

Abstract
Adenosine to inosine deamination of RNA is widespread in metazoa. Inosines are recognized as guanosines and, therefore, this RNA-editing can influence the coding potential, localization and stability of RNAs. Therefore, RNA editing contributes to the diversification of the transcriptome in a flexible manner. The editing reaction is performed by adenosine deaminases that act on RNA (ADARs), which are essential for normal life and development in many organisms. Changes in editing levels are observed during development but also in neurological pathologies like schizophrenia, depression or tumors. Frequently, changes in editing levels are not reflected by changes in ADAR levels suggesting a regulation of enzyme activity. Until now, only a few factors are known that influence the activity of ADARs. Here we present a two-stage in vivo editing screen aimed to isolate enhancers of editing. A primary, high-throughput yeast-screen is combined with a more accurate secondary screen in mammalian cells that uses a fluorescent read-out to detect minor differences in RNA-editing. The screen was successfully employed to identify DSS1/SHFM1, the RNA binding protein hnRNP A2/B1 and a 3′ UTR as enhancers of editing. By varying intracellular DSS1/SHFM1 levels, we can modulate A to I editing by up to 30%. Proteomic analysis indicates an interaction of DSS1/SHFM1 and hnRNP A2/B1 suggesting that both factors may act by altering the cellular RNP landscape. An extension of this screen to cDNAs from different tissues or developmental stages may prove useful for the identification of additional enhancers of RNA-editing.
Keywords: RNA-editing, yeast screen, DSS1, TREX-2, regulation of RNA-editing, RNA-binding
Introduction
Adenosine to inosine (A to I) conversion by adenosine deaminases that act on RNA (ADARs) increases transcriptome and proteome diversity. In this type of RNA editing, individual or multiple adenosines are deaminated in double-stranded or structured RNA regions.1 Since inosine is interpreted as guanosine by cellular machineries, editing in coding regions of mRNAs may lead to recoding of genetic information. Consistently, editing was shown to create various functional protein isoforms for several genes.1 In mammals, the most prominent substrates encode the serotonin 2C receptor (5-HT2C) or glutamate receptor (GluA2) isoforms.2-4 Editing in these substrates is essential for proper development and function of the nervous system.5,6
Recent transcriptome-wide bioinformatic and deep sequencing analysis has revealed thousands of editing sites, predominantly in structured non-coding regions of mRNAs but also within coding regions.7-10 Moreover, precursors of miRNAs have been identified as substrates for A to I editing. Editing of the double-stranded pri- and pre- miRNAs may affect their processing, stability or change the specificity of miRNAs.11-14
A to I editing is performed by members of the family of adenosine deaminases that act on RNA (ADARs). ADARs have been found in all metazoa.15,16 Three ADAR members are found in mammals: ADAR1, -2 and -3, with ADAR3 being apparently catalytically inactive.17-19 ADARs bind dsRNA via their dsRNA binding domains while the deamination reaction is performed by the deaminase domain located in the C-terminal part of all ADARs.1,20 Both ADAR1 and ADAR2 have overlapping, yet distinct substrate specificities.21-23 Site-specificity is determined by the structure and sequence of the RNA surrounding the editing site, which is recognized by the double-stranded RNA binding domains but also by the deaminase domain that displays context-specific editing preferences.21,23-29
Although ADARs are able to act without cofactors some findings indicate the existence of mechanisms that regulate editing activity. For instance, the extent of editing frequently fails to correlate with expression levels of ADARs.30-32 Also, recent studies on the tissue-specific editing of the K/E site in cyFIP2 showed no correlation between editing and ADAR2 expression.33 Moreover, hypoediting of GluA2 Q/R and 5-HT2CR sites was detected in malignant gliomas with no significant change in the expression of ADAR2.34,35 Finally, changes in editing of the serotonin 5-HT2c receptor have been reported in patients with mental disorders and animal studies have indicated that fluoxetine treatment can lead to altered editing patterns of serotonin receptor 5-HT2c.36-40
A few molecular mechanisms that can regulate editing activity have been determined. These include alternative splicing, self-editing, sumoylation, heterodimerization or ubiquitination of ADARs, or the regulated cellular sequestration of active enzyme complexes.41-48
However, so far, no systematic screen for factors that may alter editing activity has been performed. Both a yeast and a mammalian reporter system have been reported that allow the detection of editing events in those two systems.29,49 Here we present a novel, two-step screening system that allows the rapid identification of cellular factors altering RNA-editing. A high-throughput yeast screen is combined with a sensitive and accurate mammalian editing screen. The screen was successfully employed to identify enhancers of editing. Of the candidates isolated, DSS1/SHFM1 a component of the proteasome subunit and the hnRNP protein A2B1 were further characterized as enhancers of editing.
Results
Creation of a yeast strain to monitor RNA-editing
To identify factors, that influence ADAR-mediated RNA-editing a reporter system was set up in S. cerevisiae. The auxotrophy gene HIS3 was chosen as a reporter to screen for enhancers of editing. An editing substrate for both ADAR1 and ADAR2 was introduced into the 5′ region of the His3 gene, shortly downstream of the AUG initiation codon. For this, the stem-loop containing the R/G editing site of glutamate receptor subunit B50 was shortened and modified to contain an amber stop codon at the editing site (Fig. 1A and C). The construct was integrated with its own promoter. However, functional HIS3 protein expression requires editing of the transcribed RNA leading to a conversion of the amber stop codon to a tryptophan (W) codon (Fig. 1A). Thus, expression of ADAR1 or ADAR2 would lead to prototrophy in histidine biosynthesis (Fig. 1B).

Figure 1. Design of a yeast strain for the identification of enhancers of editing. (A) Principle of the reporter system: A stem-loop sequence was introduced into the 5′ coding region of the yeast His3 gene. The stem-loop harbors an amber stop codon preventing translation of His3 unless A to I editing occurs. (B) Construction of the screening strain and steps involved. The editing substrate was stably integrated. Expression of rADAR2 from a cen vector leads to His3 production and growth on medium lacking histidine. This strain was mated with a strain pretransformed with a cDNA library and candidates containing potential enhancers of editing were determined by growth on media lacking histidine. Chosen candidates were further confirmed in yeast and mammalian cells. (C) Comparison of the amber stem-loop and the GluA2 R/G editing site. The changes in the stem are indicated. (D) Test for expression of ADARs from tetracycline inducible vector with and without doxycicline induction by western blotting. (E) Comparison of growth of strains expressing or lacking rADAR2 in different selective media. The identical number of cells was incubated in a 96-well plate with different media. After 24 hrs of growth the cells were pelleted and visualized to determine growth. A strain containing a pre-edited version (W) of the amber stem-loop grows in the presence or absence of His. A strain containing the amber stem loop relies on the presence and induction of ADAR2 for efficient growth on media lacking histidine. The picture was inverted for clearer visualization. (F) Comparison of editing levels of the amber substrate. Total RNA was isolated from the strains lacking or containing rADAR2, with and without DOX induction. RT-PCR and subsequent sequencing was performed to determine editing levels. Expression of rADAR2 strongly induces editing. The edited adenosine is indicated with an asterisk.
For this, cDNAs encoding tagged versions of human (h) ADAR 1 or rat (r) ADAR2 were expressed from inducible, single-copy yeast plasmids. Interestingly, while both proteins hADAR1 and rADAR2 were easily detectable when expressed upon doxycycline induction only ADAR2 was able to edit the stem loop and to confer HIS3 prototrophy (Fig. 1D and F). Addition of different amounts of doxycycline to the media led to different levels of rADAR2 expression and consequently different levels of editing as well as yeast growth rates. Due to leakiness of the promoter minimal growth was even observable in the absence of doxycycline (Fig. 1E). Therefore, since we were aiming at isolating enhancers of RNA-editing, doxycycline induction was omitted during the following screen, relying on the low base levels of ADAR2 expression in the non-induced state. Furthermore, to allow a stringent screen for activators of ADARs, HIS3 activity was inhibited by the addition of 20mM 3-aminotriazole.
Identification of enhancers of editing
To identify cellular factors that stimulate editing, a haploid yeast strain pretransformed with a human fetal brain cDNA library was mated with the reporter strain and 3.3 × 106 clones were screened for increased growth on plates lacking histidine. 61 of the fastest growing colonies were picked for further investigation. cDNA inserts could be successfully amplified and sequenced from 52 of these candidates. Of these, 32 encoded annotated genes while 20 plasmids contained fragments derived from DNA contigs with no annotated function. Ten of the 32 sequences that showed homology to protein encoding cDNAs covered only the 3′ UTRs of these cDNAs, suggesting that the encoded RNAs might stimulate yeast growth or affect editing on their own. The remaining 22 protein coding fragments (roughly 1/3 of all stimulating clones) encoded proteins of different functions including RNA-binding proteins.
