

Abstract
The global interplay between bacteria and bacteriophages has generated many macromolecules useful in biotechnology, through the co-evolutionary see-saw of bacterial defense and viral counter-attack measures. Bacteria can protect themselves using abortive infection systems, which induce altruistic suicide in an infected cell and therefore protect the clonal population at the expense of the infected individual. Our recent paper describes how bacteriophage ΦTE successfully subverted the activity of a plasmid-borne abortive infection system. ΦTE evolved mimics of the small RNA antitoxin that naturally inhibits the active toxin component of this anti-viral mechanism. These mutant phages further manipulated the behavior of the host population, through transduction of the plasmid encoding the abortive infection system. Transductants thereby became enslaved by the abortive infection system, committing suicide in response to infection by the original phage population. In effect, the new host was infected by an “addictive altruism,” to the advantage of the resistant bacteriophage.
Keywords: bacteriophage, toxin-antitoxin, abortive infection, mimicry, transduction, Pectobacterium atrosepticum, RNA pseudoknot, phage resistance
Toxin-antitoxin systems can act as phage-resistance mechanisms
Being the most prevalent, and among the most ancient, organisms on the planet, bacteria and their bacteriophage predators have had ample opportunities to develop the tools required to wage their microbial war. Adaptive evolution has driven the production of bacterial measures to ward off bacteriophage infections. In response, bacteriophages have developed counter-measures, which in turn promote the selection of further bacterial systems, ad infinitum over the millennia. The known primary bacterial defenses have been divided into several main categories1; alteration or masking of surface features required for recognition by the bacteriophage, inhibition of DNA injection by the bacteriophage, use of restriction enzymes or CRISPR loci to degrade foreign nucleic acids, and the abortive infection systems.
These abortive infection (Abi) systems operate in a counter-intuitive fashion, causing the virus-infected bacterial cell to commit suicide. This can be considered an altruistic act to protect the clonal population.2 Most Abi systems were identified on plasmids of Gram-positive lactococci used in dairy fermentations, and were exploited as tools for the development of hyper-resistant strains for use in dairy fermentations that are inherently vulnerable to spoilage by bacteriophage infection. The mechanisms that result in cell death by these Abi systems are only now beginning to be understood at the molecular level. It is clear that these mechanisms are varied and can rely upon two or more partners. Previously, we identified one such Abi system on a plasmid of the plant pathogen Pectobacterium atrosepticum (aka Erwinia).3 We found that this system was effective against multiple phages in several Gram-negative host backgrounds, and relied upon two parts—a protein toxin and a less stable RNA antitoxin.3,4 The working model was that, upon phage infection, the antitoxin would be degraded and the toxin would be activated, thereby killing the host and, consequently the virus, during replication and morphogenesis.
Toxin-antitoxin (TA) systems5-7 were originally identified as systems which aid the maintenance of plasmids within a population, although they have also been attributed roles in stress responses,8 developmental processes, regulation of pathogenicity and the formation of persister cells,9,10 among others. Until recently, all of the known TA systems could be classified as one of two Types. In Type I, the antitoxic mRNA interacts with the toxin mRNA to prevent translation of the toxic protein. In Type II, the toxic protein is bound by an antitoxic protein. In both cases, the antitoxin is more labile and, upon activation of the system, it is preferentially degraded, allowing the toxin to take effect. This can lead to transiently reversible bacteriostasis or, over prolonged periods, cell death. The ToxIN system from P. atrosepticum was described as the paradigm of the prevalent Type III systems, wherein a specific RNA sequence is able to inhibit a toxic protein.3,11 The precise molecular details of the ToxIN Type III system were later elucidated when the structure of a heterohexameric complex, comprising ToxI antitoxic RNA bound to ToxN toxic protein, was solved crystallographically.12 The ToxI RNA observed in the complex is a 36 nucleotide unit, cleaved from its repetitive precursor by ToxN. Within this complex, the ToxI RNA takes on a convoluted tertiary pseudoknot fold allowing it to tightly bind and inhibit the ToxN endoribonuclease prior to activation.
