

Abstract
Fragile X syndrome (FXS) is the most frequent inherited form of human mental retardation. It is characterized by cognitive impairment and physical and behavioral problems and is caused by the silencing of fmr1 transcription and the absence of the fmr1 protein (FMRP). Recently, animal models of FXS have greatly facilitated the investigation of the molecular and cellular mechanisms of this loss-of-function disorder. The present study was aimed to further characterize the role of FMRP in behavior and synaptic function by using fmr1 knockout zebrafish. In adult zebrafish, we found that fmr1 knockout produces the anxiolytic-like responses of increased exploratory behavior in light/dark and open-field tests and avoidance learning impairment. Furthermore, electrophysiological recordings from telencephalic slice preparations of knockout fish displayed markedly reduced long-term potentiation and enhanced long-term depression compared to wild-type fish; however, basal glutamatergic transmission and presynaptic function at the lateral (Dl) and medial (Dm) division of the dorsal telencephalon synapse remained normal. Taken together, our study not only evaluates the mechanism of FRMP but also suggests that zebrafish have valuable potential as a complementary vertebrate model in studying the molecular pathogenesis of human fragile X syndrome.
Introduction
The onset rate of fragile X syndrome (FXS) is approximately one in 4000 males and one in 8000 females affected [1], [2]. The clinical features of FXS include attention deficits, hyperactivity, social deficits, anxiety disorder and deficits in cognitive flexibility [3]. This syndrome is most commonly caused by a triplet repeat expansion (CGG) mutation in the fmr1 gene that encodes FMRP, the fragile X mental retardation protein [4], [5], [6]. When the CGG expansions within the 5′ untranslated region (UTR) of the fmr1 gene exceed 200 repeats (the full mutation), the hypermethylation [7] and deacetylation [8] of fmr1 result, leading to the silencing of fmr1 transcription and the absence of the FMR1 protein (FMRP).
FMRP, a cytoplasmic mRNA-binding protein, is widely expressed in various tissues with the most abundant expression in the brain and testes of mammalians [9], [10]. For example, FMRP is expressed in neurons, particularly those of the hippocampus, amygdala, and in the Purkinje cells of the cerebellum [10], [11]. In addition, the functional domains of FMRP that are evolutionarily conserved between humans and Drosophila include the nuclear localization signal (NLS) and two KH domains, the nuclear export signal (NES) and an RGG box. In humans, FMRP is highly expressed in neurons of the brain, where it is suggested to play an important role in the regulation of local mRNA translation within neuronal dendritic spines and altered synaptic function [12].
Animal models of FXS have greatly facilitated the investigation of the molecular and cellular mechanism of this disorder. For example, fmr1 knockout (KO) mice were characterized as having several behavioral profiles similar to those of fragile X patients, including hyperactivity, reduced anxiety-related behavior, and learning and memory deficits [13], [14]. Synapse function is closely correlated with dendritic spine morphology and synaptic activity. Indeed, abnormalities of the dendritic spine is a significant neuroanatomical defect in FXS patients [15] and fmr1 KO mice [16]. Therefore, the findings of a spine morphological phenotype indicate a possible defect in synaptic plasticity in FXS that could result in the cognitive disability phenotype. Long-term potentiation (LTP) and long-term depression (LTD) are the key cellular mechanisms underlying learning and memory processes [17], [18]. Consistent with behavioral learning deficits, electrophysiological studies have reported the loss of LTP in the anterior cingulate cortex (ACC) and the lateral amygdala of fmr1 KO mice [19]. However, previous studies have also shown that a lack of FMRP may lead to exaggerated metabotropic glutamate receptor (mGluR) signaling and enhanced hippocampal LTD in fmr1 KO mice [20].
The zebrafish is a small tropical freshwater teleost native to South-East Asia. Zebrafish were first used for biological research purposes by Dr. George Streisinger, in 1981 [21], [22], [23]. As a relatively simple vertebrate species, the zebrafish is a popular model organism for developmental and genetic studies [24]. It is also an excellent model for biomedical research, and the advantages of this model include genomic and physiological homology with humans, external fertilization, and large numbers of fertilized eggs. Most importantly, the transparency of the zebrafish embryo allows one to visualize the processes of embryogenesis and morphogenesis in early developmental stages. Moreover, the small size of zebrafish enables cost-efficient maintenance of numerous adult individuals. It is also possible to obtain and maintain the transgenic strains of zebrafish at a low cost.
Rupp and colleagues demonstrated that the basic components of the adult zebrafish brain are very similar to those of land vertebrates [25]. Due to the accumulated genetic knowledge and tools developed for the zebrafish, many reports make use of the zebrafish to study complex behaviors such as seizures [26], addiction [27], and learning and memory [28], [29], [30], [31]. Therefore, zebrafish is a valuable model for studying the pathways and mechanisms of human neurological disorders and clinical treatments.
The adult zebrafish FMRP shares 72% amino acid identity with that of humans, and similar to humans, it is highly expressed the brain, including in the telencephalon, diencephalon, metencephalon, spinal cord, and cerebellum [32]. In 2009, den Broader and colleagues generated the fmr1 knockout (KO) zebrafish, which provided a new genetic model system to study FXS [33]. However, research analyzing the phenotypic characteristics of fmr1 KO zebrafish in adulthood is sparse. In a previous study, we reported that the telencephalon is physiologically involved in the process of fear memory formation in the inhibitory avoidance task in zebrafish [34]. The present study aimed to further characterize the effects of the loss of FMRP on cognitive phenotypes by investigating possible differences in cognitive behavior in inhibitory avoidance and synaptic plasticity at the Dl-Dm synapse of telencephalon in adult fmr1 KO zebrafish.
Materials and Methods
fmr1 knockout (KO) zebrafish
Zebrafish mutants carrying the fmr1 hu2787 allele were obtained from the Wellcome Trust Sanger Institute Zebrafish Mutant Resource. This allele carries a C432T change, which causes a premature termination at codon position 113 [33]. Fish (4–8 months of age) of both sexes were used for these experiments; fmr1 KO and control fish of the TL background were maintained according to standard procedures [35] and following guidelines approved by the Institutional Animal Care and Use Committee (IACUC) of National Taiwan Normal University (Approval number: 10030).
