

Abstract
Although well-established, the underlying mechanisms involved in obesity-related plasma adiponectin decline remain elusive. Oxidative stress is associated with obesity and insulin resistance and considered to contribute to the progression toward obesity-related metabolic disorders. In this study, we investigated the effects of 4-hydroxynonenal (4-HNE), the most abundant lipid peroxidation end product, on adiponectin production and its potential implication in obesity-related adiponectin decrease. Long-term high-fat diet feeding led to obesity in mouse, accompanied by decreased plasma adiponectin and increased adipose tissue 4-HNE content. Exposure of adipocytes to exogenous 4-HNE resulted in decreased adiponectin secretion in a dose-dependent manner, which was consistent with significantly decreased intracellular adiponectin protein abundance. In contrast, adiponectin gene expression was significantly elevated by 4-HNE treatment, which was concomitant with increased peroxisome proliferator-activated receptor gamma (PPAR-γ) gene expression and transactivity. The effect was abolished by T0070907, a PPAR-γ antagonist, suggesting that PPAR-γ activation plays a critical role in this process. To gain insight into mechanisms involved in adiponectin protein decrease, we examined the effects of 4-HNE on adiponectin protein degradation. Cycloheximide (CHX)-chase assay revealed that 4-HNE exposure accelerated adiponectin protein degradation, which was prevented by MG132, a potent proteasome inhibitor. Immunoprecipitation assay showed that 4-HNE exposure increased ubiquitinated adiponectin protein levels. These data altogether indicated that 4-HNE enhanced adiponectin protein degradation via ubiquitin–proteasome system. Finally, we demonstrated that supplementation of HF diet with betaine, an antioxidant and methyl donor, alleviated high-fat-induced adipose tissue 4-HNE increase and attenuated plasma adiponectin decline. Taken together, our findings suggest that the lipid peroxidation product 4-HNE can differentially regulates adiponectin gene expression and protein abundance and may play a mechanistic role in obesity-related plasma adiponectin decline.
Keywords: Oxidative stress, 4-HNE, Adiponectin, PPAR-γ, Proteasome, Betaine
1. Introduction
Obesity is a condition in which adipocytes accumulate a large amount of fat and become enlarged. Adipocytes are known to express and secrete a variety of biologically active molecules, so-called adipokines. Notable among these adipokines is adiponectin, which plays an important role in the suppression of the metabolic derangements that may result in type 2 diabetes, obesity, atherosclerosis and non-alcoholic fatty liver diseases (NAFLD) (Mangge et al., 2010; Matsuzawa, 2010; Phillips and Kung, 2010). Unlike most other adipokines, plasma adiponectin is decreased in obesity and associated complications (Arita et al., 1999). Although it has been intensely investigated and much progress has been made over last decade, the mechanisms involved in obesity-related adiponectin suppression remain elusive.
Oxidative stress, as the result of a combination of increased free radical production and impaired ability of cells to detoxify the radicals and to repair damaged molecules, is increased in obesity and its associated metabolic disorders and is suggested to participate in the onset and/or progression of these disease processes (Baynes and Thorpe, 1999; Furukawa et al., 2004; Pennathur and Heinecke, 2007; Whaley-Connell et al., 2011). By covalently modifying membrane-associated or intracellular proteins, oxidative stress induces a variety of cellular damage directly or indirectly through production of a variety of membrane lipid peroxidation products. Principal among these is 4-hydroxynonenal (4-HNE), which is derived from peroxidation of n-6 polyunsaturated fatty acids such as arachidonic and linoleic acids. 4-HNE reacts with amino acids, such as cysteine, lysine or histidine, and forms stable adducts with proteins, thereby modulating activities and/or expression of various proteins. At high concentrations, 4-HNE is cytotoxic to several cell types, whereas micromolar and submicromolar concentrations of 4-HNE have been shown to induce various nontoxic, cell-specific effects (Dianzani, 2003; Doorn and Petersen, 2002; Esterbauer et al., 1991). In 3T3-L1 adipocytes, 4-HNE treatment at nontoxic concentrations increased 4-HNE-insulin receptor substrate (IRS) adducts levels, leading to adipocyte insulin resistance (Demozay et al., 2008). Using these same cells, a study by Soares et al. showed that 4-HNE inhibited adiponectin gene expression and secretion in 3T3-L1 adipocytes, however, the mechanisms were not investigated (Soares et al., 2005).
Strong evidence supports that oxidative stress plays a pathologic role in obesity-related disorders; however, detailed mechanistic studies on the effects of 4-HNE on adiponectin gene expression and secretion, as well as its involvement in obesity-related decline in plasma adiponectin levels are limited. In this study, we conducted both in vivo and in vitro experiments to attempt to clarify the potential pathological role of 4-HNE in obesity-related adiponectin suppression and underlying mechanisms.
2. Materials and methods
2.1. Animal care and feeding
Male C57BL/6 mice (Charles River Laboratories, Wilmington, MA; 8 weeks old) weighing 25 ± 0.5 g (means ± SD) were housed in Biologic Resources Laboratory at the University of Illinois at Chicago. The studies were approved by the Institutional Animal Care and Use Committee, which is certified by the American Association of Accreditation of Laboratory Animal Care. For diet-induced obesity experiments, ten mice were randomly assigned into 2 groups and started on either control diet (Con) or high-fat diet (HF). Both control diet (D12450B, 10% calories as fat) and high-fat diet (D12451, 45% calories as fat) was obtained from Research Diets Inc. (New Brunswick, NJ 08901). For betaine supplementation experiments, betaine (anhydrous; Sigma, St. Louis, MO) was supplemented in the drinking water at a concentration of 1% (wt/vol) and started simultaneously with HF diet feeding. All animals accessed to diet and water ad libitum. Mice were maintained on the treatments for 12 weeks before being killed. At the end of the experiment, the mice were anesthetized with Avertin (300 mg/kg body weight) after 4 h fasting, and plasma and epididymal fat pad samples were harvested for biochemical assays.
