

Abstract
Photodynamic therapy (PDT) employs photoexcitation of a sensitizer to generate tumor-eradicating reactive oxygen species. We recently showed that irradiating breast cancer COH-BR1 cells after treating with 5-aminolevulinic acid (ALA, a pro-sensitizer) resulted in rapid upregulation of inducible nitric oxide (NO) synthase (iNOS). Apoptotic cell killing was strongly enhanced by an iNOS inhibitor (1400W), iNOS knockdown (kd), or a NO scavenger, suggesting that NO was acting cytoprotectively. Stress signaling associated with these effects was examined in this study. ALA/light-stressed COH-BR1 cells, and also breast adenocarcinoma MDA-MB-231 cells, mounted an iNOS/NO-dependent resistance to apoptosis that proved to be cGMP-independent. Immunocytochemistry and subcellular Western analysis of photostressed COH-BR1 cells revealed a cytosol-to-nucleus translocation of NF-κB which was negated by the NF-κB activation inhibitor Bay11. Bay11 also enhanced apoptosis and prevented iNOS induction, consistent with NF-κB involvement in the latter. JNK and p38 MAP kinase inhibitors suppressed apoptosis, implicating these kinases in death signaling. Post-irradiation extent and duration of JNK and p38 phosphorylation were dramatically elevated by 1400W or iNOS-kd, suggesting that these activations were suppressed by NO. Regarding pro-survival stress signaling, rapid activation of Akt was unaffected by 1400W, but prevented by Wortmannin, which also enhanced apoptosis. Thus, a link between upstream Akt activation and iNOS induction was apparent. Furthermore, p53 protein expression under photostress was elevated by iNOS-kd, whereas robust Survivin induction was abolished, consistent with p53 and Survivin being negatively and positively regulated by NO, respectively. Collectively, these findings enhance our understanding of cytoprotective signaling associated with photostress-induced NO and suggest iNOS inhibitor-based approaches for improving PDT efficacy.
Keywords: photodynamic therapy, apoptosis, nitric oxide, stress resistance signaling
Introduction
Produced naturally by nitric oxide synthase (NOS) enzymes, nitric oxide (NO) has numerous biological roles, including involvement in vasorelaxation, neurotransmission, and eradication of pathogenic microorganisms (1). When generated at high rates (e.g. by activated macrophages and neurtophils), NO can have prooxidant cytotoxic effects, whereas at low rates, it may act cytoprotectively and promote cell growth/survival (2,3). There are numerous examples of endogenous (NOS-derived) or exogenous (chemical donor-derived) NO acting in a salutary fashion on normal (non-transformed) cells. Studies with primary hepatocytes (4,5) and differentiated (neuron-like) PC12 cells (6) showed that apoptosis induced by growth factor or serum deprivation was inhibited by exogenous NO through activation of soluble guanylyl cyclase (sGC) and thence cGMP activation of protein kinase G (PKG). A more recent study (7) revealed that human keratinocyte apoptosis provoked by ultraviolet-B radiation was strongly enhanced by NOS and sGC inhibitors, but attenuated by overexpression of inducible NOS (iNOS). These findings (7) demonstrated an antiapoptotic role for endogenous NO that was mediated at least in part by cyclic GMP. However, whether this NO derived solely from constitutive NOS or whether any stress-induced NOS might have been involved was not determined. There is increasing evidence that tumor cells can also exploit NO as an anti-apoptotic/pro-survival signaling molecule (8,9). This NO may derive from the tumor cells themselves as well as from macrophages and endothelial cells in the tumor vasculature. In contrast to normal cells (4–7), relatively little is known about the NOS/NO status of tumor cells subjected to oxidative challenges, including therapeutic challenges, or whether NO might play a role in cellular resistance to the lethal effects of these conditions. Of special interest along these lines are earlier studies (10–12) showing that administration of non-specific NOS inhibitors markedly improved the responses of various mouse-borne tumors to Photofrin-sensitized photodynamic therapy (PDT). The results were mainly attributed to diminished relaxation of tumor blood vessels by NO acting in opposition to PDT's known vasoconstrictive effects (10,11). Surprisingly little else has been done to further characterize NO's anti-PDT activity in terms of (a) whether the NO derives from tumor cells per se, tumor vasculature cells, or both; (b) which of the three NOS isoforms is most important in supplying the NO; and (c) whether the NOS/NO involved is constitutive or possibly stress-upregulated. Of added importance, but not investigated up to now is how endogenous tumor NO might modulate the stress signaling events that underlie PDT. Using a PDT model comprised of breast tumor COH-BR1 cells sensitized by 5-aminolevulinic acid (ALA)-generated protoporphyrin IX (PpIX), we showed recently that apoptotic cell photokilling was markedly enhanced by iNOS inhibitors or iNOS knockdown (13,14). We found, moreover, that both iNOS and NO were rapidly upregulated in photostressed cells and that scavenging NO with a chemical trap markedly increased the apoptotic count. The implication of these findings is that if, in a clinical PDT setting, tumor cells upregulate iNOS/NO as a cytoprotective strategy, this could diminish treatment effectiveness. In order to better understand NO-mediated resistance to photokilling from a mechanistic standpoint, we have begun to investigate the pro-survival vs. pro-death signaling events associated with iNOS and NO upregulation in ALA/light-stressed COH-BR1 cells. Recent findings along these lines are described.
Materials and methods
General materials
5-Aminolevulinic acid (ALA), Hoechst-33258 (Ho), propidium iodide (PI), 8-bromoguanosine 3',5'-cyclic monophosphate (8-Br-cGMP), Wortmannin (Wo), sodium orthovanadate, β-glycerophosphate, and a primary monoclonal anti-β-actin antibody were obtained from Sigma-Aldrich (St. Louis, MO). A 10 μM stock solution of Wo in DMSO was prepared immediately before adding to cells. Invitogen Life Technologies (Grand Island, NY) supplied the Dulbecco's modified Eagles's/Kaighn's-modified Ham's nutrient F12K (DME/F12K) growth medium, fetal bovine serum, antibiotics, and geneticin. Spermine NONOate (SPNO), N-[3-(aminomethyl)benzyl]acetamidine (1400W), 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ), and Bay11-7082 (Bay11) were from Cayman Chemicals (Ann Arbor, MI). Immediately before experimental use, stock solutions of 25 mM SPNO, 1 mM 1400W, 1 mM Bay11, and 50 mM ODQ were prepared in 10 mM NaOH, pH 7.4 phosphate buffer, ethanol, and DMSO, respectively. The p38 inhibitor SB202190 and JNK inhibitor SP600125 were from Calbiochem (Gibbstown, NJ). Freshly prepared stock solutions of 1 mM SB202190 in water and 5 mM SP600125 in ethanol were used for experiments. An Annexin V-FITC cell staining kit was from Roche Applied Sciences (Indianapolis, IN). Santa Cruz Biotechnology (Santa Cruz, CA) supplied the polyclonal antibody against human iNOS, the monoclonal antibody against β-actin, and monoclonal antibody against p53. Monoclonal antibodies against human JNK, phospho-JNK, p38 (all forms), phospho-p38 (all forms), p38α, p38β, NF-κB (p65 subunit), histone H3, and α-tubulin, along with horseradish peroxidase-conjugated IgG secondary antibodies, were from Cell Signaling Technology (Danvers, MA). The antibody against human Survivin was from AbCam (Cambridge, MA), that against human phospho-Survivin was from Novus Biologicals (Littleton, CO), and the Alexa 488-conjugated secondary antibody was from Invitrogen. Pierce Chemical Co. (Rockford, IL) supplied reagents for the bicinchoninic (BCA) protein assay and for the SuperSignal West Pico chemiluminescence detection of proteins on immunoblots. The iNOS inhibitor GW274150 was kindly supplied by GlaxoSmithKline, LLC (Research Triangle Park, NC) via a material transfer agreement.