Next, we wanted to eliminate false positive clones that support yeast growth by other means than by stimulating editing (e.g., by clearing cells of the HIS3 inhibitor 3-AT). To do this, editing levels were determined in all 61 clones by direct sequencing of the RT-PCR product of the stem loop substrate (Fig. 2A). Editing levels were calculated by determining the ratio of the G peak height to the sum of A and G peak heights. This value was compared with editing levels in a control strain transformed with an empty vector. The relative change in editing induced by the various candidates is shown in (Fig. 2B). The assay was performed in duplicate to identiy those candidates that show the most reproducible effect on RNA editing irrespective of cell growth, culture density or other variables (blue and red bars in Fig. 2B).

Figure 2. Stimulation of editing by isolated candidates. Total RNA was isolated from yeast colonies showing enhanced growth on media lacking histidine. Editing of the reporter stem-loop was determined by RT-PCR and subsequent sequencing. (A) Comparison of induction of editing by the two candidates DSS1 and the translationally controlled tumor protein (TCTP) p23. The edited adenosine is indicated by an asterisk. (B) Relative editing levels of all candidates were normalized to editing levels in the control strain and plotted as % change relative to the control strain. Two independent biological replicas were performed and are depicted in red and blue. Editing levels could be determined in duplicate for 47 of the 61 candidates In 14 cases we were unable to amplify the editing substrate in duplicate. Of the 47 duplicates, 31 (66%) showed a reproducible increase in RNA-editing. The average induction of editing measured over all clones was around 14% (blue line). For further analysis only candidates exhibiting an increase in editing above 14% were considered.
In 15 of the 61 clones investigated the substrate cDNA could only be successfully amplified in one of the two replicates. Of these, 14 (93%) showed an increase in editing. In 46 clones the substrate cDNA could be amplified and sequenced in both replicates. Here, 31 clones (67%) showed an increase in editing in both replicates. In 14 clones (30%), however, editing levels were increased in one replicate while being decreased in the other (Fig. 2B). The average increase in RNA editing for all 61 clones was at 14% (blue line in Fig. 2B). The cDNAs encoding DSS1 and the translationally controlled tumor protein TCTP (p23) showed strong enhancement of editing (Fig. 2A).
For further analysis, only clones showing an increase in editing well beyond 14% were chosen for further evaluation (Fig. 2B). Clones that passed this filter again contained protein coding cDNAs, 3′UTRs and not-annotated DNA fragments (Table S1).
Stimulation of editing in mammalian tissue culture cells
Next, to eliminate clones that can only stimulate editing in the heterologous yeast system, a secondary screen in mammalian cells was established. To allow detection of the proteins and to ensure their nuclear localization as in the yeast two-hybrid vectors, selected candidates were cloned into a mammalian tissue culture expression vector downstream of an N-terminal 6xmyc-NLS peptide and tested for their impact on RNA-editing. Protein-coding cDNAs were cloned in frame and -as a control- out of frame. Non-coding fragments derived from 3′ UTRs were cloned without further alterations. When transfected in tissue culture cells the myc-tag or the myc-tag fusions could be detected both by western blotting and in situ (Fig. 3B and C). Again, the stop-codon-containing R/G site stem-loop was used as an editing reporter. To allow a fast readout of editing levels, the substrate stem-loop was cloned between the ORFs of red and green fluorescent proteins (RFP, GFP). Transfection of this reporter plasmid leads to constitutive expression of RFP. The stop codon in the stem-loop sequence prevents GFP expression in the absence of editing, while an increase in editing leads to conversion of the stop to a tryptophan codon and increasing GFP expression (Fig. 3A). ADAR2 is expressed at moderate levels in HeLa cells with endogenous substrates being edited.51 The intensity of the red and green fluorescence of single, transfected cells was measured by fluorescence-activated cell sorting (FACS).

Figure 3. Stimulation of editing in mammalian cells. (A) A reporter construct constitutively expressing RFP, and GFP only after editing of an amber stop codon (RNAG) was transfected into HeLa cell line and visualized microscopically. Shown are untransfected cells, cells transfected with RNAG and with the construct expressing a ‘pre-edited’ version of the stop codon (RNWG). (B) Expression of myc-NLS tagged DSS1 was monitored by western blotting. Only in-frame cloned DSS1 gives rise to a 40 kDa fusion protein, the empty vector and out-of-frame plasmid only allows detection of the myc-NLS fusion peptide. (C) Localization of expressed proteins. The transfected proteins detected via anti-myc show a mostly nuclear localization due to the presence of the myc-NLS in the vector sequence. (D) FACS experiments. Two graphs showing a comparison of two candidates that stimulate editing. FABP7 and DSS1 are expressed in-frame or out-of-frame (as a control). Values were taken from a minimum of 3 different experiments. A shift of the green to red fluorescence rations toward the right indicates a stronger expression of GFP and thus editing. While overexpression of DSS1 shows a strong effect on RNA-editing, FABP7 enhances editing only mildly. The effect becomes more obvious in upper gates where RFP/GFP expression is moderate to strong. The p-values for relative differences observed in gate 5 are indicated.
HeLa cells transfected with the reporter construct alone (RNAG) already show a weak green fluorescence while the pre-edited construct (RNWG) shows rather strong green fluorescence (Fig. 3A). Upon cotransfection, 3 out of the 15 candidates investigated induced a clear increase in green fluorescence. Two of them, the fatty acid binding protein FABP7 and the 3′UTR of the tyrosine kinase DYRK2 had a moderate effect on editing. The third candidate, however, encoding the DSS1/SHFM1 protein led to a strong increase in green fluorescence (Table 1 and Fig. 3D; Fig. S1 and data not shown). Others, like the 3′ UTR of WIPF2, the cDNA expressing glutathione peroxidase or the cDNA encoding TCTP showed no effect on editing in the mammalian reporter system (Fig. S1). Overexpression of hnRNP A2B1, despite having a clear effect in the yeast editing assay (Table 1) had no significant effect on editing in the mammalian reporter assay (data not shown).
Table 1. Candidates showing stimulatory effect on editing in mammalian cells.
| Name | Function | Sequence |
|---|---|---|
|
FABP7 (fatty acid-binding protein 7) |
transport of a so far unknown hydrophobic ligand potential morphogenic activity during CNS development |
coding |
|
DYRK2 (dual specificity tyrosine-phosphorylation-regulated kinase 2) |
regulation of cellular growth and/or development |
3′UTR |
|
DSS1 (26S proteasome complex subunit DSS1) |
plays a role in ubiquitin-dependent proteolysis Interacts with the C-terminal of BRCA2 |
coding |
| hnRNP A2/B1 | Involved in RNA export, interacts with hnRNPs RNA packaging into RNP particles |
coding |
The increase in green fluorescence was also accompanied by an increase in editing of the stem loop substrate inserted between the RFP and GFP reading frames, proving that increased green fluorescence is the result of increased editing (Fig. 4A).

Figure 4. DSS1 enhances RNA editing in mammalian cells. (A) Comparison of editing levels in the RNAG editing reporter in HeLa cells. RNAG mRNA was isolated from HeLa cells transfected with an empty vector or a DSS1 expressing vector. RT-PCR with subsequent sequencing was performed. Shown is a graph with mean values of 3 runs, standard deviation and p-value as determined by student’s t-test. Also, a representative electropherogram is shown. (B) Stimulation of editing in a HeK293 cell line stably expressing rat(r)ADAR2. Anti-flag staining of Hek 293 cells with and without stable expression of rADAR2. (C) Comparison of editing levels at the K/E editing site in CyFip2 mRNA by direct sequencing of RT PCR products. Graphs with mean values of 3 independent runs, standard deviation and p-value as determined by standard deviation are shown. Also, a representative electropherogram is depicted.
However, the mammalian assay also shows that some of the candidates only stimulate editing in the yeast system. In the mammalian system, in contrast, other factors might either compete with the candidate factors, or their expression might already be at a high level so that further overexpression does not lead to a change in editing levels. The latter possibility seems to be the case for at least one candidate, hnRNP A2B1, as further experiments have shown (see below).
Editing of endogenous substrates
As both the primary yeast screen but also the secondary screen in HeLa cells were based on the modified R/G stem-loop as an editing substrate we also wanted to test the impact on editing of endogenous substrates. To do this, the influence of DSS1 and hnRNP A2/B1 on editing of the endogenous mRNA encoding CyFIP2 was tested. Since this mRNA is not edited by the low endogenous levels of ADAR2 in HeLa or HeK293 cells, we had to establish a Hek 293 cell line stably expressing rat ADAR2 (Fig. 4B). Cotransfection of DSS1 into this cell line showed an average increase of CyFIP2 editing by 8% (Fig. 4C). Cotransfection of hnRNP A2/B1 only led to a marginal increase in editing that did not prove to be statistically significant (data not shown). The moderate effect observed for DSS1 may reflect the fact that only a fraction (typically 30% to 60%) of cells is transfected, while editing levels are measured on cDNAs derived from all cells. Moreover, the factors tested, may already be present at high levels in the cell lines tested. This is in contrast to yeast cells, where expression of the reporter stem-loop and the editing-stimulatory factors is completely overlapping and some of the mammalian factors are lacking altogether.