2.2. Cell culture and induction of differentiation in 3T3-L1 cells
Mouse embryo fibroblast 3T3-L1 cells were obtained from American Type Culture Collection (Manassas, VA) and grown in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% calf serum and 1% antibiotics (Cellgro, Manassas, VA) until confluence and induced to differentiation. Briefly, 2 days postconfluence (defined as day 0), cells were exposed to differentiation medium containing 0.5 mM isobutylmethylxanthine, 1 μM dexamethasone, 1.67 μM insulin (MDI; Sigma, St. Louis, MO) and 10% fetal bovine serum (FBS) for 3 days. Then, cells were transferred to DMEM with 1.67 μM insulin and 10% FBS and refed every 2 days. Maturation of adipocytes was confirmed by Oil Red O staining of lipid droplet on day 7.
2.3. Isolation and culture of mouse primary adipocytes
Primary adipocytes were isolated from male C57BL/6 mice (8–9 weeks old) as described by us previously (Song et al., 2008).
2.4. Lipid peroxidation assay
The thiobarbituric acid reactive substances (TBARS) levels in adipose tissue, as an index of lipid peroxidation, were determined by measurement of the purple color generated by the reaction of thiobarbituric acid (TBA) with malondialdehyde in spectrophotometry as described by us previously (Song et al., 2003).
2.5. Enzyme-linked immunoabsorbent assays (ELISA) for adiponectin
Adiponectin levels in plasma or in the culture medium were measured by a commercially available enzyme-linked immunosorbent assay (ELISA) kit for mouse full-length adiponectin (Linco Research, St. Charles, MO) according to the manufacturer’s instruction.
2.6. Plasma biochemical assays
The plasma biochemical assays were performed with commercially available kits: leptin, resistin (Linco Research, St. Charles, MO) and TNF-α (Waco Chemicals, Richmond, VA).
2.7. Lactate dehydrogenase (LDH) assay
The cell death of 3T3-L1 adipocytes was determined by the measurement of LDH release into the culture medium. LDH activity was determined spectrophotometrically at 340 nm using a commercially available kit (Thermo scientific, Middletown, VA).
2.8. Protein degradation assay
Adiponectin protein degradation was assayed by CHX-chase analysis. After 2-h pretreatment with cycloheximide (5ug/ml), 3T3-L1 adipocytes were exposed to 4-HNE (30 μM) for 1, 2 or 4 h and the total proteins were isolated to detect intracellular adiponectin protein abundance via Western blot.
2.9. Immunoprecipitation
The total protein from 3T3-L1 adipocytes or adipose tissue were extracted with immunoprecipitation buffer (IP buffer: 50 mM Tris–HCl, pH 7.8, 150 mM NaCl, 1% Igepal CA630) and diluted into a concentration of 1 mg/ml. Polyclonal antibodies against adiponectin were added to this mixture at a dilution of 1:100 (Cell Signaling, Danvers, MA), and the samples were incubated overnight at 4 °C with gentle agitation. At the end of incubation, 50 μl of protein A/G-Sepharose bead suspension (Santa Cruz Biotechnology, Santa Cruz, CA) was added to each sample and gently mixed for 1 h at 4 °C. Samples were centrifuged at 12,000 rpm for 30 s; the supernatant was aspirated, and the beads were washed three times in 1 ml of IP buffer. The isolated beads were resuspended in 1× sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS–PAGE) loading buffer, heated to 100 °C for 5 min, vortexed, and flash-centrifuged, and the supernatants were loaded onto a 12% SDS–polyacrylamide gel for electrophoretic separation and immunoblotting.
2.10. Western blotting detections
For immunoblot analysis, 20 μg of 3T3-L1 adipocytes, primary adipocytes extract or adipose tissue extract was denatured at 100 °C for 5 min in 1× SDS–PAGE loading buffer. Samples were separated on a 12% SDS–polyacrylamide gel for 2 h and transferred to 0.45 μm polyvinylidene difluoride membrane (PerkinElmer Life Sciences). After transfer, membranes were blocked in 5% (wt/vol) nonfat dry milk in PBS-0.1% Tween 20 and probed with anti-4-HNE (R&D Systems, Inc., Minneapolis, MN), anti-adiponectin and anti-ubiquitin (Abcam, Cambridge, MA) or anti-peroxisome proliferator-activated receptor gamma (PPAR-γ) (Abcam, Cambridge, MA) antibodies. Horseradish peroxidase-conjugated secondary antibodies and enhanced chemiluminescence substrate kit were used in the detection of specific proteins.
2.11. Real time RT-PCR
Total RNA from 3T3-L1 or primary adipocytes was isolated with a phenol–chloroform extraction. For each sample, 1.0 μg total RNA was reverse transcribed using a high-capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA). The cDNA was amplified in MicroAmp Optical 96-well reaction plates with a SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA) on an Applied Biosystems Prism 7000 sequence detection system. Relative gene expression was calculated after nomalization by a house-keeping gene (mouse 18S rRNA). All primers were purchased from SABiosciences (Frederick, MD). The catalog numbers for adiponectin and PPAR-γ are PPM05260A and PPM05108B, respectively.
2.12. PPARγ transactivity assay
PPARγ activity was assayed using an ELISA-based transactivation TransAM® PPARγ kit (Active Motif, Carlsbad, CA) following the manufacturer’s protocol. The PPARγ TransAM® kit contains a 96-well plate with immobilized oligonucleotides containing a peroxisome proliferator response element (5′-AACTAGGTCAAAGGTCA-3′).
2.13. Statistical analysis
All data were expressed as means ± SD. Statistical analysis was performed using a one-way ANOVA and was analyzed further by Newman-Keuls test for statistical difference. Differences between treatments were considered to be statistically significant at P < 0.05.