While this structural study informed us about regulation of the two components, their interplay and the mode of ToxN toxicity, it did not address the missing link between the incoming bacteriophages and use of ToxIN as an Abi system. To this end, we performed further studies on new environmental bacteriophage isolates that showed a low-level of spontaneous resistance to the ToxIN system. These phages were described as having “escaped” the Abi system, and this was caused by a heritable mutation.3,4,13 One such phage, ΦTE, was able to escape at a frequency of ~1x10−8. Furthermore, ΦTE was also shown to be a generalized transducing phage.13
Bacteriophages acquire antitoxins to influence the host response
In our recent study,13 we sequenced the full genomes of both the wild type ΦTE and several independently isolated escape phages. Having compared the sequences, it was clear that a single region of the genome had expanded within the escape phage genomes. Upon closer examination, this region within the wild type genome contained a sequence very similar to the 36 nucleotide sequence that constituted the active pseudoknot of the ToxI antitoxin. We dubbed this sequence, ‘pseudo-ToxI’. The numbers of these pseudo-ToxI repeats had expanded in the escape phage genomes, to mosaics of four or five near-exact copies. In one specific case, the escape phage had not simply expanded the genomic locus, but it had instead “hijacked” the toxI sequence by recombination to incorporate an exact copy into the phage genome. Expansion of the pseudo-ToxI copy number is vital for an escape phenotype, because the active RNA that folds into an antitoxic pseudoknot is formed from the distal part of one repeat and leads into the proximal part of the next repeat. This difference in phasing between the DNA sequence repeats and the active RNA repeats means that the wild type phage, containing a single repeat, is unable to generate active antitoxic pseudoknots.
This study described two routes, through genomic expansion, or host recombination, by which ΦTE achieves suppression of the ToxIN system.13 In the first case, the expanded repeats are expected to generate RNA pseudoknots that can actively inhibit ToxN, by mimicking the molecular fold of the cognate ToxI antitoxin. In the second, ΦTE would be able to express exact copies of ToxI, to inhibit ToxN during ΦTE replication. To demonstrate these points, we were able to confirm that both the expanded pseudo-ToxI loci and the recombined ToxI were able to inhibit ToxN function within in vivo assays of abortive infection. Finally, it was observed that the escape locus was indeed expressed during ΦTE infections.
Molecular mimics can divert and suppress host defensive systems
Mimicry has proven a useful survival tool in nature, from camouflage through to the more insidious machinations of the cuckoo. In the case of ΦTE, the bacteriophage adopts molecular mimics of the ToxI antitoxin to ensure viral replication within ToxIN-containing hosts. Bacteria adopt molecular mimicry themselves, such as in the cases of the Qnr-family or MfpA proteins, which provide resistance to quinolone antibiotics by mimicking the structure of B-form DNA and stabilizing gyrase complexes.14 Similarly, bacteriophages like T7, and other mobile genetic elements, have been observed to generate proteins that mimic B-form DNA, and thereby act as anti-restriction factors to enable replication, even in the face of host defenses.15 Our work proposed that pseudo-ToxI is a molecular mimic and our mutagenesis study, coupled with phenotypic data, strongly support the mode of action as being via direct ToxN inhibition.
As pseudo-ToxI is an RNA mimic that is able to inhibit a Type III (protein-RNA) TA system, this highlights the possibility that some bacteriophages may have the capacity to develop further molecules that could mimic other antitoxins. This would include mimics both of the protein antitoxins from Type II (protein-protein) TA systems, and regulatory antisense RNAs that inhibit translation of Type I toxin mRNA. In the former case, one can consider the Type II TA systems such as mazEF,16 along with rnlAB and the related system lsoAB,17,18 which have all been linked to Abi activity. It has been shown recently that the Dmd protein of T4 acts as a promiscuous antitoxin, inhibiting the RnlA and LsoA toxins during T4 infection.18 If the infecting T4 lacks Dmd, the RnlA and LsoA toxins hinder its replication. Though the structure is currently unknown, Dmd could prove to be a Type II antitoxin mimic. If not, it is very likely that such a molecule will be found eventually. The hok/sok Type I TA system can also provide T4 resistance19 and it is tempting to speculate that phage-encoded antisense RNAs exist that inhibit the translation of Type I toxins.
