

Abstract
Protein tyrosine phosphatases (PTPs) are key signal-transduction regulators and have emerged as potential drug targets for inhibitor design. Here we report a yeast-based assay that provides a general means of assessing the activity and/or inhibition of essentially any classical PTP in living cells. The assay uses the activity of an exogenously expressed PTP to counter the activity of a co-expressed and toxic tyrosine kinase, such that only active PTPs are capable of rescuing growth. PTP activity gives rise to both increased growth and decreased phosphotyrosine levels; cellular PTP activity can therefore be monitored by either yeast-growth curves or anti-phosphotyrosine western blots. We show that four PTPs (TCPTP, Shp2, PEST, PTPα) are capable of rescuing the effects of v-Src toxicity. Since these PTPs are chosen from four distinct sub-families, it is likely that biologically and medicinally important PTPs from other subfamilies can similarly function in the cellular PTP assay. Because many small-molecule PTP inhibitors fail to penetrate cell membranes effectively, this cell-based assay has the potential to serve as a useful screening tool for determining the cellular efficacy of candidate inhibitors in a more biologically relevant context than can be provided by an in vitro PTP assay.
Keywords: protein tyrosine phosphatases (PTPs), yeast, cell-based assay, TCPTP, Shp2, PEST, PTPα
Introduction
Tyrosine phosphorylation is one of the central post-translational control elements in metazoan signal transduction. The phosphorylation state of a given protein can govern its enzyme activity, protein-protein interactions, and cellular distribution. Phosphorylation and dephosphorylation of a target protein thus act as a “chemical switch” that allows the cell to transmit signals in a highly regulated manner. Tyrosine phosphorylation is enzymatically controlled by two large families of enzymes, the protein tyrosine kinases (PTKs) and protein tyrosine phosphatases (PTPs) [1]. Here we present a general cell-based assay that can be used to monitor the activities of PTPs in living cells.
Selective PTP inhibitors are sought after as drug candidates due to the common association of misregulated PTP activity and human disease [2–3]. Unfortunately, despite decades of efforts, relatively few highly specific and cell-permeable PTP inhibitors have been discovered. PTP-inhibitor discovery is inherently difficult due to two recurring problems: (i) lack of target specificity: PTP catalytic domains share a significant degree of sequence and structural homology with one another; and (ii) poor bioavailability: at physiological pH, most of the known PTP-binding pharmacophores contain negatively charged pTyr mimetics that lower an inhibitor’s cellular permeability. While the former challenge (conserved active sites) is one that is shared by a number of large protein families, the latter (cell-impermeability conferred by effective pharmacophores) is particular to PTPs and has contributed to the perception that PTPs represent potentially “undruggable” targets [3].
Because cell permeability is such a central challenge in PTP-inhibitor discovery, cell-based assays are critical for assessing the usefulness of any compounds that show inhibitory activity in vitro; ideally, cell-based assays would be available for every member of the PTP family. However, cellular assays that can provide direct readout of specific PTP-inhibition events are very difficult to develop because of the large number of PTPs, all of which catalyze the same biochemical reaction. This challenge is compounded by the fact that different PTPs can have overlapping expression and localization patterns, as well as overlapping pools of substrates [4–5]. Even in the case of very well studied PTP targets for which specific substrates have been identified, it is difficult to know whether an inhibitor-induced cellular event (e.g., increase in the phosphorylation level of a putative substrate upon addition of a compound to a cell culture) is due directly to inhibition of the intended target. With these challenges in mind we have set out to develop a general, easily accessible system that can be used for testing the activity of any PTP in a cellular context.
To generate such an assay, we chose a eukaryotic model system: the budding yeast, Saccharomyces cerevisiae. We chose S. cerevisiae because it has previously been shown that this organism can be genetically engineered such that its growth rate is dependent on the activity of an exogenous PTP. Overexpression of the tyrosine kinase v-Src in yeast leads to inhibition of cell growth [6–7], and previous work has shown that simultaneous expression of one particular PTP, human protein tyrosine phosphatase 1B (PTP1B), “rescues” yeast growth and decreases cellular phosphotyrosine levels [8]. It has also been shown that the resulting v-Src- and PTP1B-expressing yeast can be used to screen for PTP1B inhibitors, as inhibition of PTP1B leads to a reversal of the rescued phenotype—i.e., a suppression of cell growth (Figure 1A) [9–10].