3. Results
3.1. Plasma adiponectin decline in obese mice is associated with increased adipose tissue oxidative stress
Male C57BL/6 mice were fed with either Control diet (Con) or high-fat (HF) diet for 12 weeks. Plasma adipokines (adiponectin, resistin, and leptin) were measured. The occurrence of oxidative stress in adipose tissue was determined by measuring TBARS and 4-HNE contents in epididymal fat pads. As shown in Fig. 1, longterm HF diet feeding induced obesity in mice, demonstrated by significant increases in body weight and epididymal fat pad mass (Fig. 1A and B). Moreover, HF diet feeding enhanced oxidative stress in adipose tissue, evidenced by significantly increased adipose tissue TBARS content (Fig. 1C) and 4-HNE-conjugated protein contents (Fig. 1D). HF diet reduced plasma adiponectin (Fig. 1E) levels, whereas both resistin and leptin levels were elevated (Fig. 1F).
Fig. 1.

Long-term high-fat diet feeding caused adiposity and decreased plasma adiponectin level, which is associated with increased oxidative stress in adipose tissue. Male C57BL/6 mice were fed with control and high-fat (HF) diets. Twelve weeks later, the epididymal fat pads were isolated to measure TBARS levels by TBA assay and the total protein was extracted to examine the 4-HNE-conjugated protein contents by Western blot. The plasma was harvested for adipokines assay. HF diet for 12 weeks significantly increased body weight (A) and epididymal fat mass (B), which were associated with elevated TBA contents (C), and 4-HNE-conjugated protein contents (D) in adipose tissue in mice. HF diet reduced plasma adiponectin levels (E), while resistin and leptin (F) levels were elevated. Data are means ± SD (n = 5). *P < 0.05. HF: high-fat diet.
3.2. Exogenous 4-HNE suppresses adiponectin secretion
To determine if increased 4-HNE contents in adipocytes contributes to lowered adiponectin secretion, we first treated fully differentiated 3T3-L1 adipocytes with 4-HNE (0, 10, 30 μM) for 16 h. Adiponectin levels in the media were measured. As shown in Fig. 2, exogenous 4-HNE exposure increased intracellular 4-HNE contents in a dose-dependent manner (Fig. 2A and B). The elevation of intracellular 4-HNE contents was associated with decreased adiponectin secretion (Fig. 2C). The similar effects of 4-HNE on adiponectin secretion were also observed in mouse primary adipocytes (Fig. 2D). To exclude the potential cytotoxic effects of 4-HNE on adipocytes, we also examined LDH levels in the cell culture media with/without 4-HNE treatment. As shown in Fig. 2E, 4-HNE did not induced obvious increase in LDH release in comparison to the untreated group.
Fig. 2.

Exogenous 4-HNE suppresses adiponectin secretion. Differentiated 3T3-L1 adipocytes were exposed to 4-HNE (0, 10, 30 μM) for 16 h and the proteins were isolated to detect 4-HNE-conjugated proteins via Western blot and the media were harvested for adiponectin release assay by ELISA kit. (A & B) 4-HNE dose-dependently increased intracellular 4-HNE-conjugated protein contents. (C) 4-HNE lowered adiponectin secretion in a dose dependant manner in 3T3-L1 adipocytes. Primary adipocytes from the epididymal fat pads of mice were isolated and treated with 4-HNE (30 μM) for 16 h and the media were harvested for adiponectin release assay. (D) 4-HNE reduced adiponectin secretion in primary adipocytes. To exclude the potential cytotoxic effects of 4-HNE on adipocytes, LDH levels in the media were measured. (E) 4-HNE at the doses used in the study had no effect on LDH release in comparison to the control cells. Data are means ± SD (n = 3). *P < 0.05 compared with the untreated group.
3.3. 4-HNE increased adiponectin gene expression via activating PPAR-γ
To determine if the suppressive effect of 4-HNE on adiponectin takes place at transcriptional level, 3T3-L1 adipocytes and primary mouse adipocytes were treated with 4-HNE (30 μM) for 16 h. The total RNA was isolated and adiponectin gene expression determined by real time RT-PCR. As shown in Fig. 3, adiponectin gene expressions were significantly increased after 4-HNE exposure in both types of adipocytes (Fig. 3A). Since PPAR-γ is a dominant transcription factor in the regulation of adiponectin gene expression, we subsequently examined the effect of 4-HNE on PPAR-γ expression. As shown in Fig. 3B, PPAR-γ gene expression was markedly increased by 4-HNE. To determine if PPAR-γ transactivation contributes to 4-HNE-induced increase in adiponectin gene expression, we first measured PPAR-γ DNA binding activity in 3T3-L1 adipocytes treated with and without 4-HNE. As shown in Fig. 3C, 4-HNE increased PPAR-γ transactivity. We then treated 3T3-L1 cells with T0070907, a PPAR-γ antagonist, prior to 4-HNE exposure. As shown in Fig. 3D, the inhibition of PPAR-γ transactivation abolished 4-HNE induced increase in adiponectin gene expression.
Fig. 3.

4-HNE increase adiponectin gene expressions in adipocytes via PPAR-γ activation. Fully differentiated 3T3-L1 adipocytes or mouse primary adipocytes were treated with 4-HNE (20 μM) for 16 h. Total RNA in each group was isolated to determine the gene expression of adiponectin and PPAR-γ via real time RT-PCR. (A) 4-HNE significantly increased adiponectin gene expressions in both 3T3-L1 and primary adipocytes. (B) 4-HNE significantly increased PPAR-γ gene expression in both 3T3-L1 and primary adipocytes. After exposure to 4-HNE (20 μM) for 16 h, the nuclear proteins in 3T3-L1 adipocytes were extracted and used for ELISA assay for PPAR-γ transactivity. (C) 4-HNE significantly increased PPAR-γ DNA binding activity in 3T3-L1 adipocytes. After pretreatment with T0070907 (10 μM), a PPAR-γ antagonist, for 2 h, 3T3-L1 adipocytes were exposed to 4-HNE (20 μM) for 16 h and the total RNA in each group was isolated to determine the gene expression of adiponectin via real time RT-PCR. (D) Inhibition of PPAR-γ activation by T0070907 abolished 4-HNE-induced elevation of adiponectin expression in 3T3-L1 adipocytes. Data are means ± SD (n = 3). *P < 0.05 when compared with the untreated group.
