

Abstract
Endochondral ossification is a carefully orchestrated process mediated by promoters and inhibitors of mineralization. Phosphatases are implicated, but their identities and functions remain unclear. Mutations in the tissue-nonspecific alkaline phosphatase (TNAP) gene cause hypophosphatasia, a heritable form of rickets and osteomalacia, caused by an arrest in the propagation of hydroxyapatite (HA) crystals onto the collagenous extracellular matrix due to accumulation of extracellular inorganic pyrophosphate (PPi), a physiological TNAP substrate and a potent calcification inhibitor. However, TNAP knockout (Alpl−/−) mice are born with a mineralized skeleton and have HA crystals in their chondrocyte- and osteoblast-derived matrix vesicles (MVs). We have shown that PHOSPHO1, a soluble phosphatase with specificity for two molecules present in MVs, phosphoethanolamine and phosphocholine, is responsible for initiating HA crystal formation inside MVs and that PHOSPHO1 and TNAP have nonredundant functional roles during endochondral ossification. Double ablation of PHOSPHO1 and TNAP function leads to the complete absence of skeletal mineralization and perinatal lethality, despite normal systemic phosphate and calcium levels. This strongly suggests that the Pi needed for initiation of MV-mediated mineralization is produced locally in the perivesicular space. As both TNAP and nucleoside pyrophosphohydrolase-1 (NPP1) behave as potent ATPases and pyrophosphatases in the MV compartment, our current model of the mechanisms of skeletal mineralization implicate intravesicular PHOS-PHO1 function and Pi influx into MVs in the initiation of mineralization and the functions of TNAP and NPP1 in the extravesicular progression of mineralization.
Keywords: Biomineralization, Bone and cartilage development, Metabolic bone disease, Animal model
The System
Mineralization of cartilage, bone, and teeth occurs by a series of physicochemical and biochemical processes that together facilitate the deposition of hydroxyapatite (HA) in specific areas of the extracellular matrix (ECM). Experimental evidence has pointed to the presence of HA crystals along collagen fibrils [1] and within the lumen of chondrocyte- and osteoblast-derived matrix vesicles (MVs) [2, 3]. Investigators in the bone mineralization field are generally divided in supporting the collagen- versus the MV-mediated mechanism of mineralization. We see no incompatibility between these mechanisms. Our working model considers that early mineralization takes place inside MVs, organelles that serve as a site for Ca2+ and inorganic phosphate (Pi) accumulation to initiate the deposition of HA crystals [3–8]. In a second step, MV membranes break down and expose preformed HA to the extracellular fluid, allowing for propagation of HA deposition onto the collagenous ECM [6, 8]. This process is orchestrated by the balanced action of promoters and inhibitors of calcification including many noncollagenous matrix proteins [9, 10].
This review article will recapitulate what we know about the functional interplay of three phosphatases, tissue-nonspecific alkaline phosphatase (TNAP), phosphatase orphan 1 (PHOSPHO1), and nucleoside pyrophosphohydrolase-1 (NPP1), during the initiation of skeletal mineralization, a process of fundamental importance to all vertebrate animal species. Alterations in the function of these phosphatases lead to soft bone, including rickets or osteomalacia, spontaneous fractures, loss of teeth, as well as inappropriate calcification of soft tissues including osteoarthritis and arterial calcification (Table 1). A key step forward has been the conceptualization of the interrelated role of these phosphatases in controlling the inorganic pyrophosphate (PPi) to Pi ratio in MVs and the ECM, acting as a crucial determinant for initiation of skeletal mineralization.
Table 1.
Nomenclature of the gene name(s), protein name(s), and gene knockout symbols and a brief description of the disease phenotype for each of the murine models of phosphatase deficiency
| Gene names, human (mouse) | Protein names | Mouse model | Phenotype |
|---|---|---|---|
| ALPL (Alpl, aka Akp2) | TNAP (aka TNSALP) | Alpl−/− | Infantile hypophosphatasia
|
| ENPP1 (Enpp1) | NPP1 (aka PC-1) | Enpp1−/− |
|
| PHOSPHO1 (Phospho1) | PHOSPHO1 | Phospho1−/− (aka Phospho1-R74X) |
|
The Key Players
Alkaline phosphatases (APs, EC 3.1.3.1) have structural similarity to a large number of other enzyme families with substantially different activities, including cofactor-independent phosphoglycerate mutase, isomerases, hydrolases, and a putative lyase, which, however, all act on similar phosphocarbohydrate (or sulfocarbohydrate) substrates [11]. In humans, the AP isozyme family is composed of TNAP, encoded by the ALPL gene, and the placental, germ-cell, and intestinal (IAP) isozymes, encoded by the ALPP, ALPP2, and ALPI genes, respectively. Mice also have four active AP loci, namely, Akp2 (aka Alpl), Akp3, Akp5, and Akp6 (aka Alpi), that encode TNAP, duodenum-specific IAP, embryonic AP, and global IAP, respectively [for review, 11]. Only TNAP is implicated in biomineralization, so this review will only cover the biological function of this isozyme.
PHOSPHO1 (EC 3.1.3.75), first identified in the chick [12] as a member of the haloacid dehalogenase (HAD) superfamily of Mg2-dependent hydrolases [13], is expressed at levels 100-fold higher in mineralizing than in non-mineralizing tissues [14]. PHOSPHO1 has 42 % homology to PHOSPHO2, another member of the HAD family, that, however, has very different substrate specificity [15] and is currently not implicated in biomineralization.