Polymerase Chain Reaction (PCR)
Adult fishes were genotyped by PCR using tissue and cell genomic DNA purification kit (Genemark, Taipei, Taiwan) according to manufacturer protocol. DNA extracted from the tails. Zebrafish genomic DNAs were prepared using Tissue and Cell Genomic DNA Purification Kit (Genemark, Taipei, Taiwan) according to manufacturer protocol. Optic density (O.D.) value of each sample was measured by a Nano spectrophotometer (NanoDrop Technologies, Inc. Wilmington, DL) at 260 and 280 nm to determine the purity and concentration of DNA. Genotyping of the present study is based on dCAPS assay (derived Cleaved Amplified Polymorphic Sequence). In this assay, a mismatch in PCR primer is used to create restriction endonuclease (RE)-sensitive polymorphism based on the target mutation. To genotype the hu2787 allele, a mismatch has been introduced into the forward primer. During PCR, this mismatch creates an RsaI restriction enzyme site in the amplified product derived from the WT DNA template. The RsaI site is not present in the PCR product containing the hu2787 mutation. A 222-bp PCR product was generated using forward primer (5′-CTA AAT GAA ATC GTC ACA TTA GAG AGG GTA) and reverse primer (5′- TCCATG ACA TCC TGC ATT AG). The amplification reaction mixture (50 µL) contained 200 ng genomic DNA, 0.5 mM of each dNTP, 1 µM of each primer, 1 unit Prozyme DNA polymerase (Protech Enterprise, Taipei, Taiwan) and 1× PCR buffer. The PCR reaction conditions began with a denaturation at 94°C for 4 min, followed by 40 cycles of 94°C for 30 seconds, 60°C for 30 seconds and 72°C for 20 seconds; lastly, 5 min at 72°C. After amplification, the PCR product was digested by RsaI restriction enzyme in 1 X restriction enzyme buffer. Finally, digested PCR products were separated by electrophoresis in 3% agarose gel. The PCR products derived from the WT template were cleaved to 193-and 29-bp DNA fragments.
Western blot analysis
After the animals were scarified, the telencephalon brain region was quickly removed from the skull and homogenized using a T-PER tissue protein extraction reagent kit (Pierce Biotechnology, Inc., Rockford, IL) with the addition of the Halt Protease Inhibitor Cocktail. The protein concentration was determined by the Bradford protein assay, and an equal amount of protein (25 µg per sample) was subjected to SDS–10% PAGE. The proteins separated on the gel of the SDS–PAGE were transferred to a PVDF membrane (Millipore, Bedford, MA). For the immune-detection, the membrane was first blocked with 5% skim milk and 0.05% Tween in PBS for 1 h at room temperature. The primary antibodies used for the detection were rabbit anti-FMRP (1∶4,000; Gift from Dr. Willemsen, #758) antibodies. The membranes were incubated with primary antibodies overnight at 4°C and, subsequently, with HRP-conjugated secondary antibodies for 1 h at room temperature. Finally, the detected signals were visualized with enhanced chemiluminescence (Bioman Scientific Co. Ltd., Taiwan) and quantitatively analyzed by a LAS3000 digital imaging system (Fujifilm, Tokyo, Japan).
Behavioral procedures
Prior to the initiation of behavioral protocols, zebrafish were separated into individual tanks a few days before the experiment. To elucidate the behavioral effects of FMRP deficiency, fish (10 WT and 12 KO) were initially assessed with a light/dark test, then tested 48 h later in an inhibitory avoidance test, and finally examined in an open-field test.
Light/dark test
To examine anxiety-related behaviors, fish were tested in a light/dark test conducted in a rectangular acryl tank (7 cm height×18 cm Length×9 cm width) divided into two equally sized compartments that were demarcated by black and white coloration. The tank water level was maintained at 3 cm. Before testing, the subjects were carefully placed in the white compartment. Then, the fish were allowed to swim freely between the two compartments without a sliding door for 5 min. Behavior was recorded using a Logitech S5500 camera and Logitech Quick Capture software. The proportion of the trial that the animal spent in the white compartment was recorded. Reduced exploration of the white compartment in this test reflects high anxiety states.
Inhibitory avoidance task (IA)
Fish were tested for emotional learning in an inhibitory avoidance task as described previously [34]. Briefly, fish underwent acclimation, one-trial training and one test session 24 h later. On day one, the fish received an acclimation trial that consisted of placing the fish in the shallow chamber for 5 min; the white, opaque guillotine door was then removed, and the fish was allowed to swim freely between the two compartments for another 5 min. In the training session, the fish was placed in the shallow compartment. After 1 min, the guillotine door to the deep compartment was opened. Once the fish entered the deep compartment, the guillotine door was closed, and a mild electric shock was applied to the deep compartment for 5 s. The fish was immediately removed from the apparatus and returned to its home tank. Twenty-four hours later, latency to enter the deep compartment of the apparatus was recorded to a maximum of 300 sec.
Open-field test
At the end of the retention test, the animals were placed into a transparent cylindrical tank (20 cm in height and 22 cm in diameter) for 10 min to test their spontaneous motor activity. The water level was maintained at 4 cm. Behavior was detected using an EthoVision video tracking system (Noldus Information Technology, Leesburg, VA, U S A). The total distance swam and the mean speed was measured for statistical analyses.
Extracellular field potential recordings
Acute telencephalic slice preparation was similar to that described previously [36]. Briefly, fish were euthanized by exposing them to an ice-cold (0∼4°C), artificially oxygenated cerebrospinal fluid (aCSF) solution consisting of the following: 117 mM NaCl, 4.7 mM KCl, 1.2 mM NaH2PO4, 2.5 mM NaHCO3, 1.2 mM MgCl2, 2.5 mM CaCl2, and 11 mM d-(+)-glucose. Their brains were quickly removed under the aCSF solution. Transverse telencephalic slices (300 µm) were prepared using a vibrotome (MA752, Campden Instruments Ltd., UK) in ice-cold aCSF. Slices were then incubated in the aCSF solution, which was bubbled continuously with 95% O2/5% CO2 for at least 1 h prior to recordings at room temperature.