3.4. 4-HNE decreased intracellular adiponectin protein abundance
The facts that 4-HNE can differentially regulate adiponectin gene expression and secretion prompted us to examine the effect of 4-HNE exposure on intracellular adiponectin protein abundance. Both ELISA and Western blot assays were conducted and the results were shown in Fig. 4A and B. Similar to the effects on secretion, intracellular adiponectin concentrations in 4-HNE treated adipocytes were lower than that in control cells.
Fig. 4.
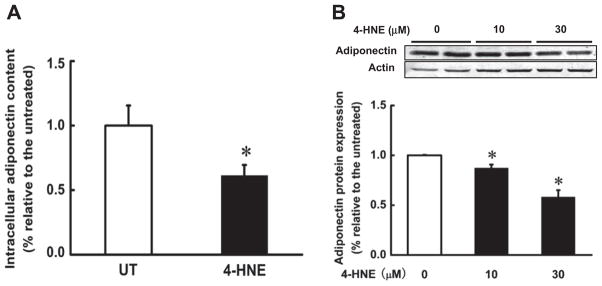
4-HNE decreased intracellular adiponectin protein abundance in 3T3-L1 adipocytes. Differentiated 3T3-L1 adipocytes were treated with 4-HNE (10 μM or 30 μM) for 16 h. The total protein in 3T3-L1 adipocytes was extracted to detect adiponectin protein abundance via ELISA assay (A) and Western blot (B). 4-HNE decreased intracellular adiponectin protein abundance in 3T3-L1 adipocytes. Data are means ± SD (n = 3). *P < 0.05 when compared with the untreated group.
3.5. 4-HNE accelerated adiponectin protein degradation via ubiquitin–proteasome system
The above observations clearly demonstrated that whereas 4-HNE increased adiponectin gene expression, the intracellular and secretary adiponectin proteins were significantly decreased. These findings raised the possibility that 4-HNE may accelerate adiponectin protein degradation. To test our hypothesis, we first examined adiponectin protein degradation rate in 3T3-L1 adipocytes treated with or without 4-HNE via CHX-chase assay. As shown in Fig. 5A, 4-HNE induced accelerated adiponectin degradation in the presence of cycloheximide. In adipocytes with no 4-HNE exposure, adiponectin protein degradation became obvious at 2 h after CHX treatment, while it became obvious at 1 h in 4-HNE exposed cells. At 4 h, adiponectin protein levels in 4-HNE treated adipocytes were markedly lower than that in control cells. Furthermore, MG-132, a specific proteasome inhibitor, rescued adiponectin degradation in both control and 4-HNE treated adipocytes (Fig. 5B), indicating that 4-HNE enhances adiponectin degradation in proteasome. Ubiquitination is the obligatory step for proteins destined to be degraded in proteasome. Therefore, we then measured ubiquitin levels of immunoprecipated adiponectin protein from 4-HNE-treated and control 3T3-L1 adipocytes. As Fig. 5C and E show, the levels of ubiquitinated adiponectin protein were significantly elevated in 4-HNE treated adipocytes. Together, these results indicate that 4-HNE reduces adiponectin protein levels by enhancing ubiquitin–proteasome mediated protein degradation. To determine if accelerated protein degradation is related to the increased formation of 4-HNE-adipoenectin adducts, 4-HNE levels in immunoprecipated adiponectin protein from 4-HNE treated and control 3T3-L1 adipocytes were measured. As shown in Fig. 5D and F, 4-HNE exposure had no effects on the adduct formation.
Fig. 5.

4-HNE accelerated adiponectin degradation via ubiquitin–proteasome system. After 2-h pretreatment with cycloheximide (5 μg/ml), 3T3-L1 adipocytes were exposed to 4-HNE (30 μM) for 1, 2 or 4 h and the total proteins were isolated to detect intracellular adiponectin protein abundance via Western blot. (A) 4-HNE reduced adiponectin protein stability. Proteasome inhibitor, MG132 (10 μM), was added to the medium 2 h before cycloheximide (5 μg/ml) treatment. Two more hours later, 3T3-L1 adipocytes were exposed to 4-HNE (30 μM) for 1, 2 or 4 h and intracellular adiponectin protein abundance was detected via Western blot. (B) MG132 abolished 4-HNE induced enhancement of adiponectin protein degradation. MG132 (10 μM) was added to the medium for 2 h before 4-HNE (30 μM) exposure. Four hours later, the proteins were isolated and immunoprecipitated by adiponectin antibody. The pull-down proteins were subjected to Western blot and probed with ubiquitin or 4-HNE antibodies to determine adiponectin-conjugated ubiquitin and 4-HNE-adiponecitn adduct levels. (C & E) 4-HNE increased adiponectin-conjugated ubiquitin contents in 3T3-L1 adipocytes in comparison to control cells. (D & F) 4-HNE exposure had no effects on 4-HNE-adiponectin adduct formation in 3T3-L1 adipocytes. Data are means ± SD (n = 3). *P < 0.05 compared with the untreated group.
3.6. Betaine supplementation alleviates adiponectin reduction in HF diet-fed mice
Betaine is a metabolic component of intracellular methionine metabolism (Fig. 7). We previously reported that betaine prevented high-fat diet induced non-alcoholic fatty liver disease via improving adipose tissue function (Wang et al., 2010a; Wang et al., 2010b). Also, we recently reported that betaine supplementation improved alcohol-induced fatty liver and liver injury via maintaining hepatic ERK1/2 activation (Wang et al., 2010a; Wang et al., 2010b). In current study, we further examined if betaine can confer beneficial effects on adipose tissue via decreasing 4-HNE contents. As shown in Fig. 6, betaine supplementation attenuated plasma adiponectin reduction in HF diet mice (Fig. 6A). This beneficial effect was associated with attenuated adipose tissue TBA (Fig. 6B) and 4-HNE (Fig. 6C and D) contents.