The nucleotide pyrophosphatase/phosphodiesterase (NPP, EC 3.1.4.1) family, that includes NPP1, comprises seven NPPs identified to date [16]. These isoenzymes, related by 24–60 % conservation in their catalytic domains and certain other modular domains, exert generally non-redundant functions via distinctions in substrates and/or subcellular localization [17]. NPP1 (previously known as plasma cell membrane glycoprotein-1, PC-1), encoded by the ENPP1 gene, is plasma membrane-bound, whereas autotaxin (NPP2) is secreted and B10 (NPP3) is abundant in intracellular spaces [17]. All three isozymes are expressed in a wide variety of tissues, including bone and cartilage; and all have the common ability to hydrolyze diesters of phosphoric acid into phosphomonoesters. NPPs have been implicated in various processes, including bone mineralization, signaling by insulin and by nucleotides, and the differentiation and motility of cells [18]. However, NPPs are known primarily as suppliers of intra- and extracellular PPi using ATP as a substrate [17]. Similar to skeletal TNAP expression, NPP1 is highly abundant on the surfaces of osteoblasts and chondrocytes as well as on the membrane of their MVs [18, 19] and is the isozyme discussed here.
The PPi/Pi Ratio
A primary inhibitor of ECM mineralization is extracellular PPi [20]. PPi is produced ectoplasmically by the enzymatic action of NPP1, which catabolizes extracellular ATP to produce PPi and AMP [17]. Intracellular PPi is also transported to the extracellular milieu by the channeling function of the ankylosis protein (ANK) [21]. TNAP plays the crucial role of restricting the concentration of extracellular PPi to maintain a PPi/Pi ratio that is permissive for normal bone mineralization [22–24]. Mice deficient in NPP1 (Enpp1−/−) or ANK (ank/ank) develop soft tissue calcification, including vascular calcification, resulting from the reduced production or transport of PPi [23–25]. In contrast, mice that completely lack TNAP function (Alpl−/−) phenocopy infantile hypophosphatasia (HPP) [26, 27] in that they are born with normally calcified skeletons but begin to display hypomineralization of the skeleton at postnatal days 6–10 that worsens until their early demise at postnatal day 20 [28, 29]. The failure of HPP bones to calcify after birth results from a block in the propagation of HA in the ECM beyond the confines of the MV membrane [4, 30] because of accumulated levels of PPi in the ECM resulting from the lack of TNAP’s pyrophosphatase function [26, 27, 31]. Genetic experiments conclusively demonstrated the antagonistic roles of TNAP and NPP1 or ANK and provided conclusive evidence of the in vivo pyrophosphatase activity of TNAP. Indeed, [Enpp1−/−; Alpl−/−] and [ank/ank; Alpl−/−] double-mutant mice showed normalized levels of extracellular PPi and rescue of the respective mineralization phenotype of single-mutant mice [23–25]. However, it is worth pointing out that the degree of improvement of the skeletal phenotype in [Enpp1−/−; Alpl−/−] double-mutant mice was site-specific, with the axial skeleton showing more improvement than the appendicular skeleton, an observation that was attributed to the higher levels of expression of NPP1 in the axial skeleton [32]. Thus, TNAP’s enzymatic degradation of PPi controls the PPi/Pi ratio to favor proliferation of HA crystals outside the MVs and along collagen fibrils. Indeed, bone mineralization is determined partly by the ability of osteoblasts to remove the physiological inhibitor of mineralization, PPi, from their surrounding ECM via expression of TNAP and by the presence of a fibrillar collagen-rich network in the bone ECM [24]. Coexpression of TNAP and a fibrillar collagenous scaffold appears to be necessary and sufficient to cause mineralization of any ECM [24].
Interestingly, chondrocyte- and osteoblast-derived MVs in both HPP patients and Alpl−/− mice retain the ability to initiate intravesicular mineral formation and contain HA crystals [4, 30], demonstrating that TNAP is not essential for the initiation of MV-mediated ECM mineralization and suggesting that other phosphatases or another mechanism might be responsible for this first step. PHOSPHO1 is expressed at levels 120-fold higher in chondrocytes in mineralizing cartilage than in nonmineralizing tissues [14, 33], shows high phosphohydrolase activity toward phosphoethanolamine and phosphocholine [34], and is active inside chondrocyte- and osteoblast-derived MVs [35]. TNAP and PHOSPHO1 were found to be coexpressed throughout the developmental stages of limb development in the chick [36], and small-molecule compounds that inhibit PHOSPHO1 activity decreased MV-mediated calcification in vitro using mouse Alpl−/− MVs [33] and in chick embryo micromass cultures [36]. Thus, we surmised that PHOSPHO1 is involved in the first step of MV-mediated initiation of mineralization during endochondral ossification.
