19 (DMSO-d6): 2.05–2.17 (m, 1H), 2.85–3.02 (m, 2H), 3.35–3.46 (m, 2H), 3.90–4.01 (m, 1H), 4.30–4.44 (m, 2H), 7.20 (d, J = 8.4, 1H), 7.37–7.53 (m, 2H), 7.79 (dd, J = 8.4, 2.0 Hz, 1H), 7.99 (d, J = 8.0 Hz, 1H), 8.09 (d, J = 7.6 Hz, 1H), 8.20 (d, J = 1.6 Hz, 1H), 9.66 (s, 1H); Compound 20 (DMSO-d6): 2.06–2.19 (m, 1H), 2.86–3.01 (m, 2H), 3.35–3.48 (m, 2H), 3.91–4.00 (m, 1H), 4.31–4.44 (m, 2H), 7.20 (d, J = 8.8 Hz, 1H), 7.37 (td, J = 8.8, 2.4 Hz, 1H), 7.76 (dd, J = 8.4, 2.0 Hz, 1H), 7.97–8.04 (m, 2H), 8.18 (d, J = 1.6 Hz, 1H), 9.67 (s, 1H); Compound 22 (CDCl3): 2.74–2.81 (m, 1H), 2.82 (s, 3H), 3.45–3.52 (m, 1H), 3.59–3.67 (m, 1H), 3.85–3.92 (m, 1H), 4.30–4.40 (m, 2H), 4.42–4.52 (m, 1H), 4.65–4.75 (m, 1H), 6.95 (d, J = 8.4 Hz, 1H), 7.49–7.56 (m, 2H), 7.63 (t, J = 8.0 Hz, 1H), 7.84 (d, J = 8.0 Hz, 1H), 8.50 (d, J = 8.4 Hz, 1H), 8.55 (s, 1H); Compound 23 (CDCl3): 2.70–2.82 (m, 1H), 2.83 (s, 3H), 3.45–3.54 (m, 1H), 3.60–3.68 (m, 1H), 3.79–3.92 (m, 1H), 4.30–4.49 (m, 3H), 4.59–4.69 (m, 1H), 6.96 (d, J = 8.4 Hz, 1H), 7.36 (td, J = 8.8, 2.4 Hz, 1H), 7.49 (dd, J = 8.4, 2.4 Hz, 1H), 7.55 (dd, J = 7.6, 2.4 Hz, 1H), 8.36–8.46 (m, 2H); Compound 24 (CDCl3): 2.60–2.72 (m, 1H), 2.79 (s, 3H), 3.40–3.52 (m, 1H), 3.63–3.75 (m, 1H), 3.80–3.88 (m, 1H), 3.91 (s, 3H), 4.23–4.41 (m, 2H), 4.43–4.61 (m, 2H), 6.97 (d, J = 8.1 Hz, 1H), 7.10 (dd, J = 8.0, 2.1 Hz, 1H), 7.32 (d, J = 2.1 Hz, 1H), 7.50 (d, J = 8.1 Hz, 1H), 7.80 (s, 1H), 8.02 (d, J =8.7 Hz, 1H).
- 29.Klunk WE, Wang YM, Huang GF, Debnath ML, Holt DP, Mathis CA. Life Sci. 2001;69:1471. doi: 10.1016/s0024-3205(01)01232-2. [DOI] [PubMed] [Google Scholar]
- 30.Kim MK, Choo IH, Lee HS, Woo JI, Chong Y. Bull. Korean Chem. Soc. 2007;28:1231. [Google Scholar]
- 31.Yang Y, Zhu L, Chen XJ, Zhang HB. J. Mol. Graph. Model. 2010;29:538. doi: 10.1016/j.jmgm.2010.10.006. [DOI] [PubMed] [Google Scholar]