Figure 1.

Using the budding yeast S. cerevisiae as a general cellular system for assaying PTP activity. (A) Previous work has shown that expression of the tyrosine kinase v-Src disrupts yeast growth (illustrated by the “Xed-out” yeast), and that co-expression of PTP1B rescues yeast growth, allowing for the cellular screening of PTP1B inhibitors [9]. (B) Potential generalizability of PTP-mediated growth rescue. As there is no physiological connection between PTP1B and v-Src, it is possible that any enzyme capable of dephosphorylating tyrosine—signified as “PTPx”—could counter v-Src activity and rescue yeast growth. If so, yeast could be used to assay cellular activities and/or inhibitors of any number of PTPs.
Is the phosphatase-mediated v-Src-countering effect described above specific to PTP1B? As there is no obvious inherent physiological connection between v-Src and PTP1B—any tyrosine-phosphatase activity could, in principle, oppose the toxic v-Src activity—we hypothesized that any number of active PTPs should be able to rescue the growth of v-Src-expressing yeast and offset v-Src-induced phosphotyrosine accumulation. If this hypothesis proves correct, then a yeast strain that co-expresses v-Src and any PTP of interest could provide an indicator of cellular PTP activity that can be monitored either by cell-growth assays or by anti-phosphotyrosine Western blots. Potentially, such an approach could be used to readily develop a cellular activity assay for any enzyme that is capable of dephosphorylating phosphotyrosine (Figure 1B).
Material and Methods
General Information
See Supplementary Material for all primer sequences used in cloning and mutagenesis, for a comprehensive list of plasmids generated for this study, and for the constitutions of buffers used in the following protocols.
Cloning and Mutagenesis
A gene encoding v-Src was cloned into p415GALL as described previously [9]. For PTP expression, a derivative of p426GAL1, pSMF043, that encodes a C-terminal six-histidine tag downstream of the multiple-cloning site was generated by inverse PCR, according to published procedures [11]. Inserts encoding the catalytic domains of human TCPTP, human Lyp, human PEST, human PTPα, human PTPH1, and mouse Shp2 were then cloned into pSMF043 as follows. PTP-encoding genes were amplified and PCR products and pSMF043 vector DNA were restriction digested and gel purified. Ligation reactions containing 400 U T4 DNA ligase (New England Biolabs), 2 μL 10×ligase buffer (New England Biolabs), ~10 ng of insert, and ~10 ng of vector in a 20 μL reaction were placed in an ice bucket overnight. Ligation products were transformed into competent DH5α E. coli and plated on LB/Agar containing 100 μg/mL ampicillin. Ampicillin-resistant colonies were isolated, and the presence of the desired inserts was confirmed by restriction analysis. An insert encoding the catalytic domain of human PTP1B was cloned into p426GAL1 essentially as described previously [9], and a C-terminal six-histidine-tag was added by Quikchange (Stratagene) insertional mutagenesis, according to the manufacturer’s instructions. “Phosphatase-dead” cysteine-to-serine mutations were introduced using Quikchange site-directed mutagenesis. The sequences of all PTP-encoding inserts were confirmed by DNA sequencing (Cornell University Life Sciences Core Laboratories Center).
Yeast Protocols
The yeast strain YPH499 (MATa ura3-52 lys2-80amber ade2-101ochre trp-Δ63 his3-Δ200 leu2-Δ1) was purchased from ATCC. Yeast transformations were carried out as described [12]. Yeast growth assays were carried out essentially as described with minor modifications [9]. Briefly, overnight 5 mL cultures of the relevant strains in 2% raffinose uracil/leucine drop-out media were grown on a rotating wheel at 30 °C. After normalization of cell concentrations, 10 μL of the cultures were added to 180 μL 4% galactose uracil/leucine drop-out media in the well of a round-bottomed 96-well plate. Two drops of paraffin oil were then added to the top of each well using a transfer pipette. The plate was then loaded into a BioTek® PowerWave 340 microplate spectrophotometer in which the OD600 was measured every five hours at 30 °C, with constant shaking between readings. Data plotted in Figures 2, 3, and 5 represent the mean OD600 of three independent wells for each strain; error bars represent the associated standard deviations.