Our recent work clearly showed that lack of PHOS-PHO1 caused changes in the endochondral growth plate and skeletal abnormalities that included decrease or loss of secondary ossification centers, decreased bone mineral density, spontaneous fractures, osteomalacia, and scoliosis [37, 38]. Phospho1−/− mice display growth plate abnormalities, spontaneous fractures, bowed long bones, osteomalacia, and scoliosis in early life. Long bones from Phospho1−/− mice deform plastically rather than fracturing during three-point bending, and Raman microscopy revealed significantly lower mineral:matrix ratios and lower carbonate substitutions in Phospho1−/− tibiae [37]. Primary cultures of Phospho1−/− tibial growth plate chondrocytes and chondrocyte-derived MVs showed reduced mineralizing ability, and plasma samples of Phospho1−/− mice showed reduced levels of TNAP and elevated PPi concentrations. Transgenic overexpression of TNAP does not correct the bone phenotype in Phospho1−/− mice, suggesting that TNAP and PHOSPHO1 act on distinct pathways. In agreement with this hypothesis, double ablation of PHOSPHO1 and TNAP function led to a dramatic phenotype: complete absence of skeletal mineralization and perinatal lethality [38]. This suggests that the availability of free Pi is not sufficient to initiate mineralization and that TNAP must be involved in generating Pi in the vicinity of MVs. Calcification, both intravesicular and extravesicular, is abolished in [Phospho1−/−; Alpl−/−] embryos despite the availability of systemic Pi in these mice. This argues that, besides the Pi produced intravesicularly by PHOSPHO1, organic phosphates might act as an additional major source of Pi, via the putative action of TNAP and/or possibly NPP1.
Indeed, a Pi-generating function has been proposed for TNAP since the discovery of this enzyme in bone by Robison [39], and its ability to hydrolyze ATP [40] as well as polyphosphates [41] has been described. We studied phosphosubstrate catalysis by osteoblast-derived MVs at physiologic pH, analyzing the hydrolysis of ATP, ADP, and PPi by isolated wild-type as well as TNAP-, NPP1- and PHOSPHO1-deficient MVs. We found that TNAP is an efficient ATPase, in addition to its established role as a pyrophosphatase in the MV compartment. We also found that, in contrast to its accepted role on the surface of osteoblasts and chondrocytes, NPP1 does not have a major PPi-generating activity at the level of MVs but acts as both an ATPase and a pyrophosphatase [42]. We also showed that TNAP and NPP1 account for all the Pi-generating ability of isolated MVs. Thus, while PHOSPHO1 modifies the PPi/Pi ratio inside MVs by releasing Pi from membrane phospholipids, TNAP has a primary role in establishing an extracellular PPi/Pi ratio conducive for ECM mineralization, via its pyrophosphatase as well as its ATPase activity. The fact that NPP1 can act as a backup phosphatase on both PPi and ATP helps to explain why Alpl−/− mice, which are null for TNAP activity, develop normal mineralization for the first 6 days of life. Thus, at least in mice, NPP1 can be considered a modifier of the HPP phenotype. This partial compensatory NPP1 activity, which is higher in the axial than in the appendicular skeleton [32], also explains how a single [Phospho1−/−; Alpl−/−] stillborn pup identified by Yadav and collaborators had residual mineralization in the axial skeleton, despite the complete absence of mineralization elsewhere [38].
Our Current Model
Analyses of [Phospho1−/−; Alpl−/−] mice provided unique clues to help put together a comprehensive model of the mechanisms of initiation of skeletal mineralization. Calcification, both intravesicular and extravesicular, is abolished in [Phospho1−/−; Alpl−/−] embryos despite the availability of systemic Pi in these mice. This argues that organic phosphates, such as ATP or ADP, might act as the major source of Pi that is required for the initiation of calcification. Chondrocytes, osteoblasts, and their derived MVs express and use phosphate transporters on their membrane for uptake of Pi [43, 44]. We must conclude that the mineralizing cells consider it efficient to invest the energy required to generate and export ATP to be used for the local generation of Pi in the immediate environment of MVs and for subsequent incorporation into MVs via Pi transporters. Thus, in the absence of both PHOSPHO1 and TNAP function there is complete lack of skeletal mineralization because there is no Pi generation from substrates attributable to the absence of TNAP’s ATPase activity and the levels of ATPase provided by NPP1 in the embryonic skeleton are clearly insufficient to allow calcification to proceed. Some calcification was observed in the axial skeleton of a single stillborn [Phospho1−/−; Alpl−/−] double knockout pup, which was likely attributable to Pi generation via the “ATPase” action of NPP1 and to the concomitant restriction of extracellular PPi concentrations by the “pyrophosphatase” activity of NPP1. This provides an explanation for why complete ablation of PHOSPHO1 function only leads to a decrease in the calcification ability of MVs but not to a complete lack of calcification. Deletion of PHOSPHO1 would suppress intravesicular generation of Pi but would leave extravesicular Pi generation via TNAP’s ATPase activity and influx via Pi transporters unaffected.
Integrating these data, it is now possible to propose an inclusive model for the initiation of skeletal mineralization that unifies a number of concepts and functions that have been considered contradictory in the past. This unified model starts with the MVs as the site of initiation of mineralization (Fig. 1). HA crystals appear inside the MVs favored by Pi accumulation resulting from a dual mechanism, i.e., PHOSPHO1-mediated intravesicular production and transporter-mediated influx of Pi produced extravesicularly primarily by TNAP’s ATPase activity or, secondarily in the absence of TNAP, by NPP1’s ATPase activity. Organophosphate compounds (ATP) and perhaps PPi are the source of Pi for this initial step of calcification. Then, extravesicular calcification is supported primarily by TNAP’s pyrophosphatase activity and secondarily by NPP1’s pyrophosphatase activity and is driven by the availability of Pi and the presence of a collagenous fibrillar scaffold and guided by other ECM mineral-binding proteins. Figure 2 shows how this model is able to explain the phenotypic abnormalities observed in the Phospho1−/−, Alpl−/−, and [Phospho1−/−; Alpl−/−] knockout mice described above. This model also takes into account the roles of both organic and inorganic phosphates in skeletal calcification, incorporates the Pi-generating and PPi-inactivating roles of TNAP, and unifies the roles of MV-mediated and collagen-mediated calcification as two separate but linked steps during endochondral ossification.