Cell culture
Wild-type (WT) COH-BR1 cells, an epithelial line derived from pleural effusion of a patient diagnosed with breast cancer (15), were obtained as a research gift from Dr. James Doroshow, City of Hope Cancer Center. Cytogenetic analysis (R.S. Esworthy, personal communication; Ref 15) confirmed that the cells were of human origin and consisted of two subpopulations, one being near diploid (56 chromosomes) and one near haploid (27–28 chromosomes). These cells lack selenoperoxidase GPx4, which detoxifies complex lipid hydroperoxides (16) and are quite sensitive to singlet oxygen-mediated photooxidative stress such as used in this study. Also used was a previously generated shRNA-based iNOS-knockdown (kd) clone of COH-BR1 cells (14). This clone was one of four generated with plasmids containing iNOS-shRNA inserts (SABiosciences, Frederick, MD). The selected clone expressed the lowest constitutive level of iNOS relative to WT cells (<20%), as determined by Western analysis (14). Human breast adenocarcinoma MDA-MB-231 cells were obtained from the ATCC repository, which provided the necessary authentication details. All cells were grown under standard culture conditions, using DME/F12K medium supplemented with 10% fetal bovine serum and antibiotics (13,14,16). Growth media for iNOS-kd cells contained geneticin (0.1 mg/ml), which was omitted 24 h before an experiment.
Cell sensitization and irradiation
Cells at 60–65% confluence were metabolically sensitized with PpIX by incubating with 1.0 mM ALA in serum-free DME/F12K medium for 45 min in the dark at 37 °C. Most of the PpIX at this point was localized in mitochondria, where it originates (14,17). Where indicated, an iNOS inhibitor (1400W or GW274150), JNK inhibitor (SP600125), p38 inhibitor (SB202190), NF-κB activation inhibitor (Bay11-7082), PI3K inhibitor (Wo), soluble guanylyl cyclase inhibitor (ODQ), or 8-Br-cGMP was added 15 min before ALA and maintained at a given concentration during all subsequent steps. The exogenous NO donor, spermine NONOate (SPNO), was used in one experiment. Immediately after sensitization, cells were switched to ALA- and serum-free medium either lacking or containing one of the above agents, as indicated. The cell dishes were irradiated at room temperature as described (14); light fluence rate was ~1.1 mW/cm2, corresponding to a delivered fluence of ~1 J/cm2 after 15 min of irradiation.
Evaluation of cell death
ALA-treated cells on coverslips were irradiated in the absence or presence of a given inhibitor (1400W, ODQ, Bay11, Wo) or NO donor (SPNO) and after 20 h of dark incubation analyzed for extent of apoptotic cell death. Cell coverslips were treated with an Annexin V-Fluos/propidium iodide (PI) staining mixture as recommended by the supplier, mounted on slides with Fluoromount G, and examined by fluorescence microscopy for apoptosis vs. necrosis (18). In some cases, this was assessed after staining cells with 5 μM Ho and 50 μM PI (14,19). Dark controls (cells treated with ALA and not irradiated) were studied alongside. Approximately 100 cells in 3–5 viewing fields were evaluated for each coverslip examined.
Immunoprecipitation and immunoblot procedures
Photostressed WT or iNOS-kd COH-BR1 cells, along with dark controls, were recovered, washed with chilled PBS, resuspended in ice-cold homogenization buffer containing protease inhibitors, and lysed (19). After centrifugation, supernatant fractions were recovered for direct Western analysis of certain proteins or immunoprecipitation of others followed by Western analysis. Levels of Akt, phospho-Akt (p-Akt), JNK, phospho-JNK (p-JNK), overall p38, overall phospho-p38 (p-p38), p53, and Survivin were assessed directly. Equal amounts of total lysate protein (19) were subjected to Laemmli SDS-PAGE using 10% acrylamide/bis-acrylamide (13.5% for Survivin determination), followed by Western blotting using primary monoclonal antibodies against Akt, p-Akt (S473), p-Akt (T308), JNK, overall p38, overall p-p38, and peroxidase-conjugated IgG secondary antibodies. Antibodies against p53 and Survivin were also used. Protein bands were detected via SuperSignal West Pico chemiluminescence and quantified using UVP LabWorks software (Upland, CA). Phosphorylation status of p38α, p38β, and Survivin was evaluated using immunoprecipitation and Western blotting. The former was carried out as described (19) using a monoclonal antibody against p38α or p38β, or against Survivin. Immunoprecipitated proteins were recovered (19) and subjected to Western analysis, using primary antibodies against p-p38 or phospho-Survivin.
Immunocytochemistry for NF-κB localization
Photostressed COH-BR1 cells on coverslips were recovered after post-irradiation dark incubation, fixed and blocked (20), then permeabilized with 0.3% Triton X-100 and incubated overnight at 4 °C with 1:50-diluted NF-κB antibody. After rinsing, the cells were incubated with Alexa 488-conjugated anti-rabbit IgG and Ho (5 μM) for 1 h at 25 °C. After another rinse, coverslips were mounted on slides, and examined using a Nikon Diaphot-200 fluorescence microscope.
Detection of NF-κB in isolated nuclear and cytosolic fractions
COH-BR1 cells were treated with ALA alone, ALA/light, and ALA/Bay11/light as described in the Cell sensitization and irradiation section. Subcellular fractionation was carried out as described previously (21) with slight modification. Four hours after treatment, cells were trypsinized, washed with PBS, resuspended in ice-cold 10 mM HEPES/10 mM KCl/1.5 mM MgCl2/0.5 mM DTT buffer (pH 7.9) and homogenized in a pre-chilled Dounce homogenizer. Homogenates were centrifuged at 230 × g for 5 min at 4 °C to pellet nuclei and other fragments. Supernatants were recovered as cytosolic fractions. Nuclear fractions were further purified by density gradient centrifugation (0.25M to 0.88M sucrose) at 2800 × g for 10 min at 4 °C. Lysates of nuclear and cytoplasmic fractions were prepared in 50 mM Tris-HCl/150 mM NaCl/1% NP-40/0.5% deoxycholate (pH 7.5). After protein determination, a sample of each fraction (100 μg protein) was analyzed by Western blotting, using antibodies against NF-κB (p65), histone H3 as a nuclear marker, and α-tubulin as a cytosolic marker.
Data analysis
Experiments dealing with determination of photostress-induced apoptosis were carried out at least in triplicate. The two-tailed Student's t-test was used for assessing the significance of perceived differences between experimental values; P < 0.05 was considered statistically significant.