Fig. 7.

Betaine is a biochemical component of intracellular methionine metabolism. Exogenous betaine supplementation can elevate intracellular SAM, a universal methyl donor, and glutathione levels through modulating intracellular methionine metabolism. SAM, S-adenosyl methionine; SAH, S-adenosyl homocysteine; MAT, methionine adenosyltransferase; BHMT, betaine-homocysteine S-methyltransferase; SAHH, SAH hydrolase; MS, methionine synthase; CBS, cystathionine beta synthase.
Fig. 6.

Betaine supplementation alleviates plasma adiponectin reduction and 4-HNE increase in adipose tissue in HF diet mice. Male C57BL/6 mice were fed HF diets with or without betaine [1% (wt/vol)] supplementation in the drinking water for 12 weeks. At last, the plasma was harvested for adiponectin assay. The epididymal fat pads were isolated to measure TBARS levels by TBA assay and the total protein in adipose tissue was extracted to detect the 4-HNE-conjugated protein content by Western blot. Betaine supplementation alleviated plasma adiponectin reduction (A), which was associated with decreased TBARS (B) and 4-HNE (C & D) contents in the adipose tissue. The 4-HNE-protein adducts levels acquired from Western blot films were expressed as the density value after all the lanes were scanned and analyzed by densitometry. Data are means ± SD (n = 5). *P < 0.05. HF: high-fat diet; BT: betaine.
4. Discussion
Adiponectin is predominantly produced and secreted by adipocytes into the circulation. Obesity-related plasma adiponectin decline is critically involved in the pathogenesis of obesity-related metabolic disorders. In the present study, we demonstrated that 4-HNE, one of the most abundant and reactive lipid peroxidation end product, increased adiponectin gene expression in both fully differentiated 3T3-L1 adipocytes and primary adipocytes isolated from mouse epididymal fat pad. 4-HNE increased PPAR-γ gene expression and its transactivity and pretreatment with T0070907, a PPAR-γ antagonist, prior to 4-HNE exposure abolished 4-HNE-mediated adiponectin gene expression increase, suggesting that PPAR-γ activation plays a mechanistic role in this process. In contrast, 4-HNE exposure led to marked reductions in both intracellular adiponectin protein contents and its secretion into the media. CHX-chase assay revealed that 4-HNE accelerated intracellular adiponectin protein degradation rate by ubiquitin–proteasome system. These data collectively suggest that 4-HNE can differentially regulate adiponectin gene expression and protein secretion in adipocytes, which may contribute to obesity-related plasma adiponectin decline.
Accumulated evidence suggests that adipose tissue oxidative stress plays a central and causal role in the pathogenesis of metabolic syndrome (Baynes and Thorpe, 1999; Furukawa et al., 2004; Pennathur and Heinecke, 2007; Whaley-Connell et al., 2011). Excessive fat accumulation increases the production of reactive oxygen species, lower cellular antioxidant levels, leading to oxidative stress in adipose tissue. Whereas various reactive oxygen species react with all cellular components, the hydroxyl radical-mediated peroxidation of polyunsaturated acyl chains of glycerophospholipids is particularly harmful as it results in the formation of lipid peroxidation production considered second messengers and the ultimate mediator of toxic effects elicited by oxidative stress. Of lipid peroxidates, 4-HNE is the most abundant and reactive aldehydic products derived from peroxidation of n-6 polyunsaturated fatty acids (Dianzani, 2003; Doorn and Petersen, 2002; Esterbauer et al., 1991). The results from our animal study are consistent with many previous clinical observations and experimental investigations showing that obesity is associated with decreased plasma adiponectin concentrations and increased production of lipid peroxidates in adipose tissue, including 4-HNE (Curtis et al., 2010; Grimsrud et al., 2007). Plasma adiponectin level is determined by complex intracellular regulatory mechanisms involved in gene expression, post-transcriptional/translational modification, and trafficking/secretion process (Liu and Liu, 2009; Wang et al., 2008). Among multiple factors, PPAR-γ plays a dominant role in transcriptional transactivating its gene expression. Using cell culture system, we clearly showed that adiponectin mRNA levels were significantly increased in response to 4-HNE exposure in both 3T3-L1 and primary adipocytes, which was concomitant with increased PPAR-γ gene expression, implying that PPAR-γ transactivation may contribute to 4-HNE-induced enhancement of adiponectin gene expression. This assumption was subsequently confirmed by direct transactivity assay and the results showing that T0070907, a PPAR-γ antagonist, abolished adiponectin mRNA increases in 4-HNE treated adipocytes. It has been well recognized that both fatty acids and their metabolites are the potential ligands for PPARs. Consistently, increased PPAR-γ expression by 4-HNE has been reported in several different cell types, including HL-60, U937, as well as osteoblastic cell lines (Almeida et al., 2009; Pizzimenti et al., 2002). Moreover, it has been reported recently that 4-HNE acted as an endogenous ligand for PPAR-δ in 3T3-L1 preadipocytes (Coleman et al., 2007). Our results, together with some others’ (Almeida et al., 2009; Pizzimenti et al., 2002), implicate that 4-HNE may function as a PPAR-γ ligand and directly activate PPAR-γ; however, the direct ligand-receptor relation remains to be established.
Emerging evidence suggests that regulatory steps involved in the post-transcriptional/translational modification and trafficking process may play a more important role in controlling plasma adiponectin levels (Liu et al., 2008). Accordingly, several proteins have been discovered recently, which are critically involved in regulating adiponectin’s trafficking and secretion at post-transcriptional/translational level (Simpson and Whitehead, 2010). The current study did not test the potential effects of 4-HNE on these pathways, however, the observations that 4-HNE decreased adiponectin levels not only in the culture media, but also inside adipocytes, suggest that 4-HNE may decrease adiponectin production via affecting protein degradation processes, instead of affecting its trafficking/secretion pathway.