Extracellular population spikes (PSs) were recorded using a 64-channel multi-electrode dish (MED64) system (Alpha MED Sciences, Tokyo, Japan) with a sample rate of 20 kHz. Recordings were performed with an 8×8 array of planar microelectrodes. Each electrode was 20×20 µm in size, and the inter-electrode spacing was 100 µm. Telencephalic slices were placed in a recording chamber and perfused with aCSF (30°C) at a flow rate of 1–2 ml/min via a peristaltic pump (Gilson Minupuls 3, Villiers Le Bel, France).A nylon mesh and a stainless steel wire were used to secure slice position and contact with electrodes during perfusion. Stimulus intensity was adjusted to evoke 40–50% of the maximal stimulation response. Test stimuli were 0.2 ms pulses every 20 s, and responses were recorded for 15 min prior to beginning the experimental treatments to assure stability of responses. Every three consecutive responses were pooled and averaged for data analysis.
Basal synaptic transmission was measured by plotting the current applied to the stimulating electrode (40–150 µA) against the amplitude of population spike responses to generate input–output curves (I/O curves). Paired-pulse facilitation was assessed by applying pairs of stimuli at varying inter-pulse intervals (20, 50, 100, 150, and 200 ms). The paired pulse ratio (PPR) was determined by calculating the ratio of the average amplitude of the second response to the first. Each trace corresponds to an average over 3 trials. After stable baseline recording, LTP was elicited by HFS protocols consisting of three stimulus trains of 100 pulses (at 100 Hz) with 20 s inter-train intervals. LTD was induced by low frequency stimulation that consisted of 1 Hz stimulation for 20 min. The magnitudes of both LTP and LTD were measured, post-induction, as an average of 10 min at the end of the recording period.
Statistical analysis
Statistical analysis was performed using SPSS version 12.0 (SPSS, Chicago). In electrophysiological experiments, each trace is the average of three consecutive responses. LTP plots were normalized to the average baseline value of each slice preparation. All values are reported as mean ± SEM. Statistical comparisons of PPF and LTP were made using the paired t-test. In all cases, p<0.05 was considered to be significant. In behavioral experiments, all values are reported as the mean ± SEM. For the analysis of inhibitory avoidance performance, the comparison among behavioral trials within the same group was carried out by using Wilcoxon test. Whereas, the results from locomotor activity studies were analyzed by Student's t test. We considered p<0.05 to be statistically significant.
Results
Genotyping results
Homozygous mutants were obtained with the expected frequency of 25%, and they had normal appearance. The sex ratio in the homozygote population was not significantly different from the other genotypes. The knockout phenotype was confirmed at the DNA level by PCR. The PCR products derived from the wild-type and fmr1 KO fish were cleaved to 193-and 222-bp DNA fragments respectively (Figure 1A). The protein level was also analyzed by Western blotting, where no FMRP protein was detectable in testes of fmr1 KO fish (Figure 1B).
Figure 1. Summary of genotyping results.
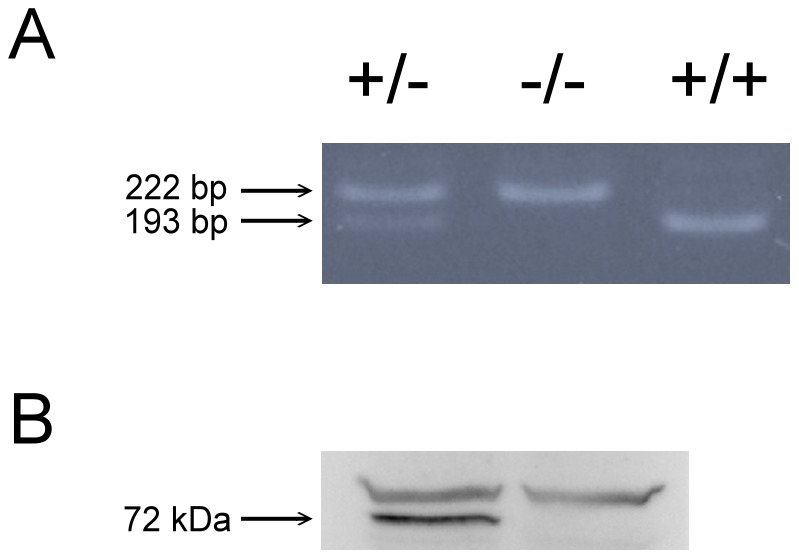
(A) Representative data obtained from genotyping of wild-type (+/+), heterozygous (+/−) and homozygous (−/−) fishes was validated by polymerase chain reaction. (B) Brain tissues were analyzed by western blot using an FMRP specific antibody. Lane 1 contains wild-type (WT) and Lane 2 contains fmr1−/− (KO). The arrow points at FMRP located. The FMRP protein is completely absent in fmr1−/−.
Anxiolytic-like responses in fmr1 KO zebrafish
The light/dark test has been proposed as a model of anxiety-like behavior in zebrafish. The time spent in white compartment and the numbers of midline crossings were analyzed for each fish. As illustrated in Figure 2, we found a significant genotypic difference in both measures. fmr1 KO fish spent more time in the white compartment (Fig. 2A, p<0.01) and had greater numbers of midline crossings compared to wild-type fish (Fig. 2B, p<0.01), indicating lower anxiety and increased locomotion in KO fishes.
Figure 2. Anxiolytic-like responses of fmr1 KO zebrafish.