Fig. 1.
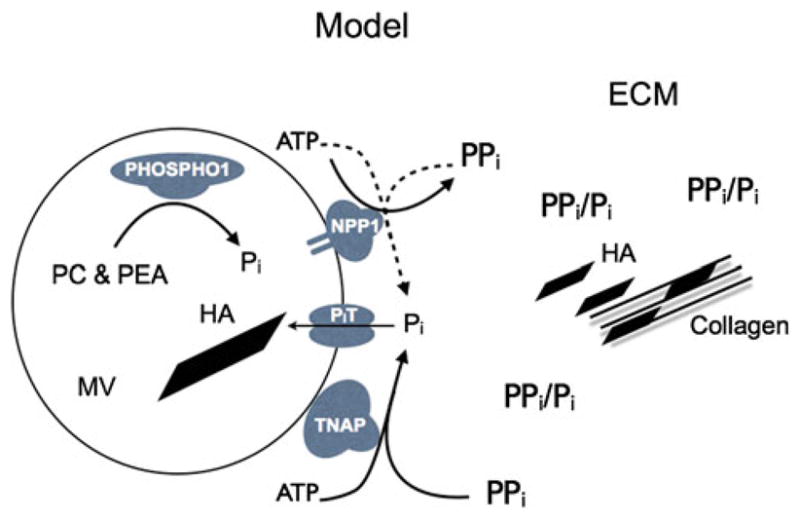
Model of initiation of skeletal mineralization including the function of PHOSPHO1, TNAP, NPP1, and phosphate transporters. The first step of MV-mediated mineralization involves the convergence of two independent biochemical pathways: intravesicular Pi generation by the enzymatic action of PHOSPHO1 and influx of Pi, generated in the perivesicular space by the activities of TNAP and NPP1, via Pi transporters. PC phosphocholine, PEA phosphoethanolamine, MV matrix vesicle, PiT phosphate transporter 1, HA hydroxyapatite, ECM extracellular matrix, Pi inorganic phosphate, PPi inorganic pyrophosphate
Fig. 2.

Summary of findings and how they can be explained by the unified model of initiation of skeletal mineralization. Original data were published in Yadav et al. [38]. Wild-type mice the first step of MV-mediated mineralization involves intravesicular Pi generation by PHOSPHO1 and influx of Pi, generated extravesicularly by TNAP and NPP1. Extravesicular propagation occurs on collagen scaffolds facilitated by the pyrophosphatase function of TNAP and NPP1. Phospho1−/− mice a growth plate and skeletal phenotype is apparent, but HA is still found inside MVs because PiT-mediated transport of Pi, generated by TNAP and NPP1, is unaffected. Akp2−/− mice rickets and osteomalacia are prominent, due to increases in extracellular PPi. HA crystals are still found inside MVs due to the Pi-generating ability of PHOSPHO1. PiT-mediated influx of Pi is greatly diminished except where NPP1 activity is high (axial skeleton). [Phospho1−/−; Alpl−/−] mice complete absence of skeletal mineralization can be explained by the absence of intravesicular Pi generation by PHOSPHO1, the lack of extravesicular Pi generation by TNAP needed for PiT-mediated influx, and accumulation of PPi in the ECM. Abbreviations as in Fig. 1
Future Challenges
We have yet to understand the intimate biochemical details of how PHOSPHO1 is implicated in intravesicular Pi generation form membrane phospholipids. Pioneering studies by Wuthier and collaborators [45] have shown that the growth and egress of nascent HA crystals from the MV lumen are accompanied by changes in the lipid composition of the MV membrane. Phosphatidylinositol, sphingomyelin, and phosphatidylethanolamine undergo rapid breakdown, while phosphatidylcholine are degraded more slowly. Given the in vitro specificity of PHOSPHO1 for phosphoethanolamine and phosphocholine [33, 34], the polar groups of phosphatidylethanolamine and phosphatidylcholine, we surmise that the physiological phospholipid changes in MVs are mediated by PHOSPHO1 action. Yet, we must elucidate if PHOSPHO1 scavenges Pi directly from these phospholipids or requires the enzymatic action of phospholipase C to release the polar groups. We are also just beginning to understand the role of NPP1 as a backup phosphatase. In this regard ENPP1 can be considered a modifier of the severity of HPP, and more work in this area will surely shed more light into the partially overlapping roles of these phosphatases/phosphodiesterases. Furthermore, the interesting suggestion that polyphosphates could be used as a readily available reservoir of Pi which, upon TNAP-mediated release, can help to concentrate Pi for either initiation or propagation of HA mineralization [41] needs to be investigated further.
The identity of the phosphate transporters implicated in the influx of Pi generated extravesicularly is yet to be determined. Pi transport in chondrocytes and osteoblasts is primarily handled by type III sodium-dependent (Na/Pi) cotransporters which have broad tissue expression [46–48]. Two related type III Na/Pi cotransporters, PiT1/Glvr1 and PiT2/Ram, are expressed by chondrocytes and osteoblasts; but literature reports and our own expression data [49] indicate that PiT1 is the major mediator of Pi flux in these cell types. There are also indications in the literature that a transporter that is not strictly Na+-dependent but responds to increases in extracellular Pi might exist, at least in the chicken [44, 45]. Thus, quite a bit of work remains to be done to identify the transporter(s) involved in this process.