Results
Photostress upregulation of cytoprotective iNOS: role of NF-κB
As shown previously (13,14), COH-BR1 cells sensitized with PpIX by treating with ALA as described in the Methods section retained most of the PpIX in mitochondria, where it originates. Exposure of sensitized cells to a 1 J/cm2 fluence of broad band visible light resulted in an 4-5-fold increase in apoptotic cell count, as assessed by Annexin V-FITC (Anx V) or Hoechst (Ho) nuclear staining after 8 h of dark incubation (Fig. 1A,B). Co-staining with propidium iodide (PI) revealed no significant necrotic cells under these conditions. When irradiation and subsequent incubation was carried out in the presence of 1400W, a competitive inhibitor of iNOS, there was a striking additional increase in apoptotic count to ~60%, i.e. 15-fold above background. Earlier work (14) revealed that MTT-assessed overall cell kill as a function of increasing light fluence was likewise enhanced by 1400W. As shown in Fig. 1B, COH-BR1 cells whose iNOS had been knocked down by at least 80% via shRNA treatment (14) also exhibited a large increase in photostress-induced apoptosis (~3-fold over ALA/light treatment alone), whereas a scrambled shRNA control exhibited no increase (not shown). Collectively, these findings confirm those reported previously (13,14) and support the notion that upregulation of iNOS and NO contributed significantly to photokilling resistance in these tumor cells. Transcription factor NF-κB has been shown to play a role in iNOS expression in various cell lines (22,23). Whether this also applied in our system was assessed by using Bay11, an inhibitor of IKK, the kinase that activates NF-κB via phosphorylation, release, and subsequent degradation of its inhibitory protein, IκB-α(24,25). As shown by the immunofluorescence micrographs in Fig. 1C, cells treated with ALA, but not irradiated, displayed NF-κB exclusively in the cytosol, as expected (24). However, after exposing cells to ALA and light (1 J/cm2), followed by 4 h of dark incubation, NF-κB was visualized mainly in the nucleus, as indicated by overlapping Ho and NF-κB signals. This translocation was strongly attenuated when irradiation and subsequent dark incubation were carried out in the presence of Bay11 (Fig. 1C). These fluorescence imaging results were confirmed by Western blot examination of pre- and post-photostress distribution of NF-κB. As shown in Fig. 1D, the protein shifted from predominantly cytosolic to predominantly nuclear after ALA/light exposure, and this shift was substantially attenuated by Bay11. In agreement with previous results (13,14), COH-BR1 cells exhibited a significant background level of iNOS protein, which remained constant after incubation with ALA for 20 h in the dark (Fig. 1D). However, when sensitized cells were irradiated, a relatively rapid and persistent elevation in iNOS protein was observed, its level at 4 h and 20 h post-irradiation being 4-fold and 5-fold greater, respectively, than that in the dark control (Fig. 1E). This upregulation was abolished when Bay11 was present during irradiation and post-irradiation incubation, indicating that NF-κB activation and nuclear translocation played a key role in photostress induction of iNOS. In addition to inhibiting NF-κB activation and nuclear translocation, Bay11 increased the extent of ALA/light-induced apoptosis in a concentration-dependent fashion, 5 μM elevating it nearly 4-fold, whereas ALA, light, or Bay11 alone did not increase apoptosis above the basal level (Fig. 1F). These findings clearly indicate that transcriptional activation of NF-κB played a key role in iNOS upregulation and that this conferred a NO-dependent hyperresistance to photokilling, consistent with previous evidence that photokilling could be enhanced by the NO scavenger cPTIO (13).
Figure 1.
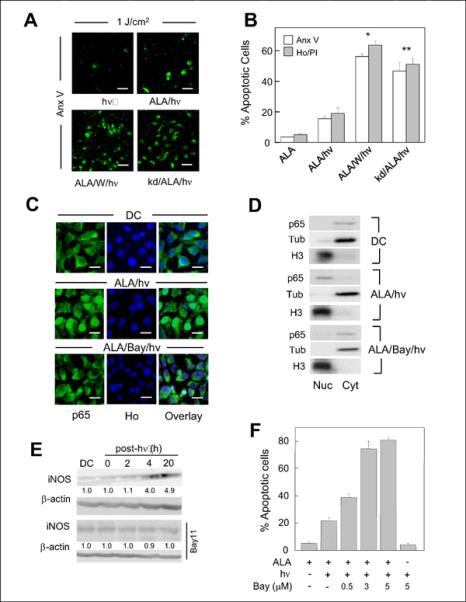
Photostress upregulation of cytoprotective iNOS: involvement of NF-κB. ALA-treated wild type (WT) COH-BR1 cells were irradiated (1 J/cm2) in the absence (ALA/hν) or presence of 10 μM 1400W (ALA/W/hν). ALA-treated iNOS-knockdown (kd) cells and non-ALA-treated WT cells were irradiated similarly (kd/ALA/h and hν, respectively). (A) Fluorescence micrographs of Annexin V-FITC (Anx V)-stained cells 8 h after irradiation. Bar: 100 μm. (B) Percentage of apoptotic cells under the above conditions, as determined by Anx V vs. PI staining (8 h post-hν) or Ho vs. PI staining (20 h post-hν). A dark control (ALA-only for 20 h) is also represented. (C) Immunocytochemical images showing effects of photostress on subcellular localization of NF-κB. ALA-treated cells were fixed 4 h after irradiation and probed with an antibody against the NF-κB p65 subunit; nuclei were detected with Ho. Also represented are cells treated with 2.5 μM Bay11 before, during and after ALA/light, and a dark control (DC). Bar: 50 μm. (D) Subcellular distribution of NF-κB determined by Western analysis. Cells exposed to ALA/light in the absence vs. presence of Bay11 (2.5 μM), along with a dark control, were homogenized 4 h after treatment and separated into nuclear (Nuc) and cytosolic (Cyt) fractions, each of which was tested for the presence of NF-κB p65 by Western blotting, histone H3 serving as a nuclear marker and α-tubulin (Tub) as a cytosolic marker. (E) iNOS level in ALA-treated cells at various times after irradiation in the absence vs. presence of 2.5 μM Bay11. Number below each lane denotes iNOS band intensity normalized to β-actin and relative to DC. Data are from one experiment representative of two for each condition. (F) Effect of Bay11 (0.5–5 μM) on extent of Ho/PI-assessed apoptosis 20 h after irradiation of ALA-treated vs. non-treated cells. Means ± SD of values from 3 separate experiments are plotted in (B) and (E). *P<0.005 vs. ALA/hν; P<0.01 vs. ALA/hν.
Determination of whether cyclic GMP plays a role in NO-mediated cytoprotection
Photostress-induced iNOS/NO might have been cytoprotective via activation of sGC with generation of cGMP, a known activator of pro-survival/anti-apoptotic PKG (26,27). At least one PDT-related study showed that exogenous NO from chemical donors protected lymphoblastoid cells against photokilling and that this could be attenuated by a sGC inhibitor (ODQ) and also by a PKG inhibitor, thus implicating cGMP in the cytoprotection (28). Using a light fluence of 2 J/cm2 on ALA-primed cells (previously shown to result in a 3-fold induction of iNOS, observed at 4 h post-h and persisting for at least another 16 h (13)), we found that ODQ at concentrations that antagonized NO protection in other cell studies (28,29) did not elevate apoptotic titer and, if anything, reduced it slightly (Supplementary Fig. S1A). On the other hand, supplying cGMP in the form of membrane-permeable 8-Br-cGMP before, during and after irradiation did diminish apoptosis by ~15% at the highest concentration used, 1 mM (Supplementary Fig. S1B). However, this effect is much smaller than that observed in other studies involving pro-survival NO (5,6,28) and is considered insignificant. On the basis of these findings, therefore, any cGMP involvement in the observed cytoprotective effects of photostress-generated NO can be ruled out.
Effects of PI3K inhibition on photostress activation of Akt, iNOS induction, and apoptotic killing
We asked whether the pro-growth/anti-apoptotic kinase Akt/PKB might be activated by ALA/light stress and, if so, how this related to iNOS/NO upregulation. As shown in Fig. 2A, Akt was rapidly activated by phosphorylation in photostressed cells, as evidenced with an antibody against phosphoserine-473 and one against phosphothreonine-308. Activation was maximal (15–18-fold above DC) from 15 min to 2 h after irradiation (1 J/cm2) and gradually subsided during longer post-irradiation incubation up to 20 h. Akt activation occurred independently of iNOS and NO upregulation because it was unaffected by 1400W (Fig. 2B). It is well known that Akt activation requires upstream activation of the oncogenic kinase PI3K (30). When PI3K was inactivated with a specific inhibitor, Wortmannin, photostress activation of Akt was markedly diminished, as was the upregulation of iNOS (Fig. 2C). Correspondingly, Wortmannin caused a strong dose-dependent increase in ALA/light-induced apoptosis, the proportion of apoptotic cells going from ~5% in the absence to ~55% in the presence of 25 nM Wortmannin (Fig. 2D).These findings clearly demonstrate that Akt activation played an important role in iNOS induction with accompanying resistance to apoptotic photokilling. Given that Akt is one of the known activators of NF-κB via phosphorylation-activation of IKK (30), this may have been a key upstream signaling event in the observed upregulation of iNOS.
Figure 2.