Figure 2.

Rescue of v-Src toxicity is not specific to PTP1B. Yeast strains expressing v-Src alone (○), v-Src and PTP1B (●), or v-Src and TCPTP (▼) were grown in a 96-well plate at 30 °C. OD600 readings for the cultures were measured every five hours.
Figure 3.

Rescue of v-Src toxicity by divergent PTPs. Yeast strains expressing v-Src alone (○), v-Src and PTP1B (●), or v-Src and a test PTP (▼; Panel A: Shp2, B: Lyp, C: PEST, D: PTPH1, E: PTPα) were grown in a 96-well plate at 30 °C. OD600 readings for the cultures were measured every five hours.
Figure 5.
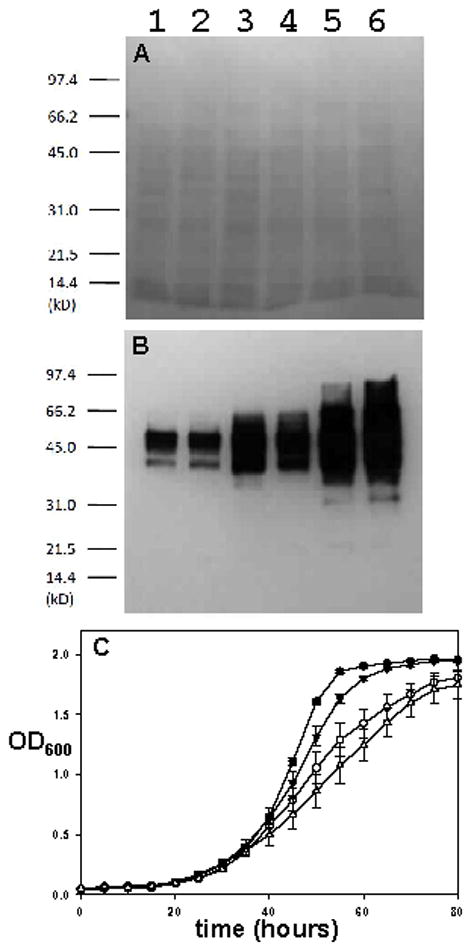
PTPs that counter v-Src toxicity reduce cellular phosphotyrosine levels. (A & B) Proteins from yeast lysates expressing v-Src with PTP1B (Lane 1), TCPTP (Lane 2), PTPα (Lane 3), PEST (Lane 4), Shp2 (Lane 5), or no PTP (Lane 6) were separated by SDS-PAGE and transferred to nitrocellulose. The resulting membrane was stained with Ponceau S (A), washed, and immunoblotted with an anti-phosphotyrosine antibody (B). (C) Yeast strains co-expressing v-Src and TCPTP (●), PTPα (○), PEST (▼), or Shp2 (△) were grown in a 96-well plate at 30 °C. OD600 readings for the cultures were measured every five hours
Western Blots
Cultures (5 mL) in 2% raffinose uracil/leucine drop-out media were inoculated with the relevant yeast strains and grown at 30 °C until the cells had reached log-phase growth (OD600 of 0.4–0.6, approximately 10–12 hours), at which point galactose (3.3%) was added. After a 20-hour induction, samples of total cellular protein from approximately 2×107 cells were prepared for electrophoresis as described [13]. Proteins were separated by SDS-PAGE and transferred to nitrocellulose. The nitrocellulose membrane was stained with Ponceau S, photographed and washed with TBS.