An important reason that the bone community is still skeptical of the role of MVs in bone mineralization stems from our current inability to explain how HA crystals formed in the MV environment can promote further propagation of mineralization onto the collagen matrix. In vivo, collagen molecules assemble into three-dimensional structures in which each molecule is in a staggered arrangement relative to its neighbors. Collagen is packed in three dimensions through strict and contiguous alignment of its hole and overlap zones. Extensive work by M. Glimcher, M. Nylen, W. Landis, and many others has indicated that the pattern of holes and overlap sites among collagen molecules provided channels or gaps in their assemblage where HA crystals could nucleate [50, and references therein]. It has been difficult to visualize how apatitic crystals formed within MVs could make their way to these collagen gaps. Other investigators, including K. Prostak, S. Lees, and H. Schwarcz, have found that up to 80 % of the HA can be external to the collagen fibers [51, and references therein]. Given that MVs display collagen-binding molecules on their membranes (TNAP, ANXA5), we can now begin to conceive possible mechanisms of transfer of MV-initiated HA crystals to the collagen fibers. This will undoubtedly become an area of active research in the near future.
In addition, the mineralization field needs to define if MV-mediated calcification and the enzymatic regulation of the PPi/Pi ratio discussed in this review are universal cellular mechanisms that precede collagen-mediated propagation of matrix mineralization in all calcifying tissues or if they are restricted to certain skeletal and dental tissues. It is very clear that odontoblasts mineralize the dentin via the production of MVs and regulation of the PPi/Pi ratio, as demonstrated through the analysis of Alpl−/− hypophosphatasia mice and the success of enzyme replacement using mineral-targeting TNAP to correct the dentin phenotype [52, 53]. Similarly, the acellular cementum is under strict regulation by the PPi/Pi ratio [54]; and this ratio is normalized, and acellular cementum corrected, in hypophosphatasia mice treated with enzyme replacement [55]. Whether cementum mineralization proceeds via MVs has not yet been determined. Neither is it clear if enamel formation and mineralization involve the function of MVs, a tissue notoriously devoid of collagen. However, enamel defects are also present in hypophosphatasia mice, and these defects are preventable by enzyme replacement with mineral-targeting TNAP [56], arguing that the PPi/Pi ratio is also involved in the regulation of enamel matrix mineralization. Finally, there is some evidence that dystrophic calcification of vascular smooth muscle cells, as occurs in chronic kidney disease, also involves the production and function of MVs [57]; and alterations in the PPi/Pi ratio are crucial in the pathophysiology of medial vascular calcification in chronic kidney disease as well as in the rare condition generalized arterial calcification of infancy caused by ENPP1 gene mutations [58, 59]. We can expect further work in this area to focus on devising pharmacological approaches to treating medial vascular calcification, a critically unmet clinical need, by targeting the functions of TNAP [58] and PHOSPHO1 [60] as a means of correcting the pathological PPi/Pi imbalance that is central to its pathogenesis.
Acknowledgments
The author’s work cited in this review has been supported by grants DE12889, AR53102, and AR47908 from the National Institutes of Health.
Footnotes
The author has stated that there is no conflict of interest.
References
- 1.Glimcher MJ. Bone: nature of the calcium phosphate crystals and cellular, structural, and physical chemical mechanisms in their formation. Rev Mineral Geochem. 2006;64:223–282. [Google Scholar]
- 2.Anderson HC. Vesicles associated with calcification in the matrix of epiphyseal cartilage. J Cell Biol. 1969;41:59–72. doi: 10.1083/jcb.41.1.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ali SY, Sajdera SW, Anderson HC. Isolation and characterization of calcifying matrix vesicles from epiphyseal cartilage. Proc Natl Acad Sci USA. 1970;67:1513–1520. doi: 10.1073/pnas.67.3.1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anderson HC, Hsu HH, Morris DC, Fedde KN, Whyte MP. Matrix vesicles in osteomalacic hypophosphatasia bone contain apatite-like mineral crystals. Am J Pathol. 1997;151:1555–1561. [PMC free article] [PubMed] [Google Scholar]
- 5.Register TC, McLean FM, Low MG, Wuthier RE. Roles of alkaline phosphatase and labile internal mineral in matrix vesicle-mediated calcification. Effect of selective release of membrane-bound alkaline phosphatase and treatment with isosmotic pH 6 buffer. J Biol Chem. 1986;261:9354–9360. [PubMed] [Google Scholar]