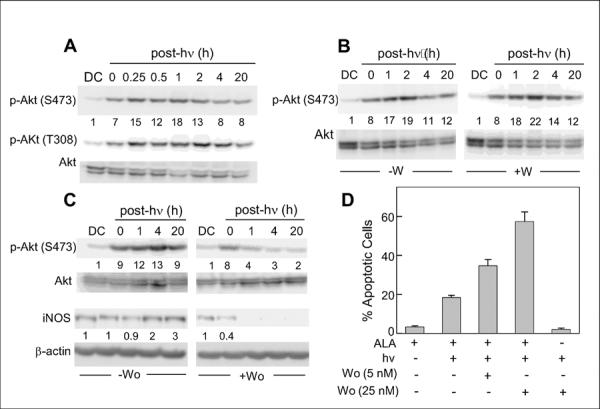
Photostress activation of Akt. (A) WT COH-BR1 cells were exposed to ALA and light (1 J/cm2), then analyzed for Akt phosphorylation by Western blotting at various post-irradiation times from 0 min up to 20 h. Antibodies detecting overall Akt and Akt phosphorylated at S473 or T308 were used. (B) Phosphorylation after exposing cells to the same photostress as in (A) in the absence (−W) or presence (+W) of 10 μM 1400W. (C) Effects of PI3K inhibitor Wortmannin on photostress activation of Akt and upregulation of iNOS. Cells were exposed to ALA/light in the absence vs. presence of 25 nM Wortmannin, then subjected to Western blot analysis for p-Akt, overall Akt, iNOS, and β-actin at the indicated post-irradiation times. Total cell protein per lane (A, B, C): ~125 μg. Number below each lane for p-Akt (S473) in panels A, B, and C indicates p-Akt band intensity normalized to total Akt and relative to the dark control (DC); number below each lane for iNOS in panel C indicates iNOS band intensity normalized to β-actin and relative to DC. Western blots are from one experiment representative of four in each case. (D) Extent of Ho-assessed apoptosis 20 h after stressing cells with ALA/light in the absence vs. presence of 5 nM or 25 nM Wortmannin. Data from 3 replicate experiments are plotted as means ± SD.
Effects of iNOS inhibition or knockdown on photostress activation of MAPK enzymes
In a previous study (19), we found that total immunodetectable JNK in COH-BR1 cells was transiently phosphorylation-activated by ALA/light stress and that this effect could be abolished by NO from an exogenous source, spermine-NONOate (SPNO) (31). Similar behavior was observed for total p38 MAPK. We asked in the present study whether photostress-upregulated NO by itself, i.e. in the absence of exogenous NO, would affect JNK and p38 similarly. As shown in Fig. 3A, ALA/light challenged cells exhibited a transient activation of the 54 kDa and 46 kDa isoforms of JNK (p-JNK formation), which was detected immediately after irradiation (1 J/cm2), reached a maximum 15–30 min later (~7-fold above DC), then subsided and was barely detectable after 4 h. Overall p38 was also activated (p-p38 formation) and exhibited a similar transient, peaking at ~3-times the control level after 30 min (Fig. 3A). The ALA-containing dark controls (DC) displayed minimal p-JNK and p-p38 throughout incubation. When photodynamic challenge and subsequent dark incubation were carried out in the presence of 1400W, a striking intensification and prolongation of JNK phosphorylation was observed such that both p-JNK bands were still elevated after 20 h (Fig. 3A). A similar prolongation of p38 phosphorylation was observed, and in this case, some elevation of p-p38 was even apparent in the DC (Fig. 3A). Knockdown of iNOS also resulted in a dramatic post-irradiation persistence of pJNK and p-p38 (Fig. 3B). Consequently, interfering with NO production in photostressed cells extended the lifetimes of these activated MAPKs and this could explain the increased apoptotic count under these conditions (Fig. 1B). JNK and p38 involvement was confirmed by showing that stress-induced apoptosis of iNOS-kd cells was markedly diminished by SP600125 (a JNK inhibitor) or SB202190 (a p38 inhibitor) (Fig. 3C). We found, furthermore, that the post-irradiation elevation/persistence of p-JNK and p-p38 caused by iNOS knockdown could be substantially reversed by introducing SPNO into the reaction system at a low, nontoxic concentration (0.1 mM) (Supplemental Fig. S2). As shown previously (13,14), SPNO could also “rescue” 1400W-treated or iNOS-kd cells from more extensive ALA/light-induced apoptosis.
Figure 3.
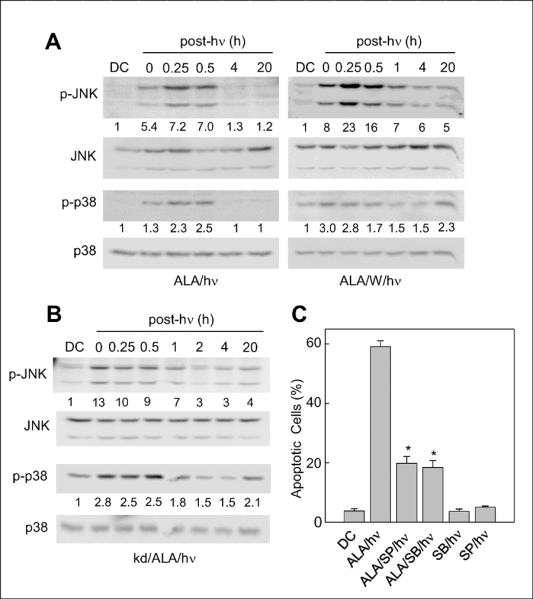
Pro-apoptotic activation of JNK and p38 in photostressed cells: effects of iNOS inhibition and iNOS knockdown. (A) ALA-treated WT COH-BR1 cells were irradiated (1 J/cm2) in the absence or presence of 10 μM 1400W, then recovered for Western blot analysis, either immediately (0 min) or after increasing periods of dark incubation ranging from 15 min to 20 h. Primary antibodies against p-JNK, overall JNK, p-p38, and overall p38 were used. (B) ALA-treated iNOS-knockdown cells were irradiated and analyzed similarly. Total cell protein per lane (A, B): 120 μg; number below each lane for p-JNK and p-p38 represents band intensity normalized to total JNK and p38, respectively, and relative to DC. Blots shown are from one experiment representative of three for each condition. (C) Effect of JNK and p38 inhibition on extent of Ho-assessed apoptosis in ALA/light-treated iNOS-kd cells at 20 h post-hν. In this case, cells were irradiated in the absence or presence of JNK inhibitor SP600125 (SP, 25 μM) or p38 inhibitor SB202190 (SB, 20 μM). A dark control with ALA (DC) and a light control with SP or SB present were also analyzed. Mean values ± SD from 3 independent experiments are plotted. *P<0.01 vs. ALA/hν.
Contrasting effects of photostress on p38α and p38β with modulation by iNOS-kd
Using immunoprecipitation and immunoblotting, we showed previously that the alpha isoform of p38 was activated by ALA/light stress, whereas the beta isoform (initially much more phosphorylated than alpha) was deactivated, and both effects were inhibited by exogenous NO from SPNO (19). Our findings were consistent with reports that these kinases affect stressed cell viability in opposing ways, p38 being pro-apoptotic and p38 anti-apoptotic (32,33). To learn whether endogenous NO might affect these MAPKs similarly, we used the same analytical approaches to distinguish their activation states in wild type versus iNOS-kd cells. As shown by the Western blot in Fig. 4A, phosphorylated p38α (p-p38α) in ALA-primed WT cells increased ~1.7-fold 15 min after irradiation, but declined below the control level 20 h later. In contrast, p-p38 in iNOS-kd cells was higher at both times, reaching ~2.5-fold at 20 h. The situation with p-p38 was strikingly different (Fig. 4B), i.e. it decayed below the control level throughout for both cell types, but appeared to do so faster in iNOS-kd cells. It is apparent from these data, which agree with those obtained with exogenous NO (19), that the most dramatic long-term negative effect of stress-induced NO was on p38α activation, whereas its effect on p38β deactivation, though evident, was less substantial.
Figure 4.