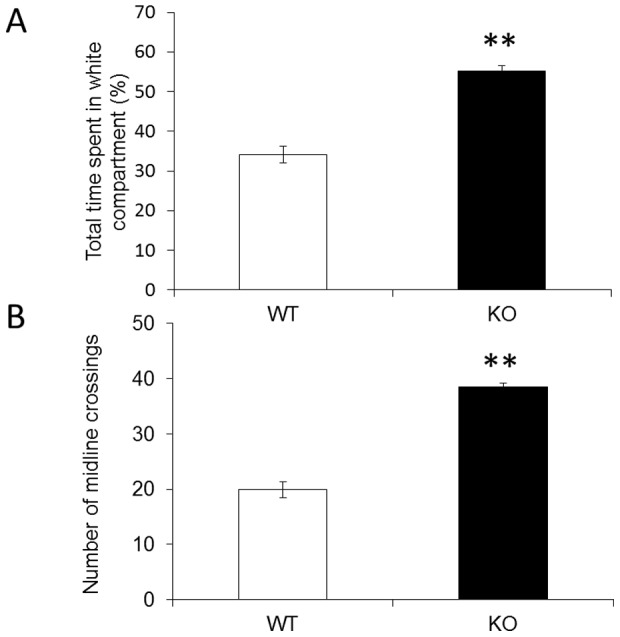
(A) Bar graphs of the time spent in the white compartment by fmr1 KO and wild-type fish. **p<0.01 compared with wild-type fish. (B) Bar graph of the number of midline crossings for fmr1 KO (n = 12) and wild-type fish (n = 10). **p<0.01 compared with wild-type fish.
Impaired inhibitory avoidance learning in fmr1 KO zebrafish
The inhibitory avoidance test has been extensively used for assessing memories of aversive experiences. In this study, fmr1 KO and wild-type fish were trained in the inhibitory avoidance learning task, and latency to enter the deep compartment was assessed 24 h after training. As illustrated in Figure 3 the difference between the latencies in the training and test sessions for wild-type was statistically significant (Fig. 3, n = 10, p<0.05). In contrast, no significant difference was observed in the fmr1 KO fishes. Additionally, the retention test was significantly different (p<0.05) between wild-type and fmr1 KO fish.
Figure 3. The inhibitory avoidance of fmr1 KO and wild-type fish.
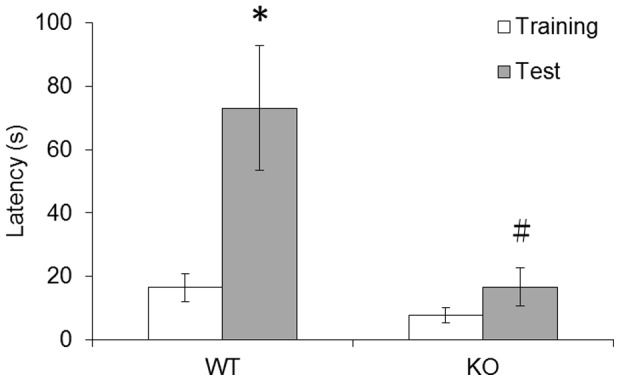
Bars indicate the mean latencies ± the SEMs to cross from the shallow to the deep compartment (in seconds) in the training and test sessions for both genotypes. *p<0.05 compared with training sessions; # p<0.05 compared with wild-type fish.
Hyperactivity in fmr1 KO zebrafish
Hyperactivity is the most common symptom of FXS patients and fmr1 KO mice. To determine whether genotypic differences in locomotor activity were present between genotypes, the total distances swam and mean speeds of fmr1 KO and wild-type fish were calculated in an open field apparatus for 5 min. As shown in Figure 4, the total distances moved and the mean speeds of fmr1 KO fish were higher than those of wild-type fish (p<0.001 for both outcomes).
Figure 4. Locomotor activity of fmr1 KO and wild-type fish.
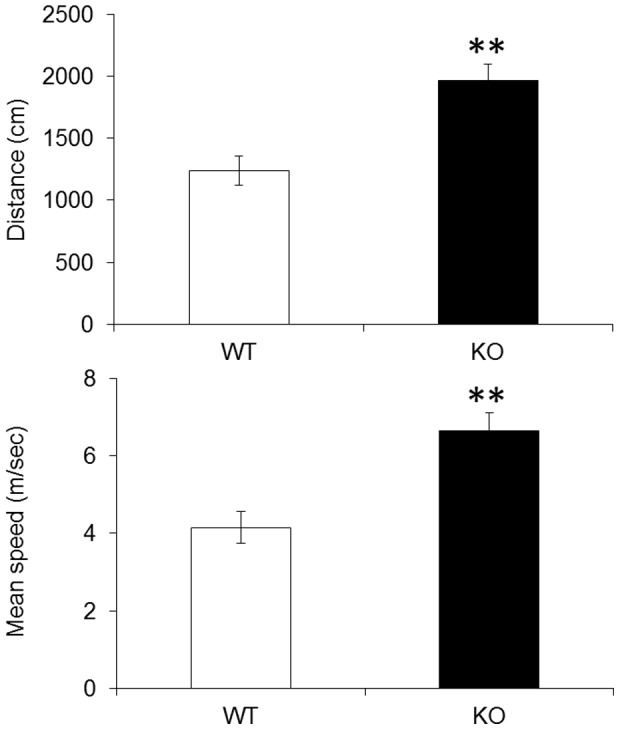
Bar graphs of the total distance moved (in cm) and mean speeds (in m/sec) of fmr1 KO and wild-type fish. **p<0.001 compared with wild-type fish.
Basal synaptic transmission and PPF in fmr1 KO zebrafish
Basal synaptic transmission at the Dl-Dm synapse was measured by field potential responses to increasing stimulation intensities. As shown in Figure 5A, the amplitude of the population spikes obtained from wild-type and fmr1 KO slices were compared, and no significant difference between genotypes was noted. Additionally, paired pulse facilitation (FFP) was measured in slices from both genotypes. As shown in Figure 5B, pairs of presynaptic fiber stimulation pulses delivered at inter-pulse intervals of 20, 50, 100, 150 and 200 milliseconds evoked nearly identical amounts of PPF in slices from wild-type and fmr1 KO zebrafish. We suggest that basal glutamatergic transmission and presynaptic function at the Dl-Dm synapse remain normal in fmr1 KO zebrafish
Figure 5. Basal synaptic function is not different between fmr1 KO and wild-type fish.