Antitoxin mimicry provides a route to subversion of TA-encoded defense systems that is independent of activation of the system. In the case of phage T4, the TA system is passively activated, as infection by T4 shuts down host gene expression and results in loss of the antitoxin.17 It is not known how ToxIN senses incoming ΦTE. This could be a passive process, as for T4 and RnlAB, or the complex could directly respond to a phage-encoded product. We are currently exploring this question. Multiple options for triggering of TA-based defenses are possible, and molecular mimicry of the antitoxin provides an escape route compatible with any of these.
Bacteriophages can enforce bacterial altruism by horizontal gene transfer
The final aspect of the work on ΦTE focused upon generalized transduction. ΦTE was shown to transfer chromosomal markers between host strains,13 so it was decided to also ascertain whether ΦTE could transduce plasmids. More importantly, could it transduce a ToxIN-encoding plasmid? Wild type ΦTE and escape phages all successfully transferred ToxIN plasmids to new hosts, which is a remarkable example of phage-mediated transfer of a phage-resistance mechanism.13 The frequency at which this occurs with wild type ΦTE is low; the phage must first acquire an escape mutation prior to generating transducing particles. By isolating escape phages within the laboratory we artificially accelerated the process. Nevertheless, in the microbial world, there are an estimated 1025 viral infections of bacteria occurring per second,20 thereby, in principle, allowing even exceedingly rare events to occur regularly. Furthermore, in the presence of a population of ToxIN-containing bacteria, the escape mutants should have a significant evolutionary advantage by preferentially replicating and outpacing the sensitive wild type phages.
It is possible to speculate how these transduction events could affect a microbial community. One such scenario is described in Figure 1. Initially, ΦTE encounters ToxIN within a host and will, in most cases, be aborted. Within a small number of infections, however, an escape phage arises. This escape phage can freely infect other ToxIN-carrying cells, and replicate unchecked, while the wild type ΦTE remains susceptible to abortion. Furthermore, the increasing populations of ΦTE escape variants could begin dissemination of the ToxIN plasmid into new host strains in the vicinity. This could then provide this new host with protection from wild type ΦTE (as well as from many other phage species), but the host would remain susceptible to the ΦTE escape phages. In this scenario, the new hosts would have been infected with a system that drives them toward altruism; the ΦTE escape phages would therefore have influenced their hosts, generating a tailor-made host protected from other competitor phages. At this point, the ΦTE escape phages might risk obliterating their host population, so it would be advantageous if a balance could be achieved. It is not immediately obvious how this may arise, but it could involve other resistance mechanisms. Is it possible that there may be a fitness cost to carrying expanded amounts of pseudo-ToxI, which limits the spread of escape phages in the environment? Alternatively, perhaps the ToxN protein may evolve so that it is no longer inhibited by pseudo-ToxI. The inevitable evolutionary “waltz” between predator and prey will continue, although the precise choreography of that interaction could depend on many physiological and environmental factors; not least proximity and relative population densities of bacteria and phage.
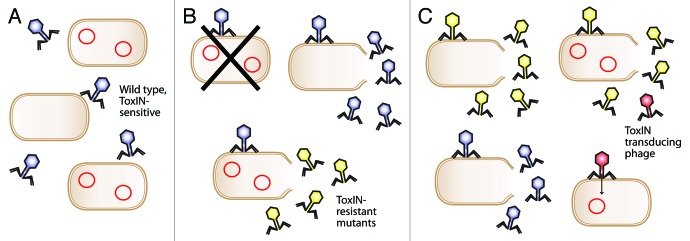
Figure 1. Evolution of ToxIN escape phages and ToxIN plasmid dissemination. (A). A mixed population of ToxIN-carrying and ToxIN-negative cells is infected by a species of ToxIN-sensitive bacteriophage (blue). ToxIN plasmids are represented by red circles. (B). The ToxIN-sensitive virus can replicate normally in a ToxIN-negative host, while infection of a ToxIN-carrying cell results in destruction of both the host and the virus. In rare cases, however, the virus spontaneously mutates and produces ToxIN-resistant progeny (yellow). (C). The ToxIN-resistant phage population can now infect and proliferate within both the ToxIN-carrying and ToxIN-negative cells, and, in the former case, may generate transducing particles at low frequency (red). This transducing particle will transfer ToxIN through the population, further reducing the proportion of cells that are suitable hosts for the original wild type bacteriophage. The escape phage thereby establishes a dominant population.