Ubiquitin–proteasome system (UPS) plays a central role in the regulation of intracellular protein degradation. Proteins targeted for the degradation by this pathway will first undergo a process called “ubiquitination”, whereby a polyubiquitin chain is covalently attached to the proteins via a series of ATP-consuming reactions. Conjugation to ubiquitin is an obligatory step for protein degradation in eukaryotes. Precise control of protein breakdown by the ubiquitin system is crucial for numerous cellular processes including cell cycle progression, cell growth and proliferation, signal transduction, and transcription (Hochstrasser, 1995; Wilkinson et al., 1980) The involvement of UPS in adiponectin protein degradation has been reported by several groups and the estimated half-life of intracellular adiponectin was approximately 3.8 h (Clasen et al., 2005; Combs et al., 2004). Given the fact that UPS is primarily responsible for the degradation of damaged proteins and that 4-HNE can modify proteins by forming covalent adduct, it is rational to postulate that 4-HNE may signal, thereby accelerate, protein degradation via adduct formation. In fact, 4-HNE regulated protein degradation via UPS has been documented (Carbone et al., 2004). In this study, we showed that 4-HNE exposure increased ubiquitination of adiponectin and 4-HNE-mediated adiponectin degradation was attenuated by proteasome inhibition. Thus, our results support that 4-HNE exposure accelerated adiponectin degradation via ubiquin-proteasome system. Unexpectedly, the increase in 4-HNE-adiponectin adduct formation was undetectable after 4-HNE exposure. These results suggest that accelerated adiponectin protein degradation in response to 4-HNE exposure is not due to the direct adiponectin protein modification; instead, 4-HNE exposure may affect other steps/enzymes involved in adiponectin protein ubiquitination process. A noteworthy hypothesis is that 4-HNE may affect, either directly or indirectly, gene expression/enzyme activity of E3 ligase for adiponectin, thereby accelerating its ubiquitination and subsequent degradation by UPS. At present, the E3 ligase that controls the ubiquitination of adiponectin hasn’t been identified. Considering the critical role of adiponectin decline in obesity-related metabolic disorders, the identification of the ligase is of great clinical significance. In this context, the future investigation in this aspect is certainly warranted.
Unlike initially considered, adipocytes are quite reactive to oxidative stress. Intracellular 4-HNE in adipocytes is detectable even under resting conditions and the free form of 4-HNE at basal condition was estimated to be in the range of 0.1–1 μM (Soares et al., 2005). However, due to its high reactivity, it is understandable that the total 4-HNE levels are supposed to be above this range. In 3T3-L1 adipocytes, the exposure to glucose oxidase doubled the intracellular 4-HNE contents (Soares et al., 2005). Moreover, long-term high-fat diet feeding resulted in ~2–3-fold increase in total adipose protein carbonylation (Grimsrud et al., 2007). Although the concentration of 4-HNE used in our study is higher than the “physiological” concentrations in several tissues and cells, it is very likely that high localized concentration of 4-HNE may be formed during chronic metabolic stress. Moreover, exogenously added 4-HNE may far less efficient than 4-HNE continuously produced with cells. In fact, in this study, 30 μM exogenous 4-HNE led to ~3-fold increase in the intracellular 4-HNE levels, suggesting that 4-HNE doses used in our in vitro study mimicked a “pathological” condition.
Betaine (trimethylglycine) is found in microorganisms, plants and animals and is a significant component of many foods, including wheat, shellfish, spinach, and sugar beets. It is also a naturally occurring metabolite of choline and an essential biochemical component of the methionine-homocysteine cycle (Craig, 2004; Kim and Kim, 2005). Betaine can serve as a precursor of glutathione via increasing intracellular concentration of S-adenosylmethionine, an endogenous allosteric activator of cystathionine beta synthase (CBS), the enzyme converting homocysteine to cysteine, a rate-liming substrate for glutathione synthesis (Fig. 7). Elevated intracellular glutathione levels after betaine supplementation have been reported in different cell types (Ganesan et al., 2010; Islam et al., 2009). In addition, betaine is also a methyl donor and plays a critical role in intracellular transmethylation reactions, whereby affects a variety of intracellular signaling pathways (Kharbanda et al., 2005; Li et al., 2011; Schwab et al., 2011). It is conceivable that betaine represent an ideal agent in improving obesity-related adiponectin dysregulation. Indeed, the previous study in our laboratory showed that betaine improved non-alcoholic fatty liver disease in a high-fat diet fed mice model via improving adipose tissue function (Wang et al., 2010a; Wang et al., 2010b). In that study, we reported that betaine supplementation alleviated high-fat diet induced decrease in plasma adiponectin levels. In current study, we examined whether the beneficial effects of betaine were associated with its effects on adipose tissue oxidative stress. We show here that betaine supplementation reduced adipose tissue oxidative stress, demonstrated by significantly decreased TBARS and 4-HNE contents. Therefore, our in vivo results corroborates that increased adipose tissue oxidative stress are critically involved in adiponectin suppression in obese subjects and betaine may be a potential nutritional therapeutic choice for prevention of obesity-related metabolic complications.
Taken together, our study demonstrated that 4-HNE can differentially regulate adiponectin gene expression and protein degradation. Our findings provide evidence to support the existence of one more layer of regulatory mechanism in adiponectin production, which may contribute, at least partially, to obesity-related plasma adiponectin decline.
Acknowledgments
Financial Support
Supported by the National Institutes of Health grants K01 AA015344 and R01 AA017442 (Z Song).
We thank Dr. Alan Diamond from the Department of Pathology for his technical support and scientific advice.