(A) Summary of the input-output curves that were created by comparing PS amplitude and stimulus intensity (40–130 µA)(n = 6). (B) Paired-pulse facilitation (FFP) was measured by applying paired stimuli and quantifying the facilitation of the second potential relative to the first as a function of the inter-pulse interval (<200 ms)(n = 7).
Synaptic plasticity in fmr1KO zebrafish
In zebrafish, FMRP is highly expressed in the telencephalon [33], an important brain region involved in synaptic plasticity and learning and memory processes. This fact raises an intriguing possibility that FMRP is involved in synaptic plasticity. We next examined whether the loss of FMRP function in zebrafish was related to modulation of synaptic plasticity; to do this, long-term potentiation (LTP) and long-term depression (LTD) were characterized. As shown in Figure 6, LTP was induced by a standard protocol with three trains of high frequency stimulation. LTP magnitude was significantly reduced in fmr1 KO zebrafish (181.0±7%, n = 9 in wild-type vs. 146.8±6%, n = 10 in fmr1 KO, p<0.05; Fig. 6). LTD is a long-lasting decrease in the synaptic response of the same synapses following prolonged low-frequency stimulation (LFS). LFS-induced LTD was enhanced in slices from fmr1 KO fish compared to slices from wild-type fish (104.3±7%, n = 4 in wild-type vs. 76.5±5%, n = 6 in fmr1KO, p<0.05; Fig. 7). These findings suggest that FMRP plays an important functional role in regulating telencephalic synaptic plasticity in zebrafish.
Figure 6. LTP was significantly reduced in fmr1 KO zebrafish.

(A) The arrow indicates delivery of HFS. Insets are representative, superimposed, single sweeps before and after LTP induction in wild-type (n = 9) and fmr1 KO (n = 10) zebrafish. (B) Summary of the averaged magnitudes of LTP. Bars correspond to the percentages of baseline PS amplitudes during the last 10 min. *p<0.05 compared with wild-type.
Figure 7. LTD was significantly enhanced in fmr1 KO zebrafish.

(A) LTD was induced by a 20 minute LFS (1 Hz) protocol. Insets are representative, superimposed, single sweeps before and after LTD induction in wild-type (n = 4) and fmr1 KO (n = 6) zebrafish. (B) Summary of the averaged magnitudes of LTD. Bars correspond to the percentages of baseline PS amplitude during the last 10 min. *p<0.05 compared with wild-type.
Discussion
Fragile X syndrome (FXS) is caused by loss of the fragile X mental retardation protein (FMRP). To understand the molecular and cellular pathogenesis of FXS, the disease has been successfully modeled in mice [14], [38], Drosophila [37] and zebrafish [33]. In the present study, using fmr1 KO zebrafish, we were able to investigate the functional role of the fmr1 gene in mediating cognitive behavior and synaptic plasticity at the Dl-Dm synapse in the telencephalon of zebrafish. Our results can be summarized as follows: (1) fmr1 KO fish exhibit anxiolytic-like behavior, impaired emotional learning, and hyperactivity, and (2) electrophysiological recordings from telencephalic slice preparations of fmr1 KO fish showed markedly reduced LTP and enhanced LTD compared with wild-type fish. This study provides the first evidence that FMRP is involved in cognitive functions and telencephalic synaptic plasticity in zebrafish and suggests that zebrafish are a new genetic model system to study Fragile X syndrome (FXS).
Previous behavioral studies have demonstrated that fmr1 KO mice replicate many of the human behavioral features of FXS, including hyperactivity, learning deficits, impaired social interaction, and abnormal anxiety-related responses [14]. Furthermore, behavioral profiles are a critical first step toward understanding the function of fmr1. Here, we performed a series of behavioral analyses on the fmr1 KO zebrafish that included the light/dark test, the inhibitory avoidance test, and the open-field test to further characterize the consequences of the absence of FMRP.
Interestingly, significant behavioral differences were detected in the light/dark test. Compared with wild-type fish, fmr1 KO fish had reduced anxiety-related responses in the light/dark test. Our results are remarkably consistent with previous studies [13], [38], [39], [40], [41] in which the loss of FMRP has been reported to be related to anxiolytic responses in mice. Moreover, fmr1 KO zebrafish show a significantly greater number of crossed lines in the lit compartment, which significantly contributed to locomotor activity. Thus, hyperactivity may be present in fmr1 KO zebrafish.
Cognitive impairment is a common symptom of FXS patients and FXS mouse models. For instance, Liu et al. (2011) noted impaired inhibitory avoidance acquisition in the fmr1 KO mice [13]. Here, using an inhibitory avoidance test, we evaluated whether Fmr1 null mutant zebrafish exhibited learning and memory impairments. Consistent with the notion that FMRP is involved in certain types of learning and memory, we found a significant impairment in the inhibitory avoidance (IA) task in fmr1 KO fishes. These results suggested that the absence of FMRP might disrupt the detection abilities of and/or the response of the brain's fear system. After the retention test in the IA task, the animals were subjected to an open-field test; activity in the open-field is often used as a measure of exploration in zebrafish. The distance traveled and the mean speed in the open-field task was significantly higher in fmr1 KO fishes. Our behavioral analyses of the fmr1 KO fish in the light/dark and open-field tests supports previously reported results [13], [14], [39], suggesting that the absence FMRP expression leads to hyperactivity or increased exploratory behavior.
According to neuroanatomical and behavioral analyses, the telencephalic pallium is a key component of the fear circuitry of teleost fish. For example, goldfish with lesions to the telencephalon have impaired avoidance conditioning [42], [43]. In a previous study, we reported that the physiological function of the telencephalon is involved in the process of fear memory formation in inhibitory avoidance tasks in zebrafish [34]. Furthermore, electrophysiological evidence has demonstrated that the intratelencephalic connections between the lateral and medial pallium, and the Dl-Dm synapse, play important roles in the synaptic plasticity of the zebrafish [36]. Because our behavioral results suggested that the lack of FMRP caused inhibitory avoidance learning deficits, we hypothesized that FMRP may play an important functional role in telencephalic synaptic plasticity.