Anti-5-Histidine Westerns
Nitrocellulose membranes were incubated for 1 hour at room temperature in 27 mL 5-His Blocking Solution, washed two times (10 minutes) with TBSTT, and washed once (10 minutes) with TBS. 5-His Blocking Solution containing 5 μL primary antibody (His·Tag® Monoclonal Antibody, 0.2 mg/mL, Novagen) was then added. Membranes were subsequently incubated overnight with rocking at 4 °C, washed twice (10 minutes) with TBSTT, once (10 minutes) with TBS, and incubated for 1 hour at room temperature in 8 mL 5-His Blocking Solution with 1.6 μL secondary antibody (goat anti-mouse IgG, HRP conjugate, 1 mg/mL, Millipore). After antibody treatment, membranes were washed 5 times (10 minutes) with TBSTT and exposed to film, which was developed using standard procedures.
Anti-Phosphotyrosine Westerns
Membranes were rocked in pY Blocking Solution at room temperature for 4 hours, after which 15 mL of pY Blocking Solution containing 10 μL primary antibody (anti-phosphotyrosine antibody, clone 4G10—IgG2bκ mouse monoclonal antibody—1 mg/mL, Millipore) was added. After overnight antibody treatment at 4 °C, membranes were washed three times (10 minutes) with pY Blocking Solution and 15 mL pY Blocking Solution with 10 μL secondary antibody (goat anti-mouse IgG, HRP conjugate, 1 mg/mL, Millipore) was added. The membranes were rocked for 1 hour at room temperature and then washed three times (5 minutes) with PBS containing 0.05% Tween®-20. Membranes were then exposed to film, which was developed using standard procedures.
Results and Discussion
Cellular readout of T-cell protein tyrosine phosphatase (TCPTP) activity
As noted above, it has been shown previously that overexpression of the tyrosine kinase v-Src in yeast leads to inhibition of cell growth, and that co-expression of the phosphatase PTP1B rescues yeast growth and decreases cellular phosphotyrosine levels [8–9]. The central assumption of developing a general yeast assay for cellular PTP activity is that the previously demonstrated PTP-induced growth rescue is not specific to PTP1B, and that other PTPs could function similarly in countering v-Src’s activity. As a first test of this idea we chose T-cell protein tyrosine phosphatase (TCPTP). TCPTP is PTP1B’s closest homologue; the two enzymes share a very high degree of homology in their PTP domains (69% identity), and they have almost identical active sites [14]. However, PTP1B and TCPTP have distinct biological functions—most dramatically shown by their completely different knockout phenotypes [15–16]—and it is by no means certain that TCPTP could functionally replace PTP1B in yeast cells that express v-Src. To test whether TCPTP could rescue yeast growth we cloned a gene encoding the TCPTP catalytic domain into a modified p426GAL1 vector and co-expressed the PTP with v-Src in living yeast cells. (All PTP domains expressed in this study were six-histidine-tagged to aid in detection and/or purification of the expressed proteins; see Material and Methods.) We found that TCPTP could indeed rescue v-Src-induced cellular toxicity. As shown in Figure 2, the TCPTP catalytic domain rescues the growth of yeast that express v-Src at a level that is comparable, albeit slightly reduced, to the rescue conferred by PTP1B. This is the first demonstration that a PTP other than PTP1B is capable of rescuing the growth inhibition caused by v-Src. More importantly, the data in Figure 2 show that the “central assumption” outlined above is at least partially correct, and that yeast-growth rescue can potentially be used to assay the cellular activity for any number of different PTPs.
Cellular readout for the activities of divergent PTPs
While the ability of TCPTP to rescue v-Src-induced toxicity is promising, it may not be terribly surprising, given the aforementioned high degree of homology between the enzymes. To more broadly test the prospect of using yeast as a cellular system for testing PTP activity, we selected five additional biologically important PTPs from four sub-families that are distinct from the PTP1B/TCPTP subfamily (NT1) and are well spread across the family of both receptor-like (R) and non-transmembrane (NT) classical PTPs [14]: Shp2, PEST, Lyp, PTPH1, and PTPα. The gene that encodes Shp2 (subtype NT2, 39% PTP-domain identity with PTP1B) has been confirmed as the first bona fide PTP proto-oncogene, as germline SHP2 mutations cause Noonan and Leopard syndromes, both of which can lead to cancer predisposition [17]. PEST (subtype NT4, 36% PTP-domain identity with PTP1B) is an ubiquitously expressed signaling molecule, which is involved in actin-cytoskeleton regulation and secondary T-cell response [18–19]. Lyp (subtype NT4, 38% PTP-domain identity with PTP1B), has been shown to be a negative regulator of T-cell activation, and gain-of-function Lyp mutations have been associated with several autoimmune diseases [20–21]. PTPH1 (subtype NT5, 36% PTP-domain identity with PTP1B) acts on the mitogen-activated protein kinase p38γ and has been found to be mutated in human colorectal cancers [22–23]. And, PTPα (subtype R4, 36% PTP-domain identity with PTP1B) is a receptor PTP that dephosphorylates and activates Src-family kinases, thereby regulating a variety of downstream pathways [24].