- 6.Anderson HC. Molecular biology of matrix vesicles. Clin Orthop Relat Res. 1995;314:266–280. [PubMed] [Google Scholar]
- 7.Golub EE. Role of matrix vesicles in biomineralization. Biochim Biophys Acta. 2009;1790:1592–1598. doi: 10.1016/j.bbagen.2009.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Anderson HC, Garimella R, Tague SE. The role of matrix vesicles in growth plate development and biomineralization. Front Biosci. 2005;10:822–837. doi: 10.2741/1576. [DOI] [PubMed] [Google Scholar]
- 9.Schinke T, McKee MD, Karsenty G. Extracellular matrix calcification: where is the action? Nat Genet. 1999;21:150–151. doi: 10.1038/5928. [DOI] [PubMed] [Google Scholar]
- 10.Giachelli CM. Inducers and inhibitors of biomineralization: lessons from pathological calcification. Orthod Craniofac Res. 2005;8:229–231. doi: 10.1111/j.1601-6343.2005.00345.x. [DOI] [PubMed] [Google Scholar]
- 11.Millán J. Mammalian alkaline phosphatases: from biology to applications in medicine and biotechnology. Wiley-VCH Verlag; Weinheim: 2006. [Google Scholar]
- 12.Houston B, Seawright E, Jefferies D, Hoogland E, Lester D, Whitehead C, Farquharson C. Identification and cloning of a novel phosphatase expressed at high levels in differentiating growth plate chondrocytes. Biochim Biophys Acta. 1999;1448:500–506. doi: 10.1016/s0167-4889(98)00153-0. [DOI] [PubMed] [Google Scholar]
- 13.Stewart AJ, Schmid R, Blindauer CA, Paisey SJ, Farquharson C. Comparative modelling of human PHOSPHO1 reveals a new group of phosphatases within the haloacid dehalogenase superfamily. Protein Eng. 2003;16:889–895. doi: 10.1093/protein/gzg126. [DOI] [PubMed] [Google Scholar]
- 14.Houston B, Stewart AJ, Farquharson C. PHOSPHO1-A novel phosphatase specifically expressed at sites of mineralisation in bone and cartilage. Bone. 2004;34:629–637. doi: 10.1016/j.bone.2003.12.023. [DOI] [PubMed] [Google Scholar]
- 15.Roberts SJ, Stewart AJ, Schmid R, Blindauer CA, Bond SR, Sadler PJ, Farquharson C. Probing the substrate specificities of human PHOSPHO1 and PHOSPHO2. Biochim Biophys Acta. 2005;1752:73–82. doi: 10.1016/j.bbapap.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 16.Stefan C, Jansen S, Bollen M. NPP-type ectophosphodiesterases: unity in diversity. Trends Biochem Sci. 2005;30:542–550. doi: 10.1016/j.tibs.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 17.Terkeltaub RA. Inorganic pyrophosphate generation and disposition in pathophysiology. Am J Physiol Cell Physiol. 2001;281:C1–C11. doi: 10.1152/ajpcell.2001.281.1.C1. [DOI] [PubMed] [Google Scholar]
- 18.Bollen M, Gijsbers R, Ceulemans H, Stalmans W, Stefan C. Nucleotide pyrophosphatases/phosphodiesterases on the move. Crit Rev Biochem Mol Biol. 2000;35:393–432. doi: 10.1080/10409230091169249. [DOI] [PubMed] [Google Scholar]
- 19.Terkeltaub R. Physiologic and pathologic functions of the NPP nucleotide pyrophosphatase/phosphodiesterase family focusing on NPP1 in calcification. Purinergic Signal. 2006;2:371–377. doi: 10.1007/s11302-005-5304-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meyer JL. Can biological calcification occur in the presence of pyrophosphate? Arch Biochem Biophys. 1984;231:1–8. doi: 10.1016/0003-9861(84)90356-4. [DOI] [PubMed] [Google Scholar]
- 21.Ho AM, Johnson MD, Kingsley DM. Role of the mouse ank gene in control of tissue calcification and arthritis. Science. 2000;289:265–270. doi: 10.1126/science.289.5477.265. [DOI] [PubMed] [Google Scholar]
- 22.Johnson KA, Hessle L, Vaingankar S, Wennberg C, Mauro S, Narisawa S, Goding JW, Sano K, Millán JL, Terkeltaub R. Osteoblast tissue-nonspecific alkaline phosphatase antagonizes and regulates PC-1. Am J Physiol Regul Integr Comp Physiol. 2000;279:R1365–R1377. doi: 10.1152/ajpregu.2000.279.4.R1365. [DOI] [PubMed] [Google Scholar]
- 23.Hessle L, Johnson KA, Anderson HC, Narisawa S, Sali A, Goding JW, Terkeltaub R, Millán JL. Tissue-nonspecific alkaline phosphatase and plasma cell membrane glycoprotein-1 are central antagonistic regulators of bone mineralization. Proc Natl Acad Sci USA. 2002;99:9445–9449. doi: 10.1073/pnas.142063399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Murshed M, Harmey D, Millán JL, McKee MD, Karsenty G. Unique coexpression in osteoblasts of broadly expressed genes accounts for the spatial restriction of ECM mineralization to bone. Genes Dev. 2005;19:1093–1104. doi: 10.1101/gad.1276205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harmey D, Hessle L, Narisawa S, Johnson KA, Terkeltaub R, Millán JL. Concerted regulation of inorganic pyrophosphate and osteopontin by akp2, enpp1, and ank: an integrated model of the pathogenesis of mineralization disorders. Am J Pathol. 2004;164:1199–1209. doi: 10.1016/S0002-9440(10)63208-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Whyte MP. Hypophosphatasia. In: Scriver C, Beaudet A, Sly W, Valle D, Childs B, Kinzler K, editors. The metabolic and molecular bases of inherited disease. McGraw-Hill; New York: 2001. pp. 5313–5329. [Google Scholar]