Comparison of p38α and p38β phosphorylation status in photostressed wild type (wt) versus iNOS-kd cells. ALA-treated COH-BR1 cells were irradiated (1 J/cm2) and after two periods of dark incubation, 15 min and 20 h, probed for phosphorylation of p38α (A) and p38β (B). ALA-treated cells kept in the dark throughout (DC) were probed alongside. The p38α and p38β isoforms were immunoprecipitated with their specific antibodies and subjected to Western analysis, using a p-p38 antibody to detect extent of phosphorylation of each isoform and the specific antibodies to detect overall levels. The latter values were used for standardizing sample loads and normalizing the amounts of phosphorylation. The plot beside each immunoblot shows densitometrically-determined levels of the indicated p-p38 relative to its DC.
Contrasting effects of iNOS-kd on p53 and Survivin expression in photostressed cells
Of added interest regarding NO's role in pro-survival vs. pro-death signaling under photostress were the responses of p53 and Survivin, which are known, respectively, to promote and suppress apoptosis in various cell systems (34,35). As shown by the Western blot in Fig. 5A, panel a, overall Survivin (Suv, ~16.5 kDa) was strongly upregulated in ALA/light-stressed COH-BR1 cells, reaching 5-times its control level at 12 h post-hQ and remaining there for at least another 8 h. Immunoprecipitation followed by Western blotting for phosphorylation-activated Survivin (p-Suv, ~21 kDa) in wild type cells revealed that it was elevated nearly 2-fold 8 h after photostress and that iNOS knock-down effectively abolished this (Fig. 5A, panel b). Analyzed alongside, stress elevation of total Survivin in wild type cells was likewise canceled by iNOS-kd (Fig. 5A, panel c). With regard to p53, direct Western analysis revealed a significant starting level of this protein in wild type cells and this remained constant for several hours after ALA/light treatment (Fig. 5B). However, iNOS-kd resulted in a significant post-irradiation upregulation of p53, which reached 2-fold at 2–4 h and subsided thereafter. By contrast, the post-irradiation elevation of total Survivin observed again in wild type cells (Fig. 5B), was abrogated by iNOS-kd. Thus, inadequate NO generation under photostress conditions resulted in strikingly different responses in p53 and Survivin expression, and these responses were consistent with the observed enhancement of apoptosis caused by iNOS-kd (Fig. 1B).
Figure 5.
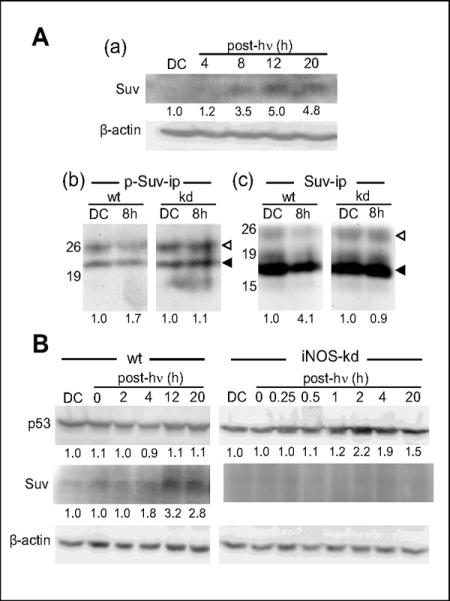
Reciprocal iNOS-dependent regulation of Survivin and p53 expression in photostressed COH-BR1 cells. (A, a) Wild type (wt) cells exposed to ALA/light (1 J/cm2) were harvested at various post-hν times; lysates were prepared and analyzed for total Survin (Suv) by Western blotting. A dark control (DC) was run alongside. (A, b) Level of phosphorylation-activated Survivin (p-Suv) in wt and iNOS-kd cells was assessed by preparing lysates at 8 h post-irradiation, immunoprecipitating (ip) Survivin, and probing for p-Suv by Western blot, using a phospho-Suv antibody. Also analyzed in the pull-down mixture was IgG light chain, which served as an internal standard for determining p-Suv level. (◁) IgG; (◀) p-Suv. (A, c) Western blots in (b) were stripped and re-probed with Suv antibody to assess total Survivin in each sample, standardized against IgG. (◁) IgG; (◀) Suv. Numbers beside blots in (b) and (c) indicate positions of protein size standards (kDa). Numbers below the 8 h bands in (b) and (c) represent densitometrically-determined band intensities standardized to IgG and normalized to DC in each case. (B) Wild type and iNOS-kd cells photostressed as in (A) were lysed at various times after irradiation and Western analyzed for p53 and Suv; numbers below bands indicate β-actin-corrected p53 and Suv levels, normalized to a DC.
Anti-apoptotic effects of iNOS in MDA-MB-231 cells
To learn whether other cancer cells besides COH-BR1 might mount an iNOS-dependent resistance to photodynamic eradication, we tested the effects of iNOS inhibitors on ALA/light-challenged MDA-MB-231 cells. As shown in Supplementary Fig. S3, apoptotic photokilling of these cells was dramatically enhanced by 1400W or GW274150 (36,37) in a dose-dependent manner, whereas L-NAME with relatively low NOS affinity/specificity had no significant effect over the concentration range used. In agreement with published IC50 values for purified iNOS (38), 1400W was more potent than GW274150, the concentration for 50% maximal apoptotic enhancement for the former being ~1 μM and for the latter ~9 μM (Fig. S3). Clearly, therefore, the anti-apoptotic effects of NO are not limited to one cancer cell type. We have not yet probed for pro-survival signaling in photostressed MDA-MB-231 cells, but general trends are expected to be similar to those of COH-BR1 cells, e.g. NF-κB-dependent iNOS upregulation and curtailed activation of JNK and p38α.
Discussion
Many tumors use NO at low constitutive levels as a pro-growth/survival signaling molecule (8,9). The NO may derive not only from macrophages and endothelial cells in tumor vasculatures, but also from tumor cells themselves. Awareness of this derives from a number of different experimental approaches, including genetic ablation or knockdown of NOS enzymes, inhibition of NOS activity, and use of NO scavengers (39,40). Among the various known effects of endogenous NO on tumors are enhanced angiogenesis and invasiveness, evasion of apoptosis, and greater resistance to chemotherapy and radiotherapy (8,9,40). As one compelling example, Sikora et al. (41) demonstrated that in vitro and in vivo treatment of human melanoma cells with the iNOS inhibitor L-nil reduced tumor cell growth and synergized with the antitumor effects of cisplatin. More recently, Eyler et al. (42), reported that glioma stem cells from human brain tumors expressed significantly more iNOS/NO than normal counterparts and proliferated more rapidly. Importantly, the tumor initiation/progression potential of these stem cells in mice was substantially reduced by iNOS inhibition or knockdown (42).
How NO might affect PDT's anti-tumor efficacy was first addressed about 12 years ago in studies involving various mouse tumor models subjected to Photofrin-sensitized PDT. In these studies, Henderson et al. (10) and Korbelik et al. (11) discovered that PDT cure rate could be significantly improved when treatment was carried out in the presence of a NOS inhibitor (L-NAME, L-NNA), and that extent of improvement correlated with the tumor's constitutive NO generating capacity. The results were interpreted in terms of competing modulations in the tumor vasculature, i.e. vasodilatory effects of endothelial NO working in opposition to PDT's well known vasoconstrictive effects (10,43). However, whether tumor cells per se might cope with PDT stress by using their own sources of NO was not considered. In addressing this question for the first time, we recently discovered that breast cancer COHBR1 cells could develop a robust hyperresistance to ALA/light-provoked apoptosis by upregulating NO through overexpression of iNOS, but not other NOS isoforms (13). This response developed rapidly after irradiation (2–3 h) and persisted for at least 20 h. Although there is considerable evidence that constitutive iNOS/NO can provide a survival/growth advantage to tumor cells (8,41,42), no other examples of cells mounting a NO-mediated defense against a therapeutic oxidative stress have been reported. Our aim in the present study was to define some of the key pro-survival vs. pro-death signaling mechanisms in photostressed cells and to better understand NO's role in the former.