Abbreviations
- 4-HNE
4-hydroxynonenal
- TBARS
thiobarbituric acid reactive substances
- UPS
ubiquitin–proteasome system
- PPAR-γ
peroxisome proliferator-activated receptor gamma
- CBS
cystathionine beta synthase
References
- Almeida M, Ambrogini E, Han L, Manolagas SC, Jilka RL. Increased lipid oxidation causes oxidative stress, increased peroxisome proliferator-activated receptor-gamma expression, and diminished pro-osteogenic Wnt signaling in the skeleton. J Biol Chem. 2009;284:27438–27448. doi: 10.1074/jbc.M109.023572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arita Y, Kihara S, Ouchi N, Takahashi M, Maeda K, Miyagawa J, Hotta K, Shimomura I, Nakamura T, Miyaoka K, Kuriyama H, Nishida M, Yamashita S, Okubo K, Matsubara K, Muraguchi M, Ohmoto Y, Funahashi T, Matsuzawa Y. Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem Biophys Res Commun. 1999;257:79–83. doi: 10.1006/bbrc.1999.0255. [DOI] [PubMed] [Google Scholar]
- Baynes JW, Thorpe SR. Role of oxidative stress in diabetic complications: a new perspective on an old paradigm. Diabetes. 1999;48:1–9. doi: 10.2337/diabetes.48.1.1. [DOI] [PubMed] [Google Scholar]
- Carbone DL, Doorn JA, Petersen DR. 4-Hydroxynonenal regulates 26S proteasomal degradation of alcohol dehydrogenase. Free Radical Biol Med. 2004;37:1430–1439. doi: 10.1016/j.freeradbiomed.2004.07.016. [DOI] [PubMed] [Google Scholar]
- Clasen R, Schupp M, Foryst-Ludwig A, Sprang C, Clemenz M, Krikov M, Thöne-Reineke C, Unger T, Kintscher U. PPARγ-activating angiotensin type-1 receptor blockers induce adiponectin. Hypertension. 2005;46:137–143. doi: 10.1161/01.HYP.0000168046.19884.6a. [DOI] [PubMed] [Google Scholar]
- Coleman JD, Prabhu KS, Thompson JT, Reddy PS, Peters JM, Peterson BR, Reddy CC, Vanden Heuvel JP. The oxidative stress mediator 4-hydroxynonenal is an intracellular agonist of the nuclear receptor peroxisome proliferator-activated receptor-beta/delta (PPARbeta/delta) Free Radical Biol Med. 2007;42:1155–1164. doi: 10.1016/j.freeradbiomed.2007.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combs TP, Pajvani UB, Berg AH, Lin Y, Jelicks LA, Laplante M, Nawrocki AR, Rajala MW, Parlow AF, Cheeseboro L, Ding YY, Russell RG, Lindemann D, Hartley A, Baker GR, Obici S, Deshaies Y, Ludgate M, Rossetti L, Scherer PE. A transgenic mouse with a deletion in the collagenous domain of adiponectin displays elevated circulating adiponectin and improved insulin sensitivity. Endocrinology. 2004;145:367–383. doi: 10.1210/en.2003-1068. [DOI] [PubMed] [Google Scholar]
- Craig SA. Betaine in human nutrition. Am J Clin Nutr. 2004;80:539–549. doi: 10.1093/ajcn/80.3.539. [DOI] [PubMed] [Google Scholar]
- Curtis JM, Grimsrud PA, Wright WS, Xu X, Foncea RE, Graham DW, Brestoff JR, Wiczer BM, Ilkayeva O, Cianflone K, Muoio DE, Arriaga EA, Bernlohr DA. Downregulation of adipose glutathione S-transferase A4 leads to increased protein carbonylation, oxidative stress, and mitochondrial dysfunction. Diabetes. 2010;59:1132–1142. doi: 10.2337/db09-1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demozay D, Mas JC, Rocchi S, Van Obberghen E. FALDH reverses the deleterious action of oxidative stress induced by lipid peroxidation product 4-hydroxynonenal on insulin signaling in 3T3–L1 adipocytes. Diabetes. 2008;57:1216–1226. doi: 10.2337/db07-0389. [DOI] [PubMed] [Google Scholar]
- Dianzani MU. 4-hydroxynonenal from pathology to physiology. Mol Aspects Med. 2003;24:263–272. doi: 10.1016/s0098-2997(03)00021-9. [DOI] [PubMed] [Google Scholar]
- Doorn JA, Petersen DR. Covalent modification of amino acid nucleophiles by the lipid peroxidation products 4-hydroxy-2-nonenal and 4-oxo-2-nonenal. Chem Res Toxicol. 2002;15:1445–1450. doi: 10.1021/tx025590o. [DOI] [PubMed] [Google Scholar]
- Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radical Biol Med. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada Y, Nakajima Y, Nakayama O, Makishima M, Matsuda M, Shimomura I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest. 2004;114:1752–1761. doi: 10.1172/JCI21625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganesan B, Buddhan S, Anandan R, Sivakumar R, AnbinEzhilan R. Antioxidant defense of betaine against isoprenaline-induced myocardial infarction in rats. Mol Biol Rep. 2010;37:1319–1327. doi: 10.1007/s11033-009-9508-4. [DOI] [PubMed] [Google Scholar]
- Grimsrud PA, Picklo MJ, Sr, Griffin TJ, Bernlohr DA. Carbonylation of adipose proteins in obesity and insulin resistance. identification of adipocyte fatty acid-binding protein as a cellular target of 4-hydroxynonenal. Mol Cell Proteomics. 2007;6:624–637. doi: 10.1074/mcp.M600120-MCP200. [DOI] [PubMed] [Google Scholar]
- Hochstrasser M. Ubiquitin, proteasomes, and the regulation of intracellular protein degradation. Curr Opin Cell Biol. 1995;7:215–223. doi: 10.1016/0955-0674(95)80031-x. [DOI] [PubMed] [Google Scholar]