Therefore, we next tested whether RNA interference against Dss1 and hnRNP A2B1 would lead to a decrease in editing. To do this, knockdown of mRNA was performed using lentiviral delivered shRNAs. At least two suitable hairpin RNAs were designed to target either of these two genes. The lentiviral system allows for efficient selection of stable clones.52 Thus clones expressing the shRNAs against either target were selected using puromycin and tested for the repression of the target mRNA relative to tubulin mRNA by qPCR. The shRNAs showing the stronger repression were used for further analysis (Fig. S2 and data not shown). For DSS1, efficiency of the shRNA was also tested on a cell line stably expressing myc-DSS1 using an antibody directed against the myc-epitope (Fig. S2).
DSS1 mRNA could be depleted by 80%. This, in turn, resulted in a decrease of cyFIP2 editing by 23%. Thus, DSS1 overexpression or repression can alter editing levels by about 30% (Fig. 5). We also depleted hnRNP A2B1 using shRNAs. Here, depletion led to a 50% decrease in hnRNP A2B1 mRNA. Correspondingly, the effect on editing was only minor, reducing editing levels by 11% (Fig. 5). The knockdown cell lines were also tested for their ability to edit the mRNA encoding filamin α (FLNA) which harbors a single editing site, leading to a Q to R codon exchange. Depletion of DSS1 again led to a 23% decrease in editing of the FLNA mRNA while depletion of hnRNP A2B1 had no effect on editing of the FLNA mRNA (Fig. 5). Interestingly, simultaneous depletion of hnRNP A2B1 and DSS1 failed to decrease RNA levels beyond those observed after depletion of DSS1 alone (data not shown).
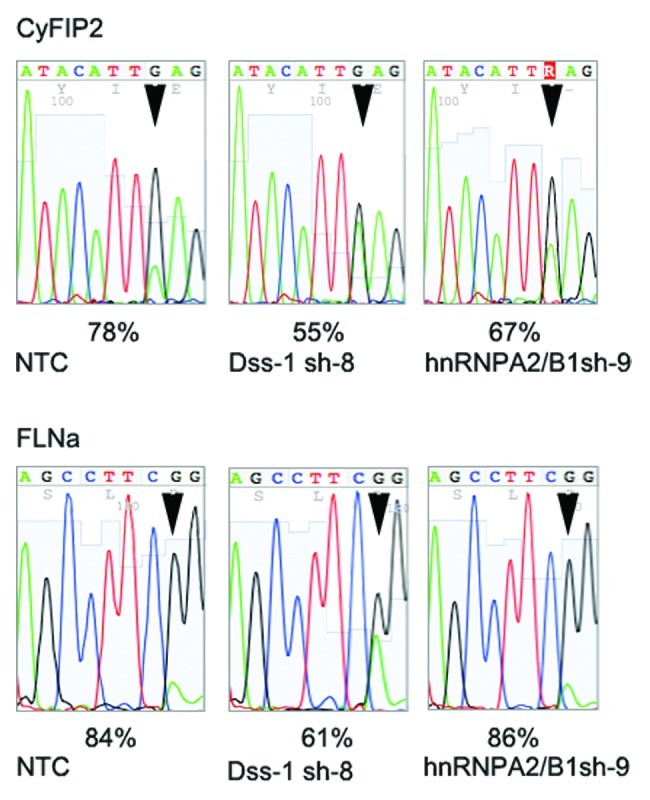
Figure 5. Depletion of DSS1 and hnRNP A2/B1 leads to a reduction in RNA-editing. Short hairpin constructs directed against DSS1 or hnRNP A2/B1 were stably integrated in HeK293 cells expressing rat ADAR2 using lentiviral gene delivery. Stable lines showing a robust knockdown of DSS1 or hnRNP A2/B1 (see Fig. S2) were established. Editing levels of CyFIP2 and FLNa mRNAs were determined by direct sequencing of RT-PCR products. Knockdown of DSS1 leads to a reduction of CyFIP2 editing by 23% while depletion of hnRNPA2B1 reduces editing by 9%. Edited positions are marked by an arrowhead. Interestingly, editing of FLNa mRNA was also reduced by 23% upon DSS1 depletion but was not affected by hnRNP A2B1 depletion.
Nuclear DSS1 stimulates RNA-editing
DSS1 is a short, 70 amino acid long, acidic protein. It was first identified as a candidate gene being deleted in patients with split hand/foot malformation type 1 (SHFM1).53 Recent evidence suggests, however, that other genes in the deleted cluster are responsible for the disease.54 DSS1 is an abundant protein that is present in the nucleus and cytoplasm and has been shown to be present in several complexes required for different cellular processes. For instance, DSS1 was shown to be part of the 26S proteasome, where it regulates interactions with a specific subset of poly-ubiquitinated p53.55-58 Moreover, several studies demonstrate an interaction between DSS1 and BRCA2.59,60 Most interestingly, the budding yeast homolog Sem1, and DSS1, were shown to be involved in splicing, 3′ end processing, and the TREX-2 mRNA export pathway, thus linking DSS1 at several levels to RNA metabolism.61-66
To determine whether the entire protein or only parts of DSS1 would be required to stimulate ADAR2 activity, deletion mutants of DSS1 were created. Since the protein is rather short, 2 expression constructs were prepared: the N- and C-terminal halves of DSS1 each consisting of 35 amino acids (Fig. 6A). The different truncations were expressed from either N- or C-terminally myc-tagged vectors, with or without an artificial NLS sequence to ensure localization of the protein. In parallel, editing of the RFP/GFP reporter construct was tested by FACS.

Figure 6. The C-terminal part of DSS1 is essential to stimulate RNA-editing. (A) Sequence and structure of DSS1. Alpha-helices, β-sheets and Asp/Glu-rich regions are shown. Additionally, N- and C-terminally truncated versions of the protein used for further experiments are indicated. (B) Localization of myc-tagged DSS1 with or without additional NLS sequence. HeLa cells expressing myc-tagged DSS1 were stained with anti-Myc and Alexa Fluor 568 conjugated antibodies. (C) Results of FACS experiments of different variants of DSS1. The GFP vs. RFP fluorescence of HeLa cells transfected with the RFP-editing stem loop-GFP substrate was compared between full-length DSS1 with or without NLS, and N-terminal (NLS-DSS1-N) and C-terminal portions (NLS-DSS1-C) of the protein expressed from vectors carrying an NLS sequence. (D) Comparison of DSS1 and ADAR2 localization. Cells stably expressing flag-rADAR2 were transfected with myc-tagged DSS1. rADAR2 shows a strong nucleolar and nuclear staining. DSS1 shows a nuclear but extranuclear staining. The localization of ADAR2 is identical in cells showing and lacking ectopic DSS1 expression, as marked by the arrowhead and arrow, respectively. Scale bar = 20µm
Full length and truncated versions of DSS1 are predominantly localized to the nucleus, irrespective of the presence of an artificial NLS (Fig. 6B). However, FACS experiments performed with the fluorescent editing reporter show that DSS1 has the strongest effect on RNA editing when an artificial NLS is added. In the absence of the artificial NLS, nuclear accumulation is still observed but with a slightly reduced stimulatory effect on RNA-editing (Fig. 6C). This suggests that it is mostly the nuclear fraction of DSS1 that stimulates ADAR2 activity.
An N-terminal truncation of DSS1 harboring an NLS only mildly reduces the stimulatory activity on RNA-editing when compared with full-length DSS1 lacking an artificial NLS. However, a clear reduction compared with full-length DSS1 with an artificial NLS can be observed. A C-terminal deletion, in contrast, almost completely abolishes the effect of DSS1. This suggests that the C-terminal part of DSS1 is more active in stimulating RNA-editing, while the N-terminal part seems rather dispensable (Fig. 6C).
To elucidate the mechanism by which DSS1 stimulates RNA editing we first tested for an interaction between rADAR2 and DSS1. However, co-immunoprecipitation experiments performed in cells stably expressing flag-tagged rADAR2 and transiently expressing myc-tagged DSS1 failed to show an interaction between the two factors (data not shown). Similarly, double immune-fluorescence staining showed no specific overlap in localization between ADAR2 and DSS1 beyond their joint presence in the nuclear compartment. While ADAR2 is predominantly localized to nucleoli but also to the nucleus, DSS1 shows an extranucleolar but nuclear staining (Fig. 6D).