To test whether these five PTPs, all of which are more distantly related to PTP1B than is TCPTP, could rescue yeast growth, we cloned the genes encoding each PTP’s catalytic domain and co-expressed each PTP with v-Src in yeast. The results for all five PTPs are shown in Figure 3 and can be summarized as follows: one PTP catalytic domain, PEST (Figure 3C), was capable of rescuing yeast growth at a level that was indistinguishable from that of PTP1B; two catalytic domains, Shp2 (Figure 3A) and PTPα (Figure 3E), rescued growth, albeit at a somewhat less robust level than that observed for PTP1B; two PTPs, Lyp (Figure 3B) and PTPH1 (Figure 3D) did not rescue yeast growth. (The apparent slight increase in growth rate shown for Lyp in Figure 3B, as compared to v-Src-only, was not observed reproducibly, and the growth rate of v-Src-only strains showed some variability from experiment to experiment within a range that encompasses the slightly increased rate in Figure 3B.)
It is not immediately apparent why PTPH1 and Lyp failed to functionally replace PTP1B. The failure is particularly curious for Lyp, given that PEST and Lyp are close homologs (64% catalytic domain identity) [14], and PEST’s catalytic domain conferred robust cell growth, comparable to that of PTP1B. To investigate whether the inability of PTPH1 and Lyp was due to an inherent inability to dephosphorylate v-Src’s cellular targets, or, alternatively, due to poor expression of these particular PTPs, we performed anti-five-histidine western blots on lysates from cultures that express the His-tagged PTPs, with PTP1B as a positive control for robust PTP expression (Figure 4). His-tagged proteins were detected, at varying levels, in all of the lysates, with the exception of the negative control (v-Src only), PTPH1, and Lyp (Figure 4B). These results suggest that the failure of PTPH1 and Lyp to increase the rate of yeast growth was due to an expression defect in the particular constructs made for PTPH1 and Lyp expression. All of the PTPs that gave detectable expression (PTP1B, PTPα, PEST, and Shp2) augmented yeast growth, suggesting that other PTPs (from these subfamilies or others) could also rescue, once a suitable expression construct is found. (The constructs for both PTPH1 and Lyp express only catalytic-domain-containing fragments of the proteins and it is possible that the resulting fragments were not stably folded in the yeast cytoplasm; attempts to find better protein “cutting points” for yeast expression were not undertaken.) Interestingly, no direct correlation was observed between the level of expression and the strength of the growth rescue, as PTPα was expressed at higher levels than PEST, but PEST conferred faster growth.
Figure 4.