Focusing initially on upstream signaling responsible for this, we observed a rapid and sustained Akt phosphorylation and iNOS upregulation in photostressed COH-BR1 cells, both of which effects were effectively abolished by a PI3K inhibitor (Wo), thus linking PDPK1-mediated Akt activation (44) to iNOS overexpression. We also showed that NF-κB translocated from the cytosol to nucleus of phtotstressed cells, i.e. it became transcriptionally activated. The fact that the IKK inhibitor Bay11 negated this as well as iNOS induction suggests that signaling for iNOS induction by stress-activated Akt occurred via NF-κB transactivation. This is supported by evidence (30,44) that Akt can phosphorylate IKK-α, which in turn activates NF-κB by phosphorylating and releasing IκB-α. While NF-κB transcriptional control of iNOS expression has been demonstrated in several other cell systems (22,45), AP-1 and Stat-1α have been implicated in other cases (46,47). Whether AP-1 or Stat-1α might also have been involved in our system is not known. ALA/light stress also caused the upregulation and phosphorylation-activation of Survivin, a member of the inhibitor of apoptosis (IAP) family (35). Survivin is expressed in many cancers (35) and has been shown to be upregulated in breast cancer and melanoma cell lines by Photofrin-sensitized photostress (48). Its expression in carcinoma cells is also known to be enhanced by low levels of endogenous or exogenous NO (49) and to be repressed by p53 (50). In agreement with this, we observed a striking reciprocal effect of iNOS-kd on Survivin and p53 expression under photostress, the former being repressed and the latter stimulated. Thus, stress-induced NO in WT cells appeared to keep p53 level in check while elevating Survivin to subdue apoptosis. The pro-survival signaling cascades proposed in this paragraph are illustrated in Fig. 6.
Figure 6.

Relevant pro-apoptotic vs. anti-apoptotic signaling cascades elicited by photodynamic stress. ROS: reactive oxygen species generated by photosensitization; singlet oxygen (1O2) is considered to be the most significant. Solid lines: based on these findings; dashed lines: proposed from published evidence of others (e.g. Refs. 48–50).
Turning our attention to the pro-death side of the signaling spectrum, we found that there was a rapid, but short-lived activation of JNK and total p38 MAPK in ALA/light-stressed COH-BR1 cells and that this was dramatically enhanced and prolonged by iNOS inhibition or knockdown in SPNO-reversible fashion, indicating that iNOS-derived NO was negatively regulating these MAPKs (see Fig. 6). In tracking the post-irradiation status of p38α and p38β in iNOS-kd vs. WT cells, we discovered that photostress-induced NO had a much greater long-term inhibitory effect on p38α phosphorylation than on p38β, which agrees with results from our previous study in which SPNO was used as a source of NO (19), i.e. before we realized that it could derive from endogenous iNOS. Our results are also consistent with those of others in demonstrating that p38α functions pro-apoptotically and p38β anti-apoptotically (32,33). We showed previously (19) that the pro-survival/growth MAPK, ERK1/2, which exhibited significant background phosphorylation in COH-BR1 cells (like p38β), was gradually deactivated by ALA/light treatment and that SPNO could prevent this or even hyperactivate ERK1/2. In addition, SPNO strongly inhibited photostress upregulation of pro-apoptotic Bax, down-regulation of anti-apoptotic Bcl-xL, and activation of pro-apoptotic Bid while elevating the level of cytoprotective heme oxygenase-1 (HO-1) (19). Although ERK1/2, HO-1, and Bcl-2 family proteins were not monitored in the present study, it is reasonable to believe that their behavior would have been similar to that observed with exogenous, i.e. supplemental NO (19).
How NO acted cytoprotectively remains to be determined. We showed previously that it does not scavenge singlet oxygen, the major cytotoxic oxidant generated by PpIX photoexcitation (51). Activation of sGC via NO ligation of its heme group, leading to cGMP activation of PKG, has also been ruled out (Supplementary Fig. S1), which contrasts with evidence from an earlier PDT-related study involving lymphoblastoid cells and exogenous NO donors (28). Other possibilities (see Fig. 6) include (a) inhibition of pro-apoptotic MAPKs such as ASK1 and JNK via S-nitrosation of cysteine residues (52,53); (b) S-nitrosation-inhibition of caspases (54); (c) S-nitrosation of Bcl-2, which inhibits its proteosomal degradation (55); and (d) S-nitrosation-inhibition of PTEN, the phosphoinositide phosphatase that negatively regulates Akt (56). We are exploring these possibilities in ongoing studies.
In summary, we have identified key pro-apoptotic vs. anti-apoptotic signaling cascades provoked by ALA/light stress in COH-BR1 cells and how iNOS/NO upregulation is linked to the latter. At least two other cancer lines, breast MDA-MB-231 and prostate PC-3 (not shown) have exhibited similar NO-dependent resistance to photokilling, suggesting that this is a general phenomenon and that the rational use of iNOS inhibitors may improve clinical PDT efficacy. These studies, along with our previous ones (13, 14), are the first to demonstrate that NO-mediated resistance to PDT stress can be induced in tumor cells per se and could have important implications on other cancer therapies based on oxidative stress.
Supplementary Material
Highlights
5-Aminolevulinic acid (ALA)-induced protoporphyrin IX in mitochondria sensitizes tumor cells to apoptotic photokilling, which is suppressed by stress-upregulated iNOS and NO.
Investigation of underling signaling activity revealed that stress NO positively effects survivin induction/activation, but negatively effects pro-apoptotic JNK and p38α activation and pro-survival p38β deactivation.
These results provide insights into how tumor cell NO can antagonize photodynamic therapy.
Acknowledgements
This work was supported by USPHS Grant CA70823 from the National Cancer Institute (to A.W.G.) and by a grant from the Wisconsin Breast Cancer Showhouse-Medical College of Wisconsin Cancer Center (to A.W.G.). The authors thank Richard G. Knowles of GlaxoSmithKline LLC for arranging to have a sample of iNOS inhibitor GW274150 sent to us via material transfer agreement. We also thank Neil Hogg and Witek Korytowski for helpful suggestions during the course of this work, and Jared Schmitt for expert technical assistance.