- 27.Whyte MP. Hypophosphatasia. In: Glorieux F, Jueppner H, Pettifor J, editors. Pediatric bone. 3. Elsevier (Academic Press); San Diego: 2012. pp. 771–794. [Google Scholar]
- 28.Narisawa S, Frohlander N, Millán JL. Inactivation of two mouse alkaline phosphatase genes and establishment of a model of infantile hypophosphatasia. Dev Dyn. 1997;208:432–446. doi: 10.1002/(SICI)1097-0177(199703)208:3<432::AID-AJA13>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 29.Fedde KN, Blair L, Silverstein J, Coburn SP, Ryan LM, Weinstein RS, Waymire K, Narisawa S, Millán JL, MacGregor GR, Whyte MP. Alkaline phosphatase knock-out mice recapitulate the metabolic and skeletal defects of infantile hypophosphatasia. J Bone Miner Res. 1999;14:2015–2026. doi: 10.1359/jbmr.1999.14.12.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Anderson HC, Sipe JB, Hessle L, Dhanyamraju R, Atti E, Camacho NP, Millán JL. Impaired calcification around matrix vesicles of growth plate and bone in alkaline phosphatase-deficient mice. Am J Pathol. 2004;164:841–847. doi: 10.1016/s0002-9440(10)63172-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Whyte MP. Hypophosphatasia and the role of alkaline phosphatase in skeletal mineralization. Endocr Rev. 1994;15:439–461. doi: 10.1210/edrv-15-4-439. [DOI] [PubMed] [Google Scholar]
- 32.Anderson HC, Harmey D, Camacho NP, Garimella R, Sipe JB, Tague S, Bi X, Johnson K, Terkeltaub R, Millán JL. Sustained osteomalacia of long bones despite major improvement in other hypophosphatasia-related mineral deficits in tissue non-specific alkaline phosphatase/nucleotide pyrophosphatase phosphodiesterase 1 double-deficient mice. Am J Pathol. 2005;166:1711–1720. doi: 10.1016/S0002-9440(10)62481-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roberts S, Narisawa S, Harmey D, Millán JL, Farquharson C. Functional involvement of PHOSPHO1 in matrix vesicle-mediated skeletal mineralization. J Bone Miner Res. 2007;22:617–627. doi: 10.1359/jbmr.070108. [DOI] [PubMed] [Google Scholar]
- 34.Roberts SJ, Stewart AJ, Sadler PJ, Farquharson C. Human PHOSPHO1 exhibits high specific phosphoethanolamine and phosphocholine phosphatase activities. Biochem J. 2004;382:59–65. doi: 10.1042/BJ20040511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stewart AJ, Roberts SJ, Seawright E, Davey MG, Fleming RH, Farquharson C. The presence of PHOSPHO1 in matrix vesicles and its developmental expression prior to skeletal mineralization. Bone. 2006;39:1000–1007. doi: 10.1016/j.bone.2006.05.014. [DOI] [PubMed] [Google Scholar]
- 36.MacRae VE, Davey MG, McTeir L, Narisawa S, Yadav MC, Millán JL, Farquharson C. Inhibition of PHOSPHO1 activity results in impaired skeletal mineralization during limb development of the chick. Bone. 2010;46:1146–1155. doi: 10.1016/j.bone.2009.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huesa C, Yadav MC, Finnila MA, Goodyear SR, Robins SP, Tanner KE, Aspden RM, Millán JL, Farquharson C. PHOSPHO1 is essential for mechanically competent mineralization and the avoidance of spontaneous fractures. Bone. 2011;48:1066–1074. doi: 10.1016/j.bone.2011.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yadav MC, Simao AM, Narisawa S, Huesa C, McKee MD, Farquharson C, Millán JL. Loss of skeletal mineralization by the simultaneous ablation of PHOSPHO1 and alkaline phosphatase function: a unified model of the mechanisms of initiation of skeletal calcification. J Bone Miner Res. 2011;26:286–297. doi: 10.1002/jbmr.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Robison R. The possible significance of hexosephosphoric esters in ossification. Biochem J. 1923;17:286–293. doi: 10.1042/bj0170286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Majeska RJ, Wuthier RE. Studies on matrix vesicles isolated from chick epiphyseal cartilage. Association of pyrophosphatase and ATPase activities with alkaline phosphatase. Biochim Biophys Acta. 1975;391:51–60. doi: 10.1016/0005-2744(75)90151-5. [DOI] [PubMed] [Google Scholar]
- 41.Omelon S, Georgiou J, Henneman ZJ, Wise LM, Sukhu B, Hunt T, Wynnyckyj C, Holmyard D, Bielecki R, Grynpas MD. Control of vertebrate skeletal mineralization by polyphosphates. PLoS One. 2009;4:e5634. doi: 10.1371/journal.pone.0005634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ciancaglini P, Yadav MC, Simao AM, Narisawa S, Pizauro JM, Farquharson C, Hoylaerts MF, Millán JL. Kinetic analysis of substrate utilization by native and TNAP-, NPP1-, or PHOS-PHO1-deficient matrix vesicles. J Bone Miner Res. 2010;25:716–723. doi: 10.1359/jbmr.091023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Suzuki A, Ghayor C, Guicheux J, Magne D, Quillard S, Kakita A, Ono Y, Miura Y, Oiso Y, Itoh M, Caverzasio J. Enhanced expression of the inorganic phosphate transporter Pit-1 is involved in BMP-2-induced matrix mineralization in osteoblast-like cells. J Bone Miner Res. 2006;21:674–683. doi: 10.1359/jbmr.020603. [DOI] [PubMed] [Google Scholar]