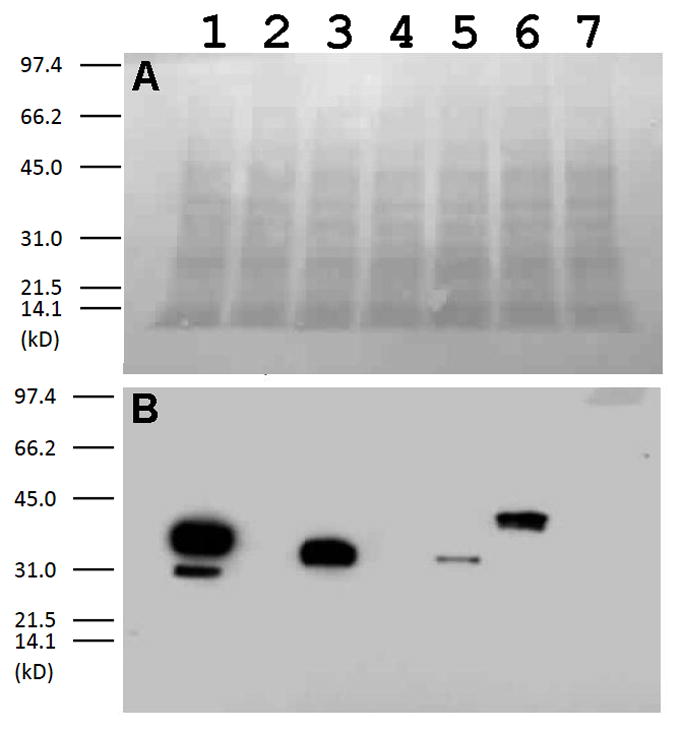
Six-histidine-tagged PTPs that fail to counter v-Src toxicity also do not express. Proteins from yeast lysates expressing v-Src with PTP1B-His6 (Lane 1), PTPH1-His6 (Lane 2), PTPα-His6 (Lane 3), Lyp-His6 (Lane 4), PEST-His6 (Lane 5), Shp2-His6 (Lane 6), or no PTP (Lane 7) were separated by SDS-PAGE and transferred to nitrocellulose. The resulting membrane was stained with Ponceau S (A), washed, and immunoblotted with an anti-five- histidine antibody (B).
Correlation of growth rescue and reduction of cellular phosphotyrosine content
Wild-type S. cerevisiae strains contain little to no detectable phosphotyrosine, whereas yeast cells that exogenously express the tyrosine kinase v-Src contain many tyrosine-phosphorylated proteins, some subset of which are presumably responsible for the v-Src-induced toxicity [6]. Co-expression of PTP1B reduces the cellular level of phosphotyrosine markedly, as compared to a strain that expresses v-Src alone [8–9]. To investigate whether the PTP domains that were found to rescue growth in the current study were exerting phosphatase activity at the molecular level, we used Western blots to analyze the phosphotyrosine levels of lysates from cells whose growth had been rescued by the PTPs TCPTP, PTPα, PEST, and Shp2. (Having confirmed that the Lyp and PTPH1 constructs did not express efficiently, these PTPs were not investigated further.)
As shown in Figure 5, all four PTPs that were found to rescue growth also reduced the amount of phosphotyrosine in the cells, compared to a v-Src only strain (Figure 5B, Lane 6). The degree of decrease in phosphotyrosine levels varied widely, however: Shp2 expression (Lane 5) reduced phosphotyrosine levels only slightly; PTPα (Lane 4) gave a somewhat more robust decrease; PEST (Lane 3) decreased phosphotyrosine levels further still; and the phosphotyrosine content of TCPTP-expressing yeast (Lane 2) was essentially indistinguishable from that of a strain expressing PTP1B (Lane 1). Side-by-side comparison of growth-assay (Figure 5C) and phosphotyrosine (Figure 5B) data derived from the identical growth experiment revealed a direct correlation between a PTP domain’s ability to reduce phosphotyrosine levels and the degree of growth-rate increase. In both analyses, the maximum degree of yeast “normalcy” (low phosphotyrosine, robust growth) was conferred by TCPTP, followed by PEST, PTPα, and Shp2, respectively. The observed correlation between phosphotyrosine decrease and growth-rate increase strongly suggest that overall PTP activity—and not the substrate specificity of the particular PTP chosen—is the major determinant of growth rescue. (Note that in the comparisons of Figures 2 and 3C, PEST appears to confer slightly stronger growth than TCPTP. Fresh transformants were prepared for each separate experiment [9] and the Figure 2/3 data derived from different transformants of ostensibly identical strains as compared to the Figure 5 experiments. We observed minor experiment-to-experiment variability in both PTP expression levels and relative growth rescue, so no firm conclusions about which PTPs are the “best” rescuers should be drawn.)