DSS1 increases cellular ADAR levels and interacts with hnRNPs
DSS1 has been reported to be a component of the lid subunit of the 26S proteasome but also as an interaction partner of several factors and complexes such as BRCA2, the TREX-2 complex or the integrator complex.65-67 We therefore tested whether overexpression of DSS1 influences ADAR2 levels.
Indeed, co expression of DSS1 led to a slight but significant increase in ADAR2 levels by 10% (Fig. S3), which might positively stimulate RNA-editing.
On the other hand, we purified DSS1 from stably transfected cells by tandem affinity purification and identified associated proteins by mass spectrometry (Fig. S4 and Table S2). While the majority of hits did correspond to components of the 26S proteasome, many hits also corresponded to hnRNP proteins A2B1, A1B, C, D and G. This finding also opens the possibility that DSS1 acts by altering the hnRNP landscape thereby altering editing patterns.
Discussion
In this study a functional in vivo A to I editing system was developed to identify factors that can stimulate RNA editing by ADAR2. A primary yeast screen that allows high throughput screening was combined with a secondary, more accurate screen in mammalian cells. The screen was successfully employed to obtain factors that increase ADAR-mediated RNA editing.
The yeast editing system has previously been used to study the impact of changes in the catalytic domain of ADAR2 on substrate preferences.68 In our study we have modified the yeast editing system using His3 as an auxotrophy marker that only allows growth of cells that have successfully converted a stop codon into a tryptophan codon via RNA-editing on plates lacking histidine. The His3 marker has the additional advantage that 3-aminotriazole can be used as a competitive inhibitor of His3, thus allowing for a variation in the stringency of screening conditions. Interestingly, of the 62 fastest growing clones isolated from the primary yeast screen only one third did contain a protein encoding cDNA. The remaining two thirds of clones either contained non specified DNA fragments or 3′ UTRs. Surprisingly, the majority of candidates had indeed a stimulatory effect on editing in the yeast reporter strain irrespective of their coding potential as determined by direct sequencing of the reporter stem loop (Fig. 2). It might be possible that overexpression of certain RNAs in yeast may alter the endogenous RNP composition and thereby stimulate RNA editing. Alternatively, the non-coding RNAs may act as a landing platform for ADAR, thereby stimulating editing of transcripts in close proximity as recently suggested for an intronic sequence in the Gabra3 encoding pre-mRNA.69 The fact that many clones that did stimulate editing in the yeast system failed to do so in the mammalian system obviously reflects the different cellular environment and different RNP compositions. About 20% of all yeast colonies that supported growth in the absence of histidine failed to show increased editing of the reporter stem loop. These colonies may have supported yeast growth by other means such as preventing cellular accumulation of 3-AT.
Despite the relatively high false-positive rate the yeast screen still provides a very useful tool to reduce millions of cDNAs to a manageable number of tens of clones within a very short time and with little effort. The secondary screen finally validates all candidates in the mammalian context. The simultaneous expression of a constitutive and an editing-dependent fluorescent marker from one RNA strongly reduces the number of false-positives in this secondary screen. Moreover, the secondary FACS-based screen bares two big advantages: the large number of cells that can be measured individually allows an adequate statistical analysis, and the speed of the system that allows to test multiple candidates in parallel within 24 h post transfection.
The variability of transfection rates in mammalian cells may still pose a problem for this screen. Cotransfection of the RFP-GFP editing reporter as well as the plasmids encoding the candidate enhancers of editing may be as low as 30%. Thus, only strong changes in editing will be detected while minor changes may go undetected In the future, creation of cell lines that also stably express the reporter plasmid may help to increase the sensitivity of the secondary screen.
We managed successfully to express rADAR2 in Saccharomyces cerevisiae and edit a substrate derived from GluA2 in vivo. Interestingly, expression of active ADAR1 was not as successful. Although ADAR1 protein was easily detected on western blots, we failed to detect editing activity, both indirectly via yeast growth or directly via sequencing of the GluA2 editing substrate. To exclude the possibility that either our human ADAR1 clone or the substrate were not suitable we purified the same tagged ADAR1 protein expressed from mammalian cells and performed in vitro editing reactions with in vitro transcribed substrate. This in vitro editing assay resulted in clearly detectable editing levels suggesting that hsADAR1 in yeast may be less active than rADAR2 (data not shown). At this stage we cannot conclude whether failure to detect editing by ADAR1 in yeast is specific for the substrate used, or alternatively, whether ADAR1 is inhibited or mislocalized in yeast.
However, the yeast system may also be missing critical cofactors or contain factors that interfere with ADAR1 activity. Clearly, further studies will be required to address this point.
The screen presented here was performed with a human fetal brain cDNA. It was shown that editing levels may vary largely in different brain regions, during development and disease progression70,31,71. Therefore, similar screens may be employed for tissue or development-specific cDNAs. It can be expected, that libraries from adult brain will contain additional cDNAs that can stimulate RNA-editing.
Of the factors isolated, DSS1 and hnRNP A2B1 were studied in more detail. Besides having been identified as part of the 26S proteasome DSS1 and its yeast homolog Sem1, were shown to be involved in mRNA export, 3′ end processing of snRNAs and BRCA2 binding.57,64-67 Our mass spectrometric analysis of proteins associated with DSS1 could confirm an association with proteasome components but also demonstrated numerous interactions with hnRNP proteins. Interestingly, hnRNP A2/B1, one of the interactors of DSS1 was itself isolated as an enhancer of editing from this screen. Therefore, DSS1 may be part of an RNP interaction network that can help to stimulate editing possibly via stabilizing or facilitating substrate folding and recognition by ADARs. Along these lines it is worth mentioning that also other RNA binding proteins and two ribosomal proteins were identified in the primary screen, further supporting the idea that alterations in the RNA-landscape can affect RNA editing. However, not all RNA binding proteins stimulate RNA-editing: In a separate screen performed in our lab RNA-binding proteins we could also isolate RNA-binding proteins that negatively influence RNA-editing.72
We could show that the C-terminal domain of DSS1 is crucial for its editing-stimulating activity. The C-terminal domain of DSS1 has also been found as crucial for the interaction with BRCA267 and TREX-2 65 but also for its stimulatory role for RNA export,64 suggesting that this domain may serve as an interaction platform for several proteins.
Depletion of DSS1 or hnRNP A2/B1 showed further a reduction of editing for endogenous substrates. Reduction of DSS1 had a strikingly similar effect for two different endogenous substrates. In contrast, reduction of hnRNP A2/B1 only reduced editing in the CyFIP2 encoding mRNA while the FLNa encoding mRNA was unaffected. This may suggest that both factors DSS1 and hnRNP A2/B1 may act via different mechanisms.
The finding that overexpression of DSS1 leads to a slight but significant increase in ADAR2 levels might also be related to its proteasome activity. Overexpression of DSS1 might stimulate degradation of an ADAR2 stabilizing factor. Alternatively, ADAR2 has been shown to be destabilized by ubiquitination.41 Overexpression of DSS1 therefore might also act in a dominant negative manner and prevent degradation of ubiquitinated ADAR2 via the proteasome. Thus, given the many pathways in which DSS1 is involved, further studies will be required to determine precisely the mechanism by which DSS1 can stimulate RNA editing.
In summary, the screen presented here demonstrates the suitability of a heterologous yeast editing system to allow a high throughput screening for regulators of editing. Only slight modification of this screen and use of a different set of markers (e.g., URA3 on FOA plates) allows for the screening of inhibitors of editing. Besides factors expressed from a cDNA library the presented screen may also be modified to screen for small molecule inhibitors or enhancers of editing.
Combination of the high-throughput yeast system with the fluorescence based mammalian editing system allows the rapid elimination of false positives and provides direct proof for the impact of candidate clones on editing in mammalian cells. Thus the toolbox presented may also be useful to rapidly verify candidate factors that may regulate editing. Extension of this screen to cDNAs from other sources may help to identify novel regulatory factors from specific tissues, developmental stages or under disease conditions.
Materials and Methods
Construction of a yeast strain carrying an editing-reporter
A stem-loop substrate based on the GluA2 R/G site was mutated by PCR to replace the arginine codon at the editing site by an amber-STOP codon. Upon editing, the amber stop is converted into a tryptophan codon allowing continuous translation of a downstream open reading frame. The stem-loop substrate was introduced in-frame 100 nucleotides downstream of the start codon into a His3 gene, using an artificially created XhoI restriction site. The His3 gene containing the editing substrate was fused to a neo resistance cassette and introduced via homologous ends into the Leu2 gene of Saccharomyces cerevisiae strain W303.73
rADAR2 expression
A Flag-tagged version of rat ADAR2 (a kind gift of R. Emeson, Vanderbilt University) was cloned into and expressed from the centromeric tetracycline inducible vector pCM251.74
Mating assay screen
The W303 strain expressing the editing substrate and rADAR2 was mated with Y187 carrying a pretransformed human fetal brain cDNA library (Clontech, CA). The oligo dT primed cDNA library had a complexity of 8.6 × 106 independent clones with an insert size ranging from 0.5 to > 3kb. cDNA inserts were cloned directed. About 3.3 × 106 clones were mated. This should cover the human exome with a probability of p = 0.97. Mated cells were selected for expression of His3 on plates lacking histidine and containing 20mM 3-aminotriazol.