In this ToxIN system, the ΦTE phage evolves variants that can suppress the programmed drive into suicide seen in the Abi process. Such mutant phages therefore act to keep the bacterial host alive long enough to enable productive viral infection, lysis and subsequent propagation in sibling bacteria in a local population. From that perspective, we can view the evolved escape mutants as controlling their bacterial hosts to ensure replication. In another sense, however, we could consider the ToxIN plasmid replicon system as the true manipulating force driving these alterations to the microbial community. ToxIN first ensures its own retention within the replicating host, through its addictive plasmid stabilization activity. It then protects the host populations from extinction by modulating the propagation of incoming viral parasites. In the event that the virus wins, ToxIN can simply hitch a ride to a new host (albeit as a low frequency event—but with considerable selective advantage in appropriate circumstances). The host and virus may be changing, but the evolutionary success of ToxIN could be guaranteed in this model.
Through this work we have been able to study the intricacies of one case of bacteriophage-host interactions, and yet the direct link between bacteriophage infection and ToxIN activation remains elusive. To this end, we are pursuing further studies on other ToxIN-resistant “escape” phages. In a world of incredible phage diversity, we predict that there will be multiple routes to ToxIN activation, and so, correspondingly, we expect that multiple escape mechanisms remain to be uncovered.
Glossary of Terms
Abortive infection
Phage-resistance mechanism whereby upon phage infection, the host cell dies and the viral life cycle is halted before completion.
Addictive altruism
Genetic loci can be termed, “addictive,” as their loss would result in the death of the individual cell. Toxin-antitoxin systems are addictive and can therefore enforce altruistic behaviors such as abortive infection.
Altruistic suicide
The specific death of an infected host cell to prevent viral spread, which in turn protects the clonal microbial population.
CRISPR
Clustered Regularly Interspaced Short Palindromic Repeats; genomic loci involved in protection from exogenous nucleic acids.
Escape phage
A mutant phage that has become resistant to a specific phage-resistance mechanism.
Molecular mimicry
The ability of one macromolecule to recapitulate the 3D shape and surface properties of another molecule, to subvert activity.
Pseudoknot
A convoluted tertiary RNA fold.
Toxin-antitoxin systems
Genetic loci formed from an active toxic element and an inhibitory antitoxic component, involved in the regulation of many prokaryotic cellular processes.
ToxIN, pseudo-ToxI
A protein-RNA toxin-antitoxin system, comprising a protein toxin (ToxN) and an antitoxic RNA (ToxI). Phage-encoded pseudo-ToxI acts as a molecular mimic of endogenous ToxI RNA.
Acknowledgments
This work was supported by grants from the Biotechnology and Biological Sciences Research Council (UK), a Commonwealth scholarship funded by the Department for Business, Innovation and Skills (UK Government) and the Cambridge Commonwealth Trust through the Commonwealth Scholarships Commission UK (FLS), and the Marsden Fund and a Rutherford Discovery Fellowship (PCF), Royal Society of New Zealand.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/bacteriophage/article/23830
References
- 1.Labrie SJ, Samson JE, Moineau S. Bacteriophage resistance mechanisms. Nat Rev Microbiol. 2010;8:317–27. doi: 10.1038/nrmicro2315. [DOI] [PubMed] [Google Scholar]