Phosphatase-dead mutants
To further ensure that the ability of the assayed PTPs to rescue yeast growth stems directly from their respective PTP activities, we generated “phosphatase-dead” (PD) mutants for the rescuing PTPs. These mutants each contain a serine residue at the position of the conserved PTP catalytic cysteine residue and therefore possess little to no phosphatase activity [25]. (The PD mutants are: C215S PTP1B, C216S TCPTP, C459S Shp2, C231S PEST, and C433S PTPα; the numbers of the mutation sites vary widely because full-length PTPs often contain protein domains other than their eponymous PTP domains [4].) We co-expressed the PD mutants with v-Src and tested their effectiveness at rescuing yeast growth, as compared to the corresponding wild-type PTPs (Figure 6). In agreement with previous results, PD PTP1B fails to give robust rescue of v-Src-expressing yeast growth (Figure 6A). As shown in Figures 6B and 6D, PD TCPTP and PD PEST yield results that are essentially identical to those of PD PTP1B, with the PD PTP-expressing strains growing at rates that are much lower than those of the corresponding wild-type-PTP-expressing strains. Interestingly, PD PTP1B, PD TCPTP, and PD PEST all grew at slightly higher rates than the v-Src-only (no PTP) control. It is possible that the observed increases in growth are due to low-level “leaky” residual activities of these mutants. Alternatively, since PD mutants are known to “trap” (stably bind, but not effectively dephosphorylate) their cellular substrates [25], it is possible that the trapping effect partially sequesters the cellular pools of inappropriately tyrosine-phosphorylated proteins, thereby mitigating their toxic effects.
Figure 6.

Robust rescue of v-Src toxicity is dependent on PTP activity. Yeast strains expressing v-Src alone (○), v-Src and a wild-type PTP domain (●; A: PTP1B, B: TCPTP, C: Shp2, D: PEST, E: PTPα), or v-Src and the corresponding phosphatase-dead mutant (▼; A: C215S PTP1B, B: C216S TCPTP, C: C459S Shp2, D: C231S PEST, E: C433S PTPα) were grown in a 96-well plate at 30 °C. OD600 readings for the cultures were measured every five hours
For Shp2 (Figure 6C) and PTPα (6E), the PD PTPs confer no growth-rate increase at all, as compared to the v-Src-only strain, an observation that is consistent with the hypothesis that the ability of the corresponding wild-type enzymes to augment growth is directly due to their PTP activities. Taken together, the PD-mutant data support the assumption that the general phenomenon of PTP-mediated growth rescue is directly due to the PTP activity of the expressed enzyme and, as observed with anti-phosphotyrosine blots (Figure 5), that the degree of rescue correlates with the level of PTP activity. For each catalytic domain expressed, abrogation of PTP activity through mutation of a single cysteine-to-serine mutation dramatically reduced the ability of the expressed proteins to augment the growth of v-Src-expressing yeast.
Conclusion
We have shown that PTPs from four different subfamilies (TCPTP, Shp2, PEST, and PTPα) are capable of rescuing the effects of v-Src toxicity in living yeast cells, providing cell-based assays for the activities of these divergent PTPs. Rescue was observed for all detectably expressed PTPs, suggesting that PTPs not investigated in the current study will also function in directly analogous yeast-based PTP assays. A direct correlation between the strength of rescue and cellular PTP activity, as measured by cellular phosphotyrosine levels, was observed, providing evidence that the cell-based assay has the potential to serve as a biologically relevant screening tool for determining the cellular efficacy of candidate TCPTP, Shp2, PEST, and PTPα inhibitors.
Supplementary Material
Acknowledgments
Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number R15GM071388. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The authors also gratefully acknowledge the Henry Dreyfus Teacher-Scholar Awards Program (A.C.B.), the Arnold and Mabel Beckman Foundation (S.M.F.), the Howard Hughes Medical Institute (L.K.H.), and Amherst College for funding, as well as Dr. Xin-Yu Zhang for cloning of the v-Src-expression vector used in the study.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hunter T. Protein kinases and phosphatases: the yin and yang of protein phosphorylation and signaling. Cell. 1995;80:225–236. doi: 10.1016/0092-8674(95)90405-0. [DOI] [PubMed] [Google