DNA from positive clones was isolated and amplified with plasmid primers 5′ CTATTCGATGATGAAGATCCACCAAACC 3′ and 5′ GTGAACTTGCGGGGTTTTTCATCTACGA 3′ and subjected to sequencing with one of the primers used for amplification.
RT-PCR and editing assay
Total RNA was isolated from yeast using the hot phenol method.75 First strand cDNA synthesis and the subsequent PCR were performed with RevertAid™ H Minus M-MuLV Reverse Transcriptase and Taq DNA polymerase (Fermentas, Lithuania) according to the manufacturer’s protocols. To determine editing levels the region containing the editing site was amplified with oligos forward: 5′ ATGACAGAGCAGAAAGCCCT 3′ and reverse: 5′ GTAATTCTGCTAGCCTCTG 3′. For direct sequencing the forward primer was used.
Oligos for amplification of the editing site in the FACS substrate were as follows: forward: 5′ GGTGGAGTTCAAGTCCATCTACATGG 3′ and reverse: 5′ GTGCAGATGAACTTCAGGGTCAGC 3′. The CyFip2 K/E editing site was amplified with primers forward: 5′ TCTACCTAATGGATGGAAATGTCAGTAA 3′ and reverse: 5′ ATCCCGGATCTGAACCATCTG 3′ primers. Again, for sequencing of PCR products the forward primer was used. Editing levels were determined by measuring peak heights. Editing ratios were calculated as the ratio of G-peak height to the total G+A peak heights.
Standard deviations of triplicate measures were calculated using student’s t-test.
Cell culture
HeLa, Hek 293 and Hek 293 stably transformed with Flag-tagged rADAR2 were cultured in DMEM (PAA, Austria) with high glucose supplemented with 10% fetal calf serum, 2mM L-glutamine, 100 U/ml penicillin and 100µg/ml streptomycin. Stable HeK 293 cells carrying rADAR2 were obtained via selection of the cells containing neomycin resistance with 200µg/ml G418. Clones that show a homogenous expression of rADAR2 were identified by immunofluorescence staining with anti-FLAG antibody.
Tissue culture transfection
cDNAs encoding candidate proteins were cloned into the tissue culture expression vector pCDNA3.1(-) (Invitrogen, CA) N-terminally fused to 6xmyc-tags and a NLS sequence. As a control, sequences were also cloned out of frame. Additionally, DSS1 was cloned without its UTR sequences, without the NLS sequence, or with a C-terminal myc-tag. Transfection was performed using Nanofectin reagent (PAA, Austria) according to manufacturer’s instructions. After 32–72 h cells were processed to obtain RNA, protein extracts or were stained to visualize protein expression.
FACS analysis
For FACS analysis a vector expressing RFP and GFP separated by a stop-codon containing substrate stem-loop was created, based on a previously described vector.51 In the absence of editing, only RFP is expressed. Upon editing, the RFP-GFP fusion is expressed. Changes in red and green fluorescent protein ratios can easily be quantified by FACS analysis. The vector expressing the candidates editing stimulatory protein was cotransfected with a substrate vector in a 4:1 ratio. After 72 h red and green fluorescence was measured on a FACScalibur flow cytometer (BD Biosciences) using CellQuest 3.3 software. Statistical assessment was performed with FlowJo 6.3.1 software. For statistic evaluation, 6 different gates were taken above background for the red channel while the green channel was wide open. The mean green florescence values from each gate were divided by the mean red fluorescence values. These normalized fluorescence values were plotted on a graph against the chosen gates.
Immunofluorescence analysis
Transfected cells grown for 32 to 48 h on acid-etched coverslips were fixed76 and myc-tagged proteins were detected with mAb 9E1077 and goat-anti mouse Alexa Fluor 568 (Invitrogen, CA) and in case of Flag-tagged rADAR2 with either rabbit anti-Flag (Sigma) or anti-ADAR2 antibody (rabbit serum made in our lab) followed by goat Alexa Fluor 488 anti-rabbit antibody (Invitrogen, CA).
Immunoprecipitation analysis
72 h after transfection cells were lysed by sonication in NET-2 buffer (150mM NaCl, 80mM Tris pH 7.4, 0.05% NP-40).78 150–200µg of the extract was added to Protein A Sepharose beads (GE Healthcare) coupled with anti-myc mAb 9E10. After 2 h of incubation on a rotating wheel, beads were washed with NET-2 and bound proteins were analyzed by SDS-PAGE and western blotting. Myc-tagged proteins were detected with mAb 9E10 and alkaline phosphatase coupled goat anti-mouse antibody (Sigma). Flag-tagged rADAR2 was detected with either rabbit anti-Flag (Sigma, St. Louis MO) or anti-ADAR2 antibody RED1 (Abcam, San Francisco, CA) followed by goat alkaline phosphatase anti-rabbit antibody (Pierce, Rockford, IL).
Tandem affinity purification
For mass spectrometric analysis of associated proteins the candidate proteins were tagged with an N-terminal myc-tag followed by a TEV protease cleavage site and a HA-epitope. The resulting myc-TEV-HA tagged cDNAs were stably transfected into HeLa, Hek, 293 or U2OS tissue culture cells. For immunoprecipitation 5mg Sepharose A beads (GE Healthcare) were incubated with NET-2 lysis buffer (50 mM Tris/HCl pH 7.4; 150 mM NaCl, 0.05% NP-40) for 1‘. The swollen beads were dissolved in 500 µl ml of 9E10 anti-c-MYC or 12CA5 anti-HA tissue culture supernatant and incubated at 4°C o/n. Antibody-coupled beads were washed with NET-2 buffer and antibodies were covalently coupled.79 Cell lysate in NET-2 buffer was cleared by two centrifugation steps. Aliquots of total lysates were taken. Supernatants were incubated with mAb 9E10 Sepharose A beads for one hour at 4°C. Beads were washed 4 times with NET-2 and two times with TEV-protease cleavage buffer (10 × 50 mM Tris/HCl pH 8, 0.5mM EDTA, 1mM DTT) before starting the cleavage reaction for 1h at RT. The supernatant of the cleaved and washed beads was collected and incubated with 12CA5 (anti HA) coupled Dynabeads for 1h at 4°C. After the second precipitation the beads were washed four times with NET-2 lysis buffer before boiling in 2 × SDS sample buffer.
Mass spectrometry
Samples were separated on a 7.5 –17% gradient SDS-PAGE gel at 1200V/h. After the run silver staining was performed.80 Protein bands of interest were cut out and stored in 1% acetic acid.
Proteins were reduced with 10 mM DTT and alkylated with 0.01 g/ml Iodacetamide in ammonium bicarbonate buffer, followed by tryptic digestion. Peptides were separated on an UltiMate 3000 HPLC system (Dionex, Thermo Fisher Scientific). Digests were loaded on a trapping column (PepMap C18, 5µm particle size, 300 μm i.d. × 5mm, Dionex) equilibrated with 0.1% TFA and separated on an analytical column (PepMap C18, 3 μm, 75 μm i.d. × 150 mm, Dionex) applying a 30’ linear gradient from 2.5% up to 40% ACN with 0.1% formic acid followed by a washing step with 80% ACN and 10% TFE. The HPLC was directly coupled to an LTQ-Orbitrap Velos mass spectrometer (Thermo Fisher Scientific) equipped with a nanoelectrospray ionization source (Proxeon, Thermo Fisher Scientific). The electrospray voltage was set to 1,500 V. The mass spectrometer was operated in the data-dependent mode: 1 full scan (m/z: 400–1,800, resolution 60,000) with lock mass enabled was followed by maximal 20 MS/MS scans. The lock mass was set at the signal of polydimethylcyclosiloxane at m/z 445.120025. Monoisotopic precursor selection was enabled; singly charged signals were excluded from fragmentation. The collision energy was set at 35%, Q-value at 0.25 and the activation time at 10 msec. Fragmented ions were set onto an exclusion list for 30 sec.
Raw spectra were interpreted by Mascot 2.2.04 (Matrix Science) using Mascot Daemon 2.2.2. Spectra were searched against the human nr-database with the following parameters: the peptide tolerance was set to 2 ppm, MS/MS tolerance was set to 0.8 Da, carbamidomethylcysteine was set as static modification, oxidation of methionine as a variable modification. Trypsin was selected as protease and two missed cleavages were allowed. MASCOT results were loaded into Scaffold (Ver. 3.00.02; Proteome Software). Peptide identifications were accepted if they could be established at a probability greater than 95%. as assigned by the Protein Prophet algorithm. Protein identifications were accepted if they could be established at a probability greater than 99% and at least two independent peptides per protein were identified.
Western blotting
To monitor expression of candidates isolated from the screen or to determine ADAR2 levels tissue culture cells were lysed in SDS sample buffer by sonication. After SDS PAGE electrophoresis proteins were blotted onto nitrocellulose (Schleicher and Schuell, Germany). Proteins were detected with primary antibodies followed by secondary alkaline phosphatase coupled antibodies and detection with NBT/BCIP. For precise quantification secondary antibodies were fluorophor coupled and detected by laser fluorimetry on a BioRad FX-Pro.
RNA interference
To generate shRNA expression cassettes targeting hnRNP A2B1 or DSS1 the pLKO.1 lentiviral vector system was used52 as described by Addgene (http://www.addgene.org/plko). Briefly, specific oligonucleotides corresponding to the following Broad TRC RNAi shRNA library (The RNAi Consortium) sequences were introduced into the Age I – EcoR I sites of pLKO.1 (Addgene plasmid # 10878):
DSS1 shRNA8:
ccggGCTGAACTAGAGAAACATGGTctcgagACCATGTTTCTCTAGTTCAGCtttttg
hnRNP A2B1 shRNA9:
ccggGCTTCTTCCTATTTGCCATGGctcgagCCATGGCAAATAGGAAGAAGCtttttg
NTC2 control shRNA: ccggCCTAAGGTTAAGTCGCCCTCGctcgagCGAGGGCGACTTAACCTTAGGtttttg
Cells transfected with non-targeting shRNA vectors NTC2, activating RISC and the RNAi pathway, but not targeting any human gene, were used as negative controls in knockdown experiments. All shRNA expression cassettes were verified by sequencing
Viral particle production and target cell infection
Described shRNA-pLKO.1 constructs were co-transfected with the packaging plasmid pPax2 (Addgene plasmid # 12260) and the envelop plasmid pMD2.G (Addgene plasmid #12259) into human embryonic kidney 293FT cells using Lipofectamine 2000 (Invitrogen). Virus was harvested 72 h post-transfection and concentrated using a PEG virus precipitation kit (BioVision). Infections of HeK293 cells stably transfected with ratADAR2 were performed in the presence of 10 μg/ml hexadimethrine bromide (a.k.a. polybrene, Sigma). Following transduction, cells were selected with 2 μg/ml puromycin.
Selected cells were used for the isolation of RNA to determine levels of RNA-editing. Silencing of targets was verified on cells stably expressing the myc-tagged targets by immunofluorescence staining or by qPCRs measuring a reduction of RNA levels of the targeted transcripts.
qPCR measurement
RNA samples were treated with DNase I (Fermentas) and 3 μg of total RNA was reverse transcribed to cDNAs using random hexamers and revert aid H minus MMuLV reverse transcriptase (Ferementas). Appropriate dilutions of cDNA were added to SYBR green PCR master mix (Promega) in the presence of a 15 nM final concentration of gene-speci□c primers in 20μl reaction mixtures. Primer selection was done using primer Quest (IDT). A BioRad iQ5 sequence detector system was used for real-time PCR ampli□cation and detection. Relative change in mRNA levels between samples was calculated by the 2–DDCT method. The following primers were used for real time PCR.
DSS1 forward: GGTCTGTTAGAGGAAGACGAC
DSS1 reverse: cgCCTAGAATGTATTAAGCAGg
Tubulin forward: GACAACACAGCCCTGAACCG
Tubulin reverse: GACTGGTCTGTAGTGAGCGG
hnRNP A2/B1 forward: AGCTTTGAAACCACAGAAGAA
hnRNP A2B1 reverse: TTGATCTTTTGCTTGCAGGA
Supplementary Material
Acknowledgments
The authors are thankful to Ron Emeson for the kind gift of a functional rat ADAR2 cDNA clone. The authors are also very thankful to the MFPL mass spectrometry unit for advice and help in peptide identification.
Funding
This work was supported by the Austrian Science Foundation, grant no. SFB 1706 and 4313 to M.F.J.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/rnabiology/article/23208
References
- 1.Nishikura K. Functions and regulation of RNA editing by ADAR deaminases. Annu Rev Biochem. 2010;79:321–49. doi: 10.1146/annurev-biochem-060208-105251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Burns CM, Chu H, Rueter SM, Hutchinson LK, Canton H, Sanders-Bush E, et al. Regulation of serotonin-2C receptor G-protein coupling by RNA editing. Nature. 1997;387:303–8. doi: 10.1038/387303a0. [DOI] [PubMed] [Google Scholar]
- 3.Higuchi M, Single FN, Köhler M, Sommer B, Sprengel R, Seeburg PH. RNA editing of AMPA receptor subunit GluR-B: a base-paired intron-exon structure determines position and efficiency. Cell. 1993;75:1361–70. doi: 10.1016/0092-8674(93)90622-W. [DOI] [PubMed] [Google Scholar]
- 4.Seeburg PH, Higuchi M, Sprengel R. RNA editing of brain glutamate receptor channels: mechanism and physiology. Brain Res Brain Res Rev. 1998;26:217–29. doi: 10.1016/S0165-0173(97)00062-3. [DOI] [PubMed] [Google Scholar]
- 5.Sprengel R, Single FN. Mice with genetically modified NMDA and AMPA receptors. Ann N Y Acad Sci. 1999;868:494–501. doi: 10.1111/j.1749-6632.1999.tb11318.x. [DOI] [PubMed] [Google Scholar]
- 6.Jepson JE, Reenan RA. RNA editing in regulating gene expression in the brain. Biochim Biophys Acta. 2008;1779:459–70. doi: 10.1016/j.bbagrm.2007.11.009. [DOI] [PubMed] [Google Scholar]
- 7.Athanasiadis A, Rich A, Maas S. Widespread A-to-I RNA editing of Alu-containing mRNAs in the human transcriptome. PLoS Biol. 2004;2:e391. doi: 10.1371/journal.pbio.0020391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim DD, Kim TT, Walsh T, Kobayashi Y, Matise TC, Buyske S, et al. Widespread RNA editing of embedded alu elements in the human transcriptome. Genome Res. 2004;14:1719–25. doi: 10.1101/gr.2855504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Levanon EY, Eisenberg E, Yelin R, Nemzer S, Hallegger M, Shemesh R, et al. Systematic identification of abundant A-to-I editing sites in the human transcriptome. Nat Biotechnol. 2004;22:1001–5. doi: 10.1038/nbt996. [DOI] [PubMed] [Google Scholar]
- 10.Li JB, Levanon EY, Yoon JK, Aach J, Xie B, Leproust E, et al. Genome-wide identification of human RNA editing sites by parallel DNA capturing and sequencing. Science. 2009;324:1210–3. doi: 10.1126/science.1170995. [DOI] [PubMed] [Google Scholar]
- 11.Yang W, Chendrimada TP, Wang Q, Higuchi M, Seeburg PH, Shiekhattar R, et al. Modulation of microRNA processing and expression through RNA editing by ADAR deaminases. Nat Struct Mol Biol. 2006;13:13–21. doi: 10.1038/nsmb1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kawahara Y, Zinshteyn B, Chendrimada TP, Shiekhattar R, Nishikura K. RNA editing of the microRNA-151 precursor blocks cleavage by the Dicer-TRBP complex. EMBO Rep. 2007;8:763–9. doi: 10.1038/sj.embor.7401011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kawahara Y, Zinshteyn B, Sethupathy P, Iizasa H, Hatzigeorgiou AG, Nishikura K. Redirection of silencing targets by adenosine-to-inosine editing of miRNAs. Science. 2007;315:1137–40. doi: 10.1126/science.1138050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vesely C, Tauber S, Sedlazeck FJ, von Haeseler A, Jantsch MF. Adenosine deaminases that act on RNA induce reproducible changes in abundance and sequence of embryonic miRNAs. Genome Res. 2012;22:1468–76. doi: 10.1101/gr.133025.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wagner RW, Yoo C, Wrabetz L, Kamholz J, Buchhalter J, Hassan NF, et al. Double-stranded RNA unwinding and modifying activity is detected ubiquitously in primary tissues and cell lines. Mol Cell Biol. 1990;10:5586–90. doi: 10.1128/mcb.10.10.5586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bass BL, Nishikura K, Keller W, Seeburg PH, Emeson RB, O’Connell MA, et al. A standardized nomenclature for adenosine deaminases that act on RNA. RNA. 1997;3:947–9. [letter] [PMC free article] [PubMed] [Google Scholar]
- 17.Melcher T, Maas S, Herb A, Sprengel R, Higuchi M, Seeburg PH. RED2, a brain-specific member of the RNA-specific adenosine deaminase family. J Biol Chem. 1996;271:31795–8. doi: 10.1074/jbc.271.50.31795. [DOI] [PubMed] [Google Scholar]
- 18.Melcher T, Maas S, Herb A, Sprengel R, Seeburg PH, Higuchi M. A mammalian RNA editing enzyme. Nature. 1996;379:460–4. doi: 10.1038/379460a0. [DOI] [PubMed] [Google Scholar]
- 19.Chen CX, Cho DS, Wang Q, Lai F, Carter KC, Nishikura K. A third member of the RNA-specific adenosine deaminase gene family, ADAR3, contains both single- and double-stranded RNA binding domains. RNA. 2000;6:755–67. doi: 10.1017/S1355838200000170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Macbeth MR, Schubert HL, Vandemark AP, Lingam AT, Hill CP, Bass BL. Inositol hexakisphosphate is bound in the ADAR2 core and required for RNA editing. Science. 2005;309:1534–9. doi: 10.1126/science.1113150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lehmann KA, Bass BL. Double-stranded RNA adenosine deaminases ADAR1 and ADAR2 have overlapping specificities. Biochemistry. 2000;39:12875–84. doi: 10.1021/bi001383g. [DOI] [PubMed] [Google Scholar]
- 22.Riedmann EM, Schopoff S, Hartner JC, Jantsch MF. Specificity of ADAR-mediated RNA editing in newly identified targets. RNA. 2008;14:1110–8. doi: 10.1261/rna.923308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eggington JM, Greene T, Bass BL. Predicting sites of ADAR editing in double-stranded RNA. Nat Commun. 2011;2:319. doi: 10.1038/ncomms1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Polson AG, Bass BL. Preferential selection of adenosines for modification by double-stranded RNA adenosine deaminase. EMBO J. 1994;13:5701–11. doi: 10.1002/j.1460-2075.1994.tb06908.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stefl R, Allain FH. A novel RNA pentaloop fold involved in targeting ADAR2. RNA. 2005;11:592–7. doi: 10.1261/rna.7276805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stefl R, Xu M, Skrisovska L, Emeson RB, Allain FH. Structure and specific RNA binding of ADAR2 double-stranded RNA binding motifs. Structure. 2006;14:345–55. doi: 10.1016/j.str.2005.11.013. [DOI] [PubMed] [Google Scholar]
- 27.Stefl R, Oberstrass FC, Hood JL, Jourdan M, Zimmermann M, Skrisovska L, et al. The solution structure of the ADAR2 dsRBM-RNA complex reveals a sequence-specific readout of the minor groove. Cell. 2010;143:225–37. doi: 10.1016/j.cell.2010.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stephens OM, Haudenschild BL, Beal PA. The binding selectivity of ADAR2’s dsRBMs contributes to RNA-editing selectivity. Chem Biol. 2004;11:1239–50. doi: 10.1016/j.chembiol.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 29.Pokharel S, Beal PA. High-throughput screening for functional adenosine to inosine RNA editing systems. ACS Chem Biol. 2006;1:761–5. doi: 10.1021/cb6003838. [DOI] [PubMed] [Google Scholar]
- 30.Jacobs MM, Fogg RL, Emeson RB, Stanwood GD. ADAR1 and ADAR2 expression and editing activity during forebrain development. Dev Neurosci. 2009;31:223–37. doi: 10.1159/000210185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wahlstedt H, Daniel C, Ensterö M, Ohman M. Large-scale mRNA sequencing determines global regulation of RNA editing during brain development. Genome Res. 2009;19:978–86. doi: 10.1101/gr.089409.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ring H, Boije H, Daniel C, Ohlson J, Ohman M, Hallböök F. Increased A-to-I RNA editing of the transcript for GABAA receptor subunit α3 during chick retinal development. Vis Neurosci. 2010;27:149–57. doi: 10.1017/S0952523810000180. [DOI] [PubMed] [Google Scholar]
- 33.Nishimoto Y, Yamashita T, Hideyama T, Tsuji S, Suzuki N, Kwak S. Determination of editors at the novel A-to-I editing positions. Neurosci Res. 2008;61:201–6. doi: 10.1016/j.neures.2008.02.009. [DOI] [PubMed] [Google Scholar]
- 34.Maas S, Patt S, Schrey M, Rich A. Underediting of glutamate receptor GluR-B mRNA in malignant gliomas. Proc Natl Acad Sci U S A. 2001;98:14687–92. doi: 10.1073/pnas.251531398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cenci C, Barzotti R, Galeano F, Corbelli S, Rota R, Massimi L, et al. Down-regulation of RNA editing in pediatric astrocytomas: ADAR2 editing activity inhibits cell migration and proliferation. J Biol Chem. 2008;283:7251–60. doi: 10.1074/jbc.M708316200. [DOI] [PubMed] [Google Scholar]
- 36.Iwamoto K, Nakatani N, Bundo M, Yoshikawa T, Kato T. Altered RNA editing of serotonin 2C receptor in a rat model of depression. Neurosci Res. 2005;53:69–76. doi: 10.1016/j.neures.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 37.Niswender CM, Herrick-Davis K, Dilley GE, Meltzer HY, Overholser JC, Stockmeier CA, et al. RNA editing of the human serotonin 5-HT2C receptor. alterations in suicide and implications for serotonergic pharmacotherapy. Neuropsychopharmacology. 2001;24:478–91. doi: 10.1016/S0893-133X(00)00223-2. [DOI] [PubMed] [Google Scholar]
- 38.Sodhi MS, Burnet PW, Makoff AJ, Kerwin RW, Harrison PJ. RNA editing of the 5-HT(2C) receptor is reduced in schizophrenia. Mol Psychiatry. 2001;6:373–9. doi: 10.1038/sj.mp.4000920. [DOI] [PubMed] [Google Scholar]
- 39.Gurevich I, Englander MT, Adlersberg M, Siegal NB, Schmauss C. Modulation of serotonin 2C receptor editing by sustained changes in serotonergic neurotransmission. J Neurosci. 2002;22:10529–32. doi: 10.1523/JNEUROSCI.22-24-10529.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gurevich I, Tamir H, Arango V, Dwork AJ, Mann JJ, Schmauss C. Altered editing of serotonin 2C receptor pre-mRNA in the prefrontal cortex of depressed suicide victims. Neuron. 2002;34:349–56. doi: 10.1016/S0896-6273(02)00660-8. [DOI] [PubMed] [Google Scholar]
- 41.Marcucci R, Brindle J, Paro S, Casadio A, Hempel S, Morrice N, et al. Pin1 and WWP2 regulate GluR2 Q/R site RNA editing by ADAR2 with opposing effects. EMBO J. 2011;30:4211–22. doi: 10.1038/emboj.2011.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu Y, Samuel CE. Editing of glutamate receptor subunit B pre-mRNA by splice-site variants of interferon-inducible double-stranded RNA-specific adenosine deaminase ADAR1. J Biol Chem. 1999;274:5070–7. doi: 10.1074/jbc.274.8.5070. [DOI] [PubMed] [Google Scholar]
- 43.Rueter SM, Dawson TR, Emeson RB. Regulation of alternative splicing by RNA editing. Nature. 1999;399:75–80. doi: 10.1038/19992. [DOI] [PubMed] [Google Scholar]
- 44.Cho DS, Yang W, Lee JT, Shiekhattar R, Murray JM, Nishikura K. Requirement of dimerization for RNA editing activity of adenosine deaminases acting on RNA. J Biol Chem. 2003;278:17093–102. doi: 10.1074/jbc.M213127200. [DOI] [PubMed] [Google Scholar]
- 45.Gallo A, Keegan LP, Ring GM, O’Connell MA. An ADAR that edits transcripts encoding ion channel subunits functions as a dimer. EMBO J. 2003;22:3421–30. doi: 10.1093/emboj/cdg327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sansam CL, Wells KS, Emeson RB. Modulation of RNA editing by functional nucleolar sequestration of ADAR2. Proc Natl Acad Sci U S A. 2003;100:14018–23. doi: 10.1073/pnas.2336131100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sallacz NB, Jantsch MF. Chromosomal storage of the RNA-editing enzyme ADAR1 in Xenopus oocytes. Mol Biol Cell. 2005;16:3377–86. doi: 10.1091/mbc.E05-01-0016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Desterro JM, Keegan LP, Jaffray E, Hay RT, O’Connell MA, Carmo-Fonseca M. SUMO-1 modification alters ADAR1 editing activity. Mol Biol Cell. 2005;16:5115–26. doi: 10.1091/mbc.E05-06-0536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gommans WM, McCane J, Nacarelli GS, Maas S. A mammalian reporter system for fast and quantitative detection of intracellular A-to-I RNA editing levels. Anal Biochem. 2010;399:230–6. doi: 10.1016/j.ab.2009.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hartner JC, Schmittwolf C, Kispert A, Müller AM, Higuchi M, Seeburg PH. Liver disintegration in the mouse embryo caused by deficiency in the RNA-editing enzyme ADAR1. J Biol Chem. 2004;279:4894–902. doi: 10.1074/jbc.M311347200. [DOI] [PubMed] [Google Scholar]
- 51.Schoft VK, Schopoff S, Jantsch MF. Regulation of glutamate receptor B pre-mRNA splicing by RNA editing. Nucleic Acids Res. 2007;35:3723–32. doi: 10.1093/nar/gkm314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moffat J, Grueneberg DA, Yang X, Kim SY, Kloepfer AM, Hinkle G, et al. A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell. 2006;124:1283–98. doi: 10.1016/j.cell.2006.01.040. [DOI] [PubMed] [Google Scholar]
- 53.Scherer SW, Poorkaj P, Allen T, Kim J, Geshuri D, Nunes M, et al. Fine mapping of the autosomal dominant split hand/split foot locus on chromosome 7, band q21.3-q22.1. Am J Hum Genet. 1994;55:12–20. [PMC free article] [PubMed] [Google Scholar]
- 54.Merlo GR, Paleari L, Mantero S, Genova F, Beverdam A, Palmisano GL, et al. Mouse model of split hand/foot malformation type I. Genesis. 2002;33:97–101. doi: 10.1002/gene.10098. [DOI] [PubMed] [Google Scholar]
- 55.Coux O, Tanaka K, Goldberg AL. Structure and functions of the 20S and 26S proteasomes. Annu Rev Biochem. 1996;65:801–47. doi: 10.1146/annurev.bi.65.070196.004101. [DOI] [PubMed] [Google Scholar]
- 56.Funakoshi M, Li X, Velichutina I, Hochstrasser M, Kobayashi H. Sem1, the yeast ortholog of a human BRCA2-binding protein, is a component of the proteasome regulatory particle that enhances proteasome stability. J Cell Sci. 2004;117:6447–54. doi: 10.1242/jcs.01575. [DOI] [PubMed] [Google Scholar]
- 57.Sone T, Saeki Y, Toh-e A, Yokosawa H. Sem1p is a novel subunit of the 26 S proteasome from Saccharomyces cerevisiae. J Biol Chem. 2004;279:28807–16. doi: 10.1074/jbc.M403165200. [DOI] [PubMed] [Google Scholar]
- 58.Wei SJ, Williams JG, Dang H, Darden TA, Betz BL, Humble MM, et al. Identification of a specific motif of the DSS1 protein required for proteasome interaction and p53 protein degradation. J Mol Biol. 2008;383:693–712. doi: 10.1016/j.jmb.2008.08.044. [DOI] [PubMed] [Google Scholar]
- 59.Duncan JA, Reeves JR, Cooke TG. BRCA1 and BRCA2 proteins: roles in health and disease. Mol Pathol. 1998;51:237–47. doi: 10.1136/mp.51.5.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yoshida K, Miki Y. Role of BRCA1 and BRCA2 as regulators of DNA repair, transcription, and cell cycle in response to DNA damage. Cancer Sci. 2004;95:866–71. doi: 10.1111/j.1349-7006.2004.tb02195.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mannen T, Andoh T, Tani T. Dss1 associating with the proteasome functions in selective nuclear mRNA export in yeast. Biochem Biophys Res Commun. 2008;365:664–71. doi: 10.1016/j.bbrc.2007.11.024. [DOI] [PubMed] [Google Scholar]
- 62.Thakurta AG, Gopal G, Yoon JH, Kozak L, Dhar R. Homolog of BRCA2-interacting Dss1p and Uap56p link Mlo3p and Rae1p for mRNA export in fission yeast. EMBO J. 2005;24:2512–23. doi: 10.1038/sj.emboj.7600713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wilmes GM, Bergkessel M, Bandyopadhyay S, Shales M, Braberg H, Cagney G, et al. A genetic interaction map of RNA-processing factors reveals links between Sem1/Dss1-containing complexes and mRNA export and splicing. Mol Cell. 2008;32:735–46. doi: 10.1016/j.molcel.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Faza MB, Kemmler S, Jimeno S, González-Aguilera C, Aguilera A, Hurt E, et al. Sem1 is a functional component of the nuclear pore complex-associated messenger RNA export machinery. J Cell Biol. 2009;184:833–46. doi: 10.1083/jcb.200810059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ellisdon AM, Dimitrova L, Hurt E, Stewart M. Structural basis for the assembly and nucleic acid binding of the TREX-2 transcription-export complex. Nat Struct Mol Biol. 2012;19:328–36. doi: 10.1038/nsmb.2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Baillat D, Hakimi MA, Näär AM, Shilatifard A, Cooch N, Shiekhattar R. Integrator, a multiprotein mediator of small nuclear RNA processing, associates with the C-terminal repeat of RNA polymerase II. Cell. 2005;123:265–76. doi: 10.1016/j.cell.2005.08.019. [DOI] [PubMed] [Google Scholar]
- 67.Yang H, Jeffrey PD, Miller J, Kinnucan E, Sun Y, Thoma NH, et al. BRCA2 function in DNA binding and recombination from a BRCA2-DSS1-ssDNA structure. Science. 2002;297:1837–48. doi: 10.1126/science.297.5588.1837. [DOI] [PubMed] [Google Scholar]
- 68.Pokharel S, Jayalath P, Maydanovych O, Goodman RA, Wang SC, Tantillo DJ, et al. Matching active site and substrate structures for an RNA editing reaction. J Am Chem Soc. 2009;131:11882–91. doi: 10.1021/ja9034076. [DOI] [PubMed] [Google Scholar]
- 69.Daniel C, Venø MT, Ekdahl Y, Kjems J, Ohman M. A distant cis acting intronic element induces site-selective RNA editing. Nucleic Acids Res. 2012;40:9876–86. doi: 10.1093/nar/gks691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gallo A, Locatelli F. ADARs: allies or enemies? The importance of A-to-I RNA editing in human disease: from cancer to HIV-1. Biol Rev Camb Philos Soc. 2011;87:95–110. doi: 10.1111/j.1469-185X.2011.00186.x. [DOI] [PubMed] [Google Scholar]
- 71.Venø M, Bramsen JB, Bendixen C, Panitz F, Holm I, Ohman M, et al. Spatio-temporal regulation of ADAR editing during development in porcine neural tissues. RNA Biol. 2012;9:1054–65. doi: 10.4161/rna.21082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tariq A, Garncarz W, Handl C, Balik A, Pusch O, Jantsch MF. RNA-interacting proteins act as site-specific repressors of ADAR2-mediated RNA editing and fluctuate upon neuronal stimulation. Nucleic Acids Res. 2012 doi: 10.1093/nar/gks1353. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Baudin-Baillieu A, Guillemet E, Cullin C, Lacroute F. Construction of a yeast strain deleted for the TRP1 promoter and coding region that enhances the efficiency of the polymerase chain reaction-disruption method. Yeast. 1997;13:353–6. doi: 10.1002/(SICI)1097-0061(19970330)13:4<353::AID-YEA86>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 74.Garí E, Piedrafita L, Aldea M, Herrero E. A set of vectors with a tetracycline-regulatable promoter system for modulated gene expression in Saccharomyces cerevisiae. Yeast. 1997;13:837–48. doi: 10.1002/(SICI)1097-0061(199707)13:9<837::AID-YEA145>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 75.Köhrer K, Domdey H. Preparation of high molecular weight RNA. Methods Enzymol. 1991;194:398–405. doi: 10.1016/0076-6879(91)94030-G. [DOI] [PubMed] [Google Scholar]
- 76.Fu XD, Maniatis T. Factor required for mammalian spliceosome assembly is localized to discrete regions in the nucleus. Nature. 1990;343:437–41. doi: 10.1038/343437a0. [DOI] [PubMed] [Google Scholar]
- 77.Evan GI, Lewis GK, Ramsay G, Bishop JM. Isolation of monoclonal antibodies specific for human c-myc proto-oncogene product. Mol Cell Biol. 1985;5:3610–6. doi: 10.1128/mcb.5.12.3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Steitz JA. Immunoprecipitation of ribonucleoproteins using autoantibodies. Methods Enzymol. 1989;180:468–81. doi: 10.1016/0076-6879(89)80118-1. [DOI] [PubMed] [Google Scholar]
- 79.Harlow E, Lane D. Immunoaffinity purification: binding of antigen to antibody-bead matrix in a suspension. CSH Protoc 2006; 2006. [DOI] [PubMed] [Google Scholar]
- 80.Stochaj WR, Berkelman T, Laird N. Mass spectrometry-compatible silver staining. CSH Protoc. 2007;2007:t4742. doi: 10.1101/pdb.prot4742. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.