- Islam MM, Hoque MA, Okuma E, Jannat R, Banu MN, Jahan MS, Nakamura Y, Murata Y. Proline and glycinebetaine confer cadmium tolerance on tobacco bright yellow-2 cells by increasing ascorbate-glutathione cycle enzyme activities. Biosci Biotechnol Biochem. 2009;73:2320–2323. doi: 10.1271/bbb.90305. [DOI] [PubMed] [Google Scholar]
- Kharbanda KK, Rogers DD, Mailliard ME, Siford GL, Barak AJ, Beckenhauer HC, Sorrell MF, Tuma DJ. A comparison of the effects of betaine and S-adenosylmethionine on ethanol-induced changes in methionine metabolism and steatosis in rat hepatocytes. J Nutr. 2005;135:519–524. doi: 10.1093/jn/135.3.519. [DOI] [PubMed] [Google Scholar]
- Kim SK, Kim YC. Effects of betaine supplementation on hepatic metabolism of sulfur-containing amino acids in mice. J Hepatol. 2005;42:907–913. doi: 10.1016/j.jhep.2005.01.017. [DOI] [PubMed] [Google Scholar]
- Li J, Li XM, Caudill M, Malysheva O, Bardag-Gorce F, Oliva J, French BA, Gorce E, Morgan K, Kathirvel E, Morgan T, French SW. Betaine feeding prevents the blood alcohol cycle in rats fed alcohol continuously for 1month using the rat intragastric tube feeding model. Exp Mol Pathol. 2011;91:540–547. doi: 10.1016/j.yexmp.2011.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Liu F. Transcriptional and post-translational regulation of adiponectin. Biochem J. 2009;425:41–52. doi: 10.1042/BJ20091045. [DOI] [PubMed] [Google Scholar]
- Liu M, Zhou L, Xu A, Lam KS, Wetzel MD, Xiang R, Zhang J, Xin X, Dong LQ, Liu F. A disulfide-bond A oxidoreductase-like protein (DsbA-L) regulates adiponectin multimerization. Proc Natl Acad Sci USA. 2008;105:18302–18307. doi: 10.1073/pnas.0806341105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangge H, Almer G, Truschnig-Wilders M, Schmidt A, Gasser R, Fuchs D. Inflammation, adiponectin, obesity and cardiovascular risk. Curr Med Chem. 2010;17:4511–4520. doi: 10.2174/092986710794183006. [DOI] [PubMed] [Google Scholar]
- Matsuzawa Y. Adiponectin: a key player in obesity related disorders. Curr Pharm Des. 2010;16:1896–1901. doi: 10.2174/138161210791208893. [DOI] [PubMed] [Google Scholar]
- Pennathur S, Heinecke JW. Mechanisms for oxidative stress in diabetic cardiovascular disease. Antioxid Redox Signal. 2007;9:955–969. doi: 10.1089/ars.2007.1595. [DOI] [PubMed] [Google Scholar]
- Phillips SA, Kung JT. Mechanisms of adiponectin regulation and use as a pharmacological target. Curr Opin Pharmacol. 2010;10:676–683. doi: 10.1016/j.coph.2010.08.002. [DOI] [PubMed] [Google Scholar]
- Pizzimenti S, Laurora S, Briatore F, Ferretti C, Dianzani MU, Barrera G. Synergistic effect of 4-hydroxynonenal and PPAR ligands in controlling human leukemic cell growth and differentiation. Free Radical Biol Med. 2002;32:233–245. doi: 10.1016/s0891-5849(01)00798-5. [DOI] [PubMed] [Google Scholar]
- Schwab U, Alfthan G, Aro A, Uusitupa M. Long-term effect of betaine on risk factors associated with the metabolic syndrome in healthy subjects. Eur J Clin Nutr. 2011;65:70–76. doi: 10.1038/ejcn.2010.230. [DOI] [PubMed] [Google Scholar]
- Simpson F, Whitehead JP. Adiponectin–it’s all about the modifications. Int J Biochem Cell Biol. 2010;42:785–788. doi: 10.1016/j.biocel.2009.12.021. [DOI] [PubMed] [Google Scholar]
- Soares AF, Guichardant M, Cozzone D, Bernoud-Hubac N, Bouzaïdi-Tiali N, Lagarde M, Géloën A. Effects of oxidative stress on adiponectin secretion and lactate production in 3T3–L1 adipocytes. Free Radical Biol Med. 2005;38:882–889. doi: 10.1016/j.freeradbiomed.2004.12.010. [DOI] [PubMed] [Google Scholar]
- Song Z, Zhou Z, Deaciuc I, Chen T, McClain CJ. Inhibition of adiponectin production by homocysteine: a potential mechanism for alcoholic liver disease. Hepatology. 2008;47:867–879. doi: 10.1002/hep.22074. [DOI] [PubMed] [Google Scholar]
- Song Z, Zhou Z, Chen T, Hill D, Kang J, Barve S, McClain CJ. S-adenosylmethionine (SAMe) protects against acute alcohol induced hepatotoxicity in mice. J Nutr Biochem. 2003;14:591–597. doi: 10.1016/s0955-2863(03)00116-5. [DOI] [PubMed] [Google Scholar]
- Wang Y, Lam KS, Yau MH, Xu A. Post-translational modifications of adiponectin: mechanisms and functional implications. Biochem J. 2008;409:623–633. doi: 10.1042/BJ20071492. [DOI] [PubMed] [Google Scholar]
- Wang Z, Yao T, Pini M, Zhou Z, Fantuzzi G, Song Z. Betaine improved adipose tissue function in mice fed a high-fat diet: a mechanism for hepatoprotective effect of betaine in nonalcoholic fatty liver disease. Am J Physiol Gastrointest Liver Physiol. 2010a;298:G634–G642. doi: 10.1152/ajpgi.00249.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Yao T, Song Z. Involvement and mechanism of DGAT2 upregulation in the pathogenesis of alcoholic fatty liver disease. J Lipid Res. 2010b;51:3158–3165. doi: 10.1194/jlr.M007948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whaley-Connell A, McCullough PA, Sowers JR. The role of oxidative stress in the metabolic syndrome. Rev Cardiovasc Med. 2011;12:21–29. doi: 10.3909/ricm0555. [DOI] [PubMed] [Google Scholar]
- Wilkinson KD, Urban MK, Haas AL. Ubiquitin is the ATP-dependent proteolysis factor I of rabbit reticulocytes. J Biol Chem. 1980;255:7529–7532. [PubMed] [Google Scholar]