- 2.Chopin MC, Chopin A, Bidnenko E. Phage abortive infection in lactococci: variations on a theme. Curr Opin Microbiol. 2005;8:473–9. doi: 10.1016/j.mib.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 3.Fineran PC, Blower TR, Foulds IJ, Humphreys DP, Lilley KS, Salmond GP. The phage abortive infection system, ToxIN, functions as a protein-RNA toxin-antitoxin pair. Proc Natl Acad Sci U S A. 2009;106:894–9. doi: 10.1073/pnas.0808832106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blower TR, Fineran PC, Johnson MJ, Toth IK, Humphreys DP, Salmond GP. Mutagenesis and functional characterisation of the RNA and protein components of the toxIN abortive infection and toxin-antitoxin locus of Erwinia. J Bacteriol. 2009;191:6029–39. doi: 10.1128/JB.00720-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gerdes K, Christensen SK, Løbner-Olesen A. Prokaryotic toxin-antitoxin stress response loci. Nat Rev Microbiol. 2005;3:371–82. doi: 10.1038/nrmicro1147. [DOI] [PubMed] [Google Scholar]
- 6.Hayes F, Van Melderen L. Toxins-antitoxins: diversity, evolution and function. Crit Rev Biochem Mol Biol. 2011;46:386–408. doi: 10.3109/10409238.2011.600437. [DOI] [PubMed] [Google Scholar]
- 7.Blower TR, Salmond GP, Luisi BF. Balancing at survival’s edge: the structure and adaptive benefits of prokaryotic toxin-antitoxin partners. Curr Opin Struct Biol. 2011;21:109–18. doi: 10.1016/j.sbi.2010.10.009. [DOI] [PubMed] [Google Scholar]
- 8.Wang X, Wood TK. Toxin-antitoxin systems influence biofilm and persister cell formation and the general stress response. Appl Environ Microbiol. 2011;77:5577–83. doi: 10.1128/AEM.05068-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maisonneuve E, Shakespeare LJ, Jørgensen MG, Gerdes K. Bacterial persistence by RNA endonucleases. Proc Natl Acad Sci U S A. 2011;108:13206–11. doi: 10.1073/pnas.1100186108. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 10.Gerdes K, Maisonneuve E. Bacterial persistence and toxin-antitoxin loci. Annu Rev Microbiol. 2012;66:103–23. doi: 10.1146/annurev-micro-092611-150159. [DOI] [PubMed] [Google Scholar]
- 11.Blower TR, Short FL, Rao F, Mizuguchi K, Pei XY, Fineran PC, et al. Identification and classification of bacterial Type III toxin-antitoxin systems encoded in chromosomal and plasmid genomes. Nucleic Acids Res. 2012;40:6158–73. doi: 10.1093/nar/gks231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blower TR, Pei XY, Short FL, Fineran PC, Humphreys DP, Luisi BF, et al. A processed noncoding RNA regulates an altruistic bacterial antiviral system. Nat Struct Mol Biol. 2011;18:185–90. doi: 10.1038/nsmb.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blower TR, Evans TJ, Przybilski R, Fineran PC, Salmond GP. Viral evasion of a bacterial suicide system by RNA-based molecular mimicry enables infectious altruism. PLoS Genet. 2012;8:e1003023. doi: 10.1371/journal.pgen.1003023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hegde SS, Vetting MW, Roderick SL, Mitchenall LA, Maxwell A, Takiff HE, et al. A fluoroquinolone resistance protein from Mycobacterium tuberculosis that mimics DNA. Science. 2005;308:1480–3. doi: 10.1126/science.1110699. [DOI] [PubMed] [Google Scholar]
- 15.McMahon SA, Roberts GA, Johnson KA, Cooper LP, Liu H, White JH, et al. Extensive DNA mimicry by the ArdA anti-restriction protein and its role in the spread of antibiotic resistance. Nucleic Acids Res. 2009;37:4887–97. doi: 10.1093/nar/gkp478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hazan R, Engelberg-Kulka H. Escherichia coli mazEF-mediated cell death as a defense mechanism that inhibits the spread of phage P1. Mol Genet Genomics. 2004;272:227–34. doi: 10.1007/s00438-004-1048-y. [DOI] [PubMed] [Google Scholar]
- 17.Koga M, Otsuka Y, Lemire S, Yonesaki T. Escherichia coli rnlA and rnlB compose a novel toxin-antitoxin system. Genetics. 2011;187:123–30. doi: 10.1534/genetics.110.121798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Otsuka Y, Yonesaki T. Dmd of bacteriophage T4 functions as an antitoxin against Escherichia coli LsoA and RnlA toxins. Mol Microbiol. 2012;83:669–81. doi: 10.1111/j.1365-2958.2012.07975.x. [DOI] [PubMed] [Google Scholar]
- 19.Pecota DC, Wood TK. Exclusion of T4 phage by the hok/sok killer locus from plasmid R1. J Bacteriol. 1996;178:2044–50. doi: 10.1128/jb.178.7.2044-2050.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lima-Mendez G, Toussaint A, Leplae R. Analysis of the phage sequence space: the benefit of structured information. Virology. 2007;365:241–9. doi: 10.1016/j.virol.2007.03.047. [DOI] [PubMed] [Google Scholar]