LTP is an enduring enhancement of synaptic efficacy that has been reported as a cellular model for learning, memory and neuronal plasticity in mammals [44], [45], [46], [47] and zebrafish [36], [48]. Recently, abnormal LTP of excitatory transmission has been found in the hippocampus [49], cortex [50], [51], and amygdala [19] of fmr1 KO mice. In the present study, we found that the lack of FMRP results in a reduction in LTP at the Dl-Dm synapse, but absence of FMRP did not affect basal transmission function. These findings are in line with previous studies that have described reductions in hippocampal LTP of fmr1 KO mice [52]; these studies suggest that the absence of FMRP may contribute to reduced hippocampal LTP in fmr1 KO mice via reduction of NMDAR-mediated currents [53]. In addition, FMRP is required for NMDA receptor-dependent LTP in the mouse prefrontal cortex [19]. Our previous work showed that telencephalic LTP at Dl-Dm synapses is NMDAR-dependent [36]. Therefore, we suggest that the reduction of LTP in the present study may be mediated through an NMDAR-dependent pathway.
In contrast to LTP, long-term depression (LTD) is a long-lasting weakening of synaptic strength that is due to the internalization of AMPA receptors. A recent finding that is of importance for understanding the mechanisms by which FMRP deficiency exerts its effects is the reliable observation of LTD enhancement in the hippocampus of fmr1 KO mice. For example, a lack of FMRP produced enhanced metabotropic glutamate receptor-dependent LTD [54], [55]. Using low frequency stimulation (LFS) protocol, we found that the lack of FMRP significantly enhanced LTD at Dl-Dm synapses in fmr1 KO fish compared to wild-type fish. These results may be explained by the mGluR theory of fragile X mental retardation, which suggests that an exaggerated mGluR pathway may lead to an increase in the internalization of AMPAR, as AMPAR trafficking is a driving process for enhanced LTD [56]. The influence of the absence FMRP expression on LTP and LTD leads us to suggest that FMRP may be involved in synaptic function and plasticity.
In summary, we used zebrafish as a system model, taking advantage of available transgenic lines and developmentally very similar to mammals. In addition, it is easy to breed in large numbers and relatively cheap to maintain. Which made it an idea model for prescreen potential therapeutic in vivo. We found abnormal behaviors in fmr1 KO fish that are consistent with findings in humans with defective fmr1 and fmr1 KO mice. These findings include anxiolytic-like responses such as increased exploratory behavior in light/dark and open-field tests and avoidance learning impairments. In addition, our recent study has revealed abnormal telencephalon synaptic function (in both LTP and LTD) that may be linked to the behavioral defects displayed in fmr1 KO fish. Thus, this study suggests that zebrafish is valuable as a potential complementary vertebrate model for the study of the molecular pathogenesis of human fragile X syndrome.
Acknowledgments
For providing the zebrafish knockout alleles-fmr1 hu2787- we thank the Hubrecht laboratory and the Sanger Institute Zebrafish Mutation Resource (ZF-MODELS Integrated Project; contract number LSHG-CT-2003-503496). We also thank Dr. Rob Willemsen for the kind gift of zebrafish FMR-1 antibody.
Funding Statement
This work was supported by a research grant from National Science Council, Taiwan NSC97-2320-B-003-001-MY3, NSC98-2321-B-003-004 and 100-2320-B-003-004. The authors gratitude also goes to the Academic Paper Editing Clinic, National Taiwan Normal University. For providing the zebrafish knockout alleles-fmr1hu2787 the authors thank the Hubrecht laboratory and the Sanger Institute Zebrafish Mutation Resource, (ZF-MODELS Integrated Project; contract number LSHG-CT-2003-503496; funded by the European Commission) also sponsored by the Wellcome Trust [grant number WT 077047/Z/05/Z]. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Turner G, Webb T, Wake S, Robinson H (1996) Prevalence of fragile X syndrome. Am J Med Genet 64: 196–197. [DOI] [PubMed] [Google Scholar]
- 2. Garber K, Smith KT, Reines D, Warren ST (2006) Transcription, translation and fragile X syndrome. Curr Opin Genet Dev 16: 270–275. [DOI] [PubMed] [Google Scholar]
- 3. Reiss AL, Hall SS (2007) Fragile X syndrome: assessment and treatment implications. Child Adolesc Psychiatr Clin N Am 16: 663–675. [DOI] [PubMed] [Google Scholar]
- 4. Fu YH, Kuhl DP, Pizzuti A, Pieretti M, Sutcliffe JS, et al. (1991) Variation of the CGG repeat at the fragile X site results in genetic instability: resolution of the Sherman paradox. Cell 67: 1047–1058. [DOI] [PubMed] [Google Scholar]
- 5. Oberle I, Rousseau F, Heitz D, Kretz C, Devys D, et al. (1991) Instability of a 550-base pair DNA segment and abnormal methylation in fragile X syndrome. Science 252: 1097–1102. [DOI] [PubMed] [Google Scholar]
- 6. Verkerk AJ, Pieretti M, Sutcliffe JS, Fu YH, Kuhl DP, et al. (1991) Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 65: 905–914. [DOI] [PubMed] [Google Scholar]
- 7. Sutcliffe JS, Nelson DL, Zhang F, Pieretti M, Caskey CT, et al. (1992) DNA methylation represses FMR-1 transcription in fragile X syndrome. Hum Mol Genet 1: 397–400. [DOI] [PubMed] [Google Scholar]