- 44.Wu LN, Sauer GR, Genge BR, Valhmu WB, Wuthier RE. Effects of analogues of inorganic phosphate and sodium ion on mineralization of matrix vesicles isolated from growth plate cartilage of normal rapidly growing chickens. J Inorg Biochem. 2003;94:221–235. doi: 10.1016/s0162-0134(03)00003-5. [DOI] [PubMed] [Google Scholar]
- 45.Wu LN, Genge BR, Kang MW, Arsenault AL, Wuthier RE. Changes in phospholipid extractability and composition accompany mineralization of chicken growth plate cartilage matrix vesicles. J Biol Chem. 2002;277:5126–5133. doi: 10.1074/jbc.M107899200. [DOI] [PubMed] [Google Scholar]
- 46.Nielsen LB, Pedersen FS, Pedersen L. Expression of type III sodium-dependent phosphate transporters/retroviral receptors mRNAs during osteoblast differentiation. Bone. 2001;28:160–166. doi: 10.1016/s8756-3282(00)00418-x. [DOI] [PubMed] [Google Scholar]
- 47.Yoshiko Y, Candeliere GA, Maeda N, Aubin JE. Osteoblast autonomous Pi regulation via Pit1 plays a role in bone mineralization. Mol Cell Biol. 2007;27:4465–4474. doi: 10.1128/MCB.00104-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Beck L, Leroy C, Beck-Cormier S, Forand A, Salaun C, Paris N, Bernier A, Urena-Torres P, Prie D, Ollero M, Coulombel L, Friedlander G. The phosphate transporter PiT1 (Slc20a1) revealed as a new essential gene for mouse liver development. PLoS One. 2010;5:e9148. doi: 10.1371/journal.pone.0009148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Polewski MD, Johnson KA, Foster M, Millán JL, Terkeltaub R. Inorganic pyrophosphatase induces type I collagen in osteoblasts. Bone. 2010;46:81–90. doi: 10.1016/j.bone.2009.08.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Silver FH, Landis WJ. Deposition of apatite in mineralizing vertebrate extracellular matrices: a model of possible nucleation sites on type I collagen. Connect Tissue Res. 2011;52:242–254. doi: 10.3109/03008207.2010.551567. [DOI] [PubMed] [Google Scholar]
- 51.McNally EA, Schwarcz HP, Botton GA, Arsenault AL. A model for the ultrastructure of bone based on electron microscopy of ion-milled sections. PLoS One. 2012;7:e29258. doi: 10.1371/journal.pone.0029258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Millán JL, Narisawa S, Lemire I, Loisel TP, Boileau G, Leonard P, Gramatikova S, Terkeltaub R, Camacho NP, McKee MD, Crine P, Whyte MP. Enzyme replacement therapy for murine hypophosphatasia. J Bone Miner Res. 2008;23:777–787. doi: 10.1359/JBMR.071213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Foster BL, Nagatomo KJ, Tso HW, Tran AB, Niciti FH, Narisawa S, McKee MD, Millan JL, Somerman MJ. Tooth root dentin mineralization defects in a mouse model of hypophosphatasia. J Bone Miner Res. 2012 doi: 10.1002/jbmr.1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Foster BL, Nagatomo KJ, Nociti FH, Jr, Fong H, Dunn D, Tran AB, Wang W, Narisawa S, Millán JL, Somerman MJ. Central role of pyrophosphate in acellular cementum formation. PLoS One. 2012;7(6):e38393. doi: 10.1371/journal.pone.0038393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McKee MD, Nakano Y, Masica DL, Gray JJ, Lemire I, Heft R, Whyte MP, Crine P, Millán JL. Enzyme replacement therapy prevents dental defects in a model of hypophosphatasia. J Dent Res. 2011;90:470–476. doi: 10.1177/0022034510393517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yadav MC, de Oliveira RC, Foster BL, Fong H, Cory E, Narisawa S, Sah RL, Somerman M, Whyte MP, Millán JL. Enzyme replacement prevents enamel defects in hypophosphatasia mice. J Bone Miner Res. 2012;27:1722–1734. doi: 10.1002/jbmr.1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kapustin AN, Davies JD, Reynolds JL, McNair R, Jones GT, Sidibe A, Schurgers LJ, Skepper JN, Proudfoot D, Mayr M, Shanahan CM. Calcium regulates key components of vascular smooth muscle cell-derived matrix vesicles to enhance mineralization. Circ Res. 2011;109:e1–e12. doi: 10.1161/CIRCRESAHA.110.238808. [DOI] [PubMed] [Google Scholar]
- 58.Narisawa S, Harmey D, Yadav MC, O’Neill WC, Hoylaerts MF, Millán JL. Novel inhibitors of alkaline phosphatase suppress vascular smooth muscle cell calcification. J Bone Miner Res. 2007;22:1700–1710. doi: 10.1359/jbmr.070714. [DOI] [PubMed] [Google Scholar]
- 59.Lomashvili KA, Garg P, Narisawa S, Millán JL, O’Neill WC. Upregulation of alkaline phosphatase and pyrophosphate hydrolysis: potential mechanism for uremic vascular calcification. Kidney Int. 2008;73:1024–1030. doi: 10.1038/ki.2008.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kiffer-Moreira T, Yadav MC, Zhu D, Narisawa S, Sheen C, Stec B, Cosford ND, Dahl R, Farquharson C, Hoylaerts MF, MacRae VE, Millán JL. Pharmacological inhibition of PHOSPHO1 suppresses vascular smooth muscle cell calcification. J Bone Miner Res. 2012 doi: 10.1002/jbmr.1733. [DOI] [PMC free article] [PubMed] [Google Scholar]