Research support is from two existing grants to AWG: NIH/NCI and WBCS/MCW Cancer Center
Abbreviations
- ALA
5-aminolevulinic acid
- Akt
protein kinase-B
- 8-Br-cGMP
8-bromoguanosine 3',5'-cyclic monophosphate
- Ho
Hoechst-33258
- iNOS
inducible nitric oxide synthase
- IκB
inhibitor of NF-κB
- Iκκ
IκB kinase
- JNK
c-Jun-N-terminal kinase
- 1400W
N-[3-(aminomethyl)benzyl]acetamidine
- MAPK
mitogen-activated protein kinase
- MTT
3-(4,5-dimethylthiazolyl-2-yl)-2-5-diphenyltetrazolium bromide
- NF-κB
nuclear factor-kappa B
- ODQ
1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one
- PI
propidium iodide
- PDPK1
3-phosphoinositide-dependent protein kinase-1
- PDT
photodynamic therapy
- PI3K
phosphoinositide-3-kinase
- PpIX
protoporphyrin IX
- p38
p38 mitogen-activated protein kinase
- sGC
soluble guanylyl cyclase
- SPNO
spermine-NONOate
- Suv
survivin
- Wo
Wortmannin
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Moncada SR, Palmer RMJ, Higgs EA. Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacol Rev. 1991;43:109–43. [PubMed] [Google Scholar]
- 2.Wink DA, Mitchell JB. Chemical biology of nitric oxide: insights into the regulatory, cytotoxic, and cytoprotective mechanisms of nitric oxide. Free Radic Biol Med. 1998;25:434–56. doi: 10.1016/s0891-5849(98)00092-6. [DOI] [PubMed] [Google Scholar]
- 3.Wink DA, Hines HB, Cheng RYS, Switzer CH, Flores-Santana W, Vitek MP, et al. Nitric oxide redox mechanisms in the immune response. J Leukoc Biol. 2011;89:873–91. doi: 10.1189/jlb.1010550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim Y-M, deVera ME, Watkins SC, Billiar TR. Nitric oxide protects cultured rat hepatocytes from tumor necrosis factor-alpha-induced apoptotia by inducing heat shock protein-70. J Biol Chem. 1997;272:1402–11. doi: 10.1074/jbc.272.2.1402. [DOI] [PubMed] [Google Scholar]
- 5.Kim Y-M, Talanian RV, Billiar TR. Nitric oxide inhibits apoptosis by preventing increases in caspase-3-like activity via two distinct mechanisms. J Biol Chem. 1997;272:31138–48. doi: 10.1074/jbc.272.49.31138. [DOI] [PubMed] [Google Scholar]
- 6.Kim Y-M, Chung H-T, Kim S-S, Han J-A, Yoo Y-M, Kim K-M, et al. Nitric oxide protects PC-12 cells from serum deprivation-induced apoptosis by cGMP-dependent inhibition of caspase signaling. J.Neurosci. 1999;19:6740–47. doi: 10.1523/JNEUROSCI.19-16-06740.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weller R, Schwentker A, Billiar TR, Vodovotz Y. Autologous nitric oxide protects mouse and human keratinocytes from ultraviolet B radiation-induced apoptosis. Am J Physiol Cell Physiol. 2003;284:C1140–48. doi: 10.1152/ajpcell.00462.2002. [DOI] [PubMed] [Google Scholar]
- 8.Fukumura D, Kashiwagi S, Jain RK. The role of nitric oxide in tumor progression. Nat Rev Cancer. 2006;6:521–34. doi: 10.1038/nrc1910. [DOI] [PubMed] [Google Scholar]
- 9.Wink DA, Ridnour LA, Hussain SP, Harris CC. The reemergence of nitric oxide and cancer. Nitric Oxide. 2008;19:65–7. doi: 10.1016/j.niox.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Henderson BW, Sitnik-Busch TM, Vaughn LA. Potentiation of photodynamic therapy antitumor activity in mice by nitric oxide synthase inhibition is fluence rate-dependent. Photochem Photobiol. 1999;70:64–71. [PubMed] [Google Scholar]
- 11.Korbelik M, Parkins CS, Shibuya H, Cecic I, Stratford MRL, Chaplin DJ. Nitric oxide production by tumor tissue: impact on the response to photodynamic therapy. Br J Cancer. 2000;82:1835–43. doi: 10.1054/bjoc.2000.1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reeves KJ, Reed MW, Brown NJ. The role of nitric oxide in the treatment of tumors with aminolevulinic acid-induced photodynamic therapy. J Photochem Photobiol B. 2010;101:224–32. doi: 10.1016/j.jphotobiol.2010.07.007. [DOI] [PubMed] [Google Scholar]
- 13.Bhowmick R, Girotti AW. Cytoprotective induction of nitric oxide synthase in a cellular model of 5-aminolevulinic acid-based photodynamic therapy. Free Radic Biol Med. 2010;48:1296–01. doi: 10.1016/j.freeradbiomed.2010.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bhowmick R, Girotti AW. Rapid upregulation of cytoprotective nitric oxide in breast tumor cells subjected to a photodynamic therapy-like oxidative challenge. Photochem Photobiol. 2011;87:378–86. doi: 10.1111/j.1751-1097.2010.00877.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Esworthy RS, Baker MA, Chu F-F. Expression of selenium-dependent glutathione peroxidase in human breast tumor cell lines. Cancer Res. 1995;55:957–62. [PubMed] [Google Scholar]
- 16.Hurst R, Korytowski W, Kriska T, Esworthy RS, Chu F-F, Girotti AW. Hyperresistance to cholesterol hydroperoxide-induced peroxidative injury and apoptotic death in a tumor cell line that overexpresses glutathione peroxidase isotype-4. Free Radic Biol Med. 2001;31:1051–65. doi: 10.1016/s0891-5849(01)00685-2. [DOI] [PubMed] [Google Scholar]
- 17.Peng Q, Berg K, Moan J, Kongshaug M, Nesland JM. 5-Aminolevulinic acid-based photodynamic therapy; principles and experimental research. Photochem Photobiol. 1997;65:235–51. doi: 10.1111/j.1751-1097.1997.tb08549.x. [DOI] [PubMed] [Google Scholar]
- 18.van Engeland M, Nieland LJW, Ramaekers FC, Schutte B, Reutelingsperger CP. Annexin V-affinity asay: a review on an apoptosis detection system based on phosphatidylserine exposure. Cytometry. 1998;31:1–9. doi: 10.1002/(sici)1097-0320(19980101)31:1<1::aid-cyto1>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 19.Bhowmick R, Girotti AW. Signaling events in apoptotic photokilling of 5-aminolevulinic acid-treated tumor cells: inhibitory effects of nitric oxide. Free Radic Biol Med. 2009;47:731–40. doi: 10.1016/j.freeradbiomed.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bui NT, Livolsi A, Peyron J-F, Prehn JHM. Activation of nuclear factor B and bcl-x survival gene expression by nerve growth factor requires tyrosine phosphorylation of IκBα. J Cell Biol. 2001;152:753–63. doi: 10.1083/jcb.152.4.753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Abmayr SM, Yao T, Parmely T, Workman JL. Preparation of nuclear and cytoplasmic extracts from mammalian cells. Current Protocols in Molecular Biology. 2006:1–10. doi: 10.1002/0471142727.mb1201s75. [DOI] [PubMed] [Google Scholar]
- 22.Xie Q-W, Kashiwabara Y, Nathan C. Role of transcription factor NF-κB/Rel in induction of nitric oxide synthase. J Biol Chem. 1994;269:4705–8. [PubMed] [Google Scholar]
- 23.Lowenstein CJ, Padalko E. iNOS (NOS2) at a glance. J Cell Sci. 2004;117:2865–7. doi: 10.1242/jcs.01166. [DOI] [PubMed] [Google Scholar]
- 24.Baker RG, Hayden MS, Ghosh S. NF-κB, inflammation, and metabolic disease. Cell Metabolism. 2011;13:11–22. doi: 10.1016/j.cmet.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shen H-M, Tergaonkar V. NF-κB signaling in carcinogenesis and as a potential molecular target for cancer therapy. Apoptosis. 2009;14:348–63. doi: 10.1007/s10495-009-0315-0. [DOI] [PubMed] [Google Scholar]
- 26.Hofmann F, Feil R, Kleppisch T, Schlossmann J. Function of cGMP-dependent protein kinases as revealed by gene deletion. Physiol Rev. 2006;86:1–23. doi: 10.1152/physrev.00015.2005. [DOI] [PubMed] [Google Scholar]