- 8. Coffee B, Zhang F, Warren ST, Reines D (1999) Acetylated histones are associated with FMR1 in normal but not fragile X-syndrome cells. Nat Genet 22: 98–101. [DOI] [PubMed] [Google Scholar]
- 9. Hinds HL, Ashley CT, Sutcliffe JS, Nelson DL, Warren ST, et al. (1993) Tissue specific expression of FMR-1 provides evidence for a functional role in fragile X syndrome. Nat Genet 3: 36–43. [DOI] [PubMed] [Google Scholar]
- 10. Devys D, Lutz Y, Rouyer N, Bellocq JP, Mandel JL (1993) The FMR-1 protein is cytoplasmic, most abundant in neurons and appears normal in carriers of a fragile X premutation. Nat Genet 4: 335–340. [DOI] [PubMed] [Google Scholar]
- 11. Abitbol M, Menini C, Delezoide AL, Rhyner T, Vekemans M, et al. (1993) Nucleus basalis magnocellularis and hippocampus are the major sites of FMR-1 expression in the human fetal brain. Nat Genet 4: 147–153. [DOI] [PubMed] [Google Scholar]
- 12. Bassell GJ, Warren ST (2008) Fragile X syndrome: loss of local mRNA regulation alters synaptic development and function. Neuron 60: 201–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liu ZH, Chuang DM, Smith CB (2011) Lithium ameliorates phenotypic deficits in a mouse model of fragile X syndrome. Int J Neuropsychopharmacol 14: 618–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bakker CE, Verheij C, Willemsen R, van der Helm R, Oerlemans F, et al. (1994) Fmr1 knockout mice: a model to study fragile X mental retardation. The Dutch-Belgian Fragile X Consortium. Cell 78: 23–33. [PubMed] [Google Scholar]
- 15. Hinton VJ, Brown WT, Wisniewski K, Rudelli RD (1991) Analysis of neocortex in hree males with the fragile X syndrome. Am J Med Genet 41: 289–294. [DOI] [PubMed] [Google Scholar]
- 16. Comery TA, Harris JB, Willems PJ, Oostra BA, Irwin SA, et al. (1997) Abnormal dendritic spines in fragile X knockout mice: maturation and pruning deficits. Proc Natl Acad Sci U S A 94: 5401–5404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Maren S, Fanselow MS (1995) Synaptic plasticity in the basolateral amygdala induced by hippocampal formation stimulation in vivo. J Neurosci 15: 7548–7564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Brigman JL, Wright T, Talani G, Prasad-Mulcare S, Jinde S, et al. (2010) Loss of GluN2B-containing NMDA receptors in CA1 hippocampus and cortex impairs long-term depression, reduces dendritic spine density, and disrupts learning. J Neurosci 30: 4590–4600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhao MG, Toyoda H, Ko SW, Ding HK, Wu LJ, et al. (2005) Deficits in trace fear memory and long-term potentiation in a mouse model for fragile X syndrome. J Neurosci 25: 7385–7392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Huber KM, Gallagher SM, Warren ST, Bear MF (2002) Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc Natl Acad Sci U S A 99: 7746–7750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Streisinger G, Walker C, Dower N, Knauber D, Singer F (1981) Production of clones of homozygous diploid zebra fish (Brachydanio rerio). Nature 291: 293–296. [DOI] [PubMed] [Google Scholar]
- 22. Souza BR, Tropepe V (2011) The role of dopaminergic signalling during larval zebrafish brain development: a tool for investigating the developmental basis of neuropsychiatric disorders. Rev Neurosci 22: 107–119. [DOI] [PubMed] [Google Scholar]
- 23. Chen PY, Manninga H, Slanchev K, Chien M, Russo JJ, et al. (2005) The developmental miRNA profiles of zebrafish as determined by small RNA cloning. Gene Dev 19: 1288–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vogel G (2008) Developmental biology. Lights! Camera! Action! Zebrafish embryos caught on film. Science 322: 176. [DOI] [PubMed] [Google Scholar]
- 25. Rupp B, Wullimann MF, Reichert H (1996) The zebrafish brain: a neuroanatomical comparison with the goldfish. Anat Embryol 194: 187–203. [DOI] [PubMed] [Google Scholar]
- 26. Alfaro JM, Ripoll-Gomez J, Burgos JS (2011) Kainate administered to adult zebrafish causes seizures similar to those in rodent models. Eur J Neurosci 33: 1252–1255. [DOI] [PubMed] [Google Scholar]
- 27. Ninkovic J, Folchert A, Makhankov YV, Neuhauss SC, Sillaber I, et al. (2006) Genetic identification of AChE as a positive modulator of addiction to the psychostimulant D-amphetamine in zebrafish. J Neurobiol 66: 463–475. [DOI] [PubMed] [Google Scholar]
- 28. Kim YH, Lee Y, Kim D, Jung MW, Lee CJ (2010) Scopolamine-induced learning impairment reversed by physostigmine in zebrafish. Neurosci Res 67: 156–161. [DOI] [PubMed] [Google Scholar]
- 29. Rawashdeh O, de Borsetti NH, Roman G, Cahill GM (2007) Melatonin suppresses nighttime memory formation in zebrafish. Science 318: 1144–1146. [DOI] [PubMed] [Google Scholar]
- 30. Vuaden FC, Savio LE, Piato AL, Pereira TC, Vianna MR, et al. (2012) Long-term methionine exposure induces memory impairment on inhibitory avoidance task and alters acetylcholinesterase activity and expression in zebrafish (Danio rerio). Neurochem Res 37: 1545–1553. [DOI] [PubMed] [Google Scholar]
- 31. Blank M, Guerim LD, Cordeiro RF, Vianna MR (2009) A one-trial inhibitory avoidance task to zebrafish: rapid acquisition of an NMDA-dependent long-term memory. Neurobiol Learn Mem 92: 529–534. [DOI] [PubMed] [Google Scholar]
- 32. van 't Padje S, Engels B, Blonden L, Severijnen LA, Verheijen F, et al. (2005) Characterisation of Fmrp in zebrafish: evolutionary dynamics of the fmr1 gene. Dev Genes Evol 215: 198–206. [DOI] [PubMed] [Google Scholar]
- 33. den Broeder MJ, van der Linde H, Brouwer JR, Oostra BA, Willemsen R, et al. (2009) Generation and characterization of FMR1 knockout zebrafish. PLoS One 4: e7910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ng MC, Hsu CP, Wu YJ, Wu SY, Yang YL, et al. (2012) Effect of MK-801-induced impairment of inhibitory avoidance learning in zebrafish via inactivation of extracellular signal-regulated kinase (ERK) in telencephalon. Fish Physiol Biochem 38: 1099–1106. [DOI] [PubMed] [Google Scholar]
- 35. Westerfield M (1993) The zebrafish book: a guide for the laboratory use of zebrafish (Brachydanio rerio): University of Oregon Press Eugene, OR. [Google Scholar]
- 36. Ng MC, Tang TH, Ko MC, Wu YJ, Hsu CP, et al. (2012) Stimulation of the lateral division of the dorsal telencephalon induces synaptic plasticity in the medial division of adult zebrafish. Neurosci Lett 512: 109–113. [DOI] [PubMed] [Google Scholar]
- 37. Pan L, Woodruff E III, Liang P, Broadie K (2008) Mechanistic relationships between Drosophila fragile X mental retardation protein and metabotropic glutamate receptor A signaling. Mol Cell Neurosci 37: 747–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Eadie BD, Zhang WN, Boehme F, Gil-Mohapel J, Kainer L, et al. (2009) Fmr1 knockout mice show reduced anxiety and alterations in neurogenesis that are specific to the ventral dentate gyrus. Neurobiol Dis 36: 361–373. [DOI] [PubMed] [Google Scholar]
- 39. Peier AM, McIlwain KL, Kenneson A, Warren ST, Paylor R, et al. (2000) (Over)correction of FMR1 deficiency with YAC transgenics: behavioral and physical features. Hum Mol Genet 9: 1145–1159. [DOI] [PubMed] [Google Scholar]
- 40. Goebel-Goody SM, Wilson-Wallis ED, Royston S, Tagliatela SM, Naegele JR, et al. (2012) Genetic manipulation of STEP reverses behavioral abnormalities in a fragile X syndrome mouse model. Genes Brain Behav 11: 586–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. de Diego-Otero Y, Romero-Zerbo Y, el Bekay R, Decara J, Sanchez L, et al. (2009) Alpha-tocopherol protects against oxidative stress in the fragile X knockout mouse: an experimental therapeutic approach for the Fmr1 deficiency. Neuropsychopharm 34: 1011–1026. [DOI] [PubMed] [Google Scholar]
- 42. Portavella M, Torres B, Salas C (2004) Avoidance response in goldfish: emotional and temporal involvement of medial and lateral telencephalic pallium. J Neurosci 24: 2335–2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Portavella M, Vargas JP, Torres B, Salas C (2002) The effects of telencephalic pallial lesions on spatial, temporal, and emotional learning in goldfish. Brain Res Bull 57: 397–399. [DOI] [PubMed] [Google Scholar]
- 44. Bliss TV, Collingridge GL (1993) A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361: 31–39. [DOI] [PubMed] [Google Scholar]
- 45. Nicoll RA, Malenka RC (1995) Contrasting properties of two forms of long-term potentiation in the hippocampus. Nature 377: 115–118. [DOI] [PubMed] [Google Scholar]
- 46. Voronin LL (1983) Long-term potentiation in the hippocampus. Neurosci 10: 1051–1069. [DOI] [PubMed] [Google Scholar]
- 47. Kitajima T, Hara K (1990) A model of the mechanisms of long-term potentiation in the hippocampus. Biol Cybern 64: 33–39. [DOI] [PubMed] [Google Scholar]
- 48. Nam RH, Kim W, Lee CJ (2004) NMDA receptor-dependent long-term potentiation in the telencephalon of the zebrafish. Neurosci Lett 370: 248–251. [DOI] [PubMed] [Google Scholar]
- 49. Shang Y, Wang H, Mercaldo V, Li X, Chen T, et al. (2009) Fragile X mental retardation protein is required for chemically-induced long-term potentiation of the hippocampus in adult mice. J Neurochem 111: 635–646. [DOI] [PubMed] [Google Scholar]
- 50. Li J, Pelletier MR, Perez Velazquez JL, Carlen PL (2002) Reduced cortical synaptic plasticity and GluR1 expression associated with fragile X mental retardation protein deficiency. Mol Cell Neurosci 19: 138–151. [DOI] [PubMed] [Google Scholar]
- 51. Wilson BM, Cox CL (2007) Absence of metabotropic glutamate receptor-mediated plasticity in the neocortex of fragile X mice. Proc Natl Acad Sci U S A 104: 2454–2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hu H, Qin Y, Bochorishvili G, Zhu Y, van Aelst L, et al. (2008) Ras signaling mechanisms underlying impaired GluR1-dependent plasticity associated with fragile X syndrome. J Neurosci 28: 7847–7862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yun SH, Trommer BL (2011) Fragile X mice: reduced long-term potentiation and N-Methyl-D-Aspartate receptor-mediated neurotransmission in dentate gyrus. J Neurosci Res 89: 176–182. [DOI] [PubMed] [Google Scholar]
- 54. Nosyreva ED, Huber KM (2006) Metabotropic receptor-dependent long-term depression persists in the absence of protein synthesis in the mouse model of fragile X syndrome. J Neurophysiol 95: 3291–3295. [DOI] [PubMed] [Google Scholar]
- 55. Zhang J, Hou L, Klann E, Nelson DL (2009) Altered hippocampal synaptic plasticity in the FMR1 gene family knockout mouse models. J Neurophysiol 101: 2572–2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Nakamoto M, Nalavadi V, Epstein MP, Narayanan U, Bassell GJ, et al. (2007) Fragile X mental retardation protein deficiency leads to excessive mGluR5-dependent internalization of AMPA receptors. Proc Natl Acad Sci U S A 104: 15537–15542. [DOI] [PMC free article] [PubMed] [Google Scholar]