- 27.Fraser M, Chan SL, Chan SS, Fiscus RR, Tsang BK. Regulation of p53 and suppression of apoptosis by the soluble guanylyl cyclase/cGMP pathway in human ovarian cancer cells. Oncogene. 2006;25:2203–12. doi: 10.1038/sj.onc.1209251. [DOI] [PubMed] [Google Scholar]
- 28.Gomes ER, Almeida RD, Carvalho AP, Duarte CB. Nitric oxide modulates tumor cell death induced by photodynamic therapy through a cGMP-dependent mechanism. Photochem Photobiol. 2002;76:423–30. doi: 10.1562/0031-8655(2002)076<0423:nomtcd>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 29.Lucas KA, Pitari GM, Kazerounian S, Ruiz-Stewart I, Park J, Schulz S, et al. Guanylyl cyclases and signaling by cyclic GMP. Pharmacol Rev. 2000;52:375–14. [PubMed] [Google Scholar]
- 30.Stephens L, Williams R, Hawkins P. Phosphoinositide 3-kinases as drug targets in cancer. Curr Opin Pharmacol. 2005;5:357–65. doi: 10.1016/j.coph.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 31.Keefer L, Nims RW, Davies KM, Wink DA. “NONOates” (1-substituted diazen-1-ium-1,2-diolates) as nitric oxide donors: convenient nitric oxide dosage forma. Methods Enzymol. 1996;268:281–93. doi: 10.1016/s0076-6879(96)68030-6. [DOI] [PubMed] [Google Scholar]
- 32.Wada T, Penninger JM. Mitogen-activated protein kinases in apoptosis regulation. Oncogene. 2004;23:2838–49. doi: 10.1038/sj.onc.1207556. [DOI] [PubMed] [Google Scholar]
- 33.Porras A, Zuluaga S, Black E, Valladares A, Alvarez A, Ambrosino C, et al. p38α Mitogen-activated protein kinase sensitizes cells to apoptosis induced by different stimuli. Mol Biol Cell. 2004;15:922–33. doi: 10.1091/mbc.E03-08-0592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fridman JS, Lowe SW. Control of apoptosis by p53. Oncogene. 2003;22:9030–40. doi: 10.1038/sj.onc.1207116. [DOI] [PubMed] [Google Scholar]
- 35.Altieri DC. Survivin, cancer networks and pathway-directed drug discovery. Nat. Rev. Cancer. 2008;8:61–70. doi: 10.1038/nrc2293. [DOI] [PubMed] [Google Scholar]
- 36.De Alba J, Clayton NM, Collins SD, Colthup P, Chessell I, Knowles RG. GW274150, a novel and highly selective inhibitor of the inducible isoform of nitric oxide synthase (iNOS), shows analgesic effects in rat models of inflammatory and neuropathic pain. Pain. 2006;120:170–81. doi: 10.1016/j.pain.2005.10.028. [DOI] [PubMed] [Google Scholar]
- 37.Singh D, Richards D, Knowles RG, Schwartz S, Woodcock A, Langley S, O'Connor BJ. Selective inducible nitric oxide synthase inhibition has no effect of allergen challenge in asthma. Am J Respir Crit Care Med. 2007;176:988–93. doi: 10.1164/rccm.200704-588OC. [DOI] [PubMed] [Google Scholar]
- 38.Alderton WK, Cooper CE, Knowles RG. Nitric oxide syhthases: structure, function and inhibition. Biochem J. 2001;357:593–15. doi: 10.1042/0264-6021:3570593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wink DA, Vodovotz Y, Laval J, Laval F, Dewhirst MW, Mitchell JB. The multifaceted roles of nitric oxide in cancer. Carcinogenesis. 1998;19:711–21. doi: 10.1093/carcin/19.5.711. [DOI] [PubMed] [Google Scholar]
- 40.Crowell JA, Steele VE, Sigman CC, Fay JR. Is inducible nitric oxide synthase a target for chemoprevention? Mol Cancer Ther. 2003;2:815–23. [PubMed] [Google Scholar]
- 41.Sikora AG, Gelbard A, Davies MA, Sano D, Ekmekcioglu S, Kwon J, et al. Targeted inhibition of inducible nitric oxide synthase inhibits growth of human melanoma in vivo and synergizes with chemotherapy. Clin Cancer Res. 2010;16:1834–44. doi: 10.1158/1078-0432.CCR-09-3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Eyler CE, Wu Q, Yan K, MacSwords JM, Chandler-Militello D, Misuraca KL, et al. Glioma stem cell proliferation and tumor growth are promoted by nitric oxide synthase 2. Cell. 2011;146:53–66. doi: 10.1016/j.cell.2011.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fingar VH, Henderson BW. Drug and light dose dependence of photodynamic therapy: a study of tumor and normal tissue response. Photochem Photobiol. 1987;46:837–41. doi: 10.1111/j.1751-1097.1987.tb04856.x. [DOI] [PubMed] [Google Scholar]
- 44.Jiang B-H, Liu L-Z. PI3K/PTEN signaling in tumorigenesis and angiogenesis. Biochim Biophys Acta. 2008;1784:150–8. doi: 10.1016/j.bbapap.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 45.Saura M, Zaragoza C, Bao C, McMillan A, Lowenstein CJ. Interaction of interferon regulatory factor-1 and nuclear factor kappa B during activation of inducible nitric oxide synthase transcription. J. Mol. Biol. 1999;289:459–71. doi: 10.1006/jmbi.1999.2752. [DOI] [PubMed] [Google Scholar]
- 46.Cho MK, Suh SH, Kim SG. Jun B/AP-1 and NF-κB-mediated induction of nitric oxide synthase by bovine type I collagen in serum-stimulated murine macrophages. Nitric Oxide. 2002;6:319–32. doi: 10.1006/niox.2001.0415. [DOI] [PubMed] [Google Scholar]
- 47.Gao J, Morrison DC, Parmely D, Russell SW, Murphy WJ. An interferon-gamma-activated site (GAS) is necessary for full expression of the mouse iNOS gene in response to interferon-gamma and lipoxygenase. J. Biol. Chem. 1997;272:1226–30. doi: 10.1074/jbc.272.2.1226. [DOI] [PubMed] [Google Scholar]
- 48.Ferrario A, Rucker N, Wong S, Luna M, Gomer CJ. Survivin, a member of the inhibitor of apoptosis family, is induced by photodynamic therapy and is a target for improving treatment response. Cancer Res. 2007;67:4989–95. doi: 10.1158/0008-5472.CAN-06-4785. [DOI] [PubMed] [Google Scholar]
- 49.Fetz V, Bier C, Habtemichael N, Schuon R, Schweitzer A, Kunkel M, et al. Inducible NO synthase confers chemoresistance in head and neck cancer by modulating survivin. Int J Cancer. 2009;124:2033–41. doi: 10.1002/ijc.24182. [DOI] [PubMed] [Google Scholar]
- 50.Mirza A, McGuirk M, Hockenberry TN, Wu Q, Ashar H, Black S, et al. Human survivin is negatively regulated by wild-type p53 and participates in p53-dependent apoptotic pathway. Oncogene. 2002;21:2613–22. doi: 10.1038/sj.onc.1205353. [DOI] [PubMed] [Google Scholar]
- 51.Niziolek M, Korytowski W, Girotti AW. Chain-breaking antioxidant and cytoprotective action of nitric oxide in photodynamically stressed tumor cells. Photochem Photobiol. 2003;78:262–270. doi: 10.1562/0031-8655(2003)078<0262:caacao>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 52.Park HS, Wu J-W, Cho JH, Kim M-S, Huh S-H, Ryoo K, et al. Inhibition of apoptosis signal-regulating kinase 1 by nitric oxide through a thiol redox mechanism. J Biol Chem. 2004;279:7484–90. doi: 10.1074/jbc.M304183200. [DOI] [PubMed] [Google Scholar]
- 53.Park HS, Huh S-H, Kim M-S, Lee S-H, Choi E-J. Nitric oxide negatively regulates c-Jun N-terminal kinast/stress activated protein by means of S-nitrosylation. Proc Natl Acad Sci USA. 2000;97:14382–87. doi: 10.1073/pnas.97.26.14382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li C-Q, Wogan GN. Nitric oxide as a modulator of apoptosis. Cancer Lett. 2005;226:1–15. doi: 10.1016/j.canlet.2004.10.021. [DOI] [PubMed] [Google Scholar]
- 55.Azad N, Vallyathan V, Wang L, Tantishaiyakul V, Stehlik C, Leonard SS, Rojanasakul Y. S-nitrosylation of Bcl-2 inhibits its ubiquitin-proteosomal degradation. J. Biol. Chem. 2006;281:34124–34. doi: 10.1074/jbc.M602551200. [DOI] [PubMed] [Google Scholar]
- 56.Kwak Y-D, Ma T, Diao S, Zhang X, Chen Y, Hsu J, et al. NO signaling and S-nitrosylation regulate PTEN inhibition in neurodegeneration. Mol Neurodegener. 2010;5:49–60. doi: 10.1186/1750-1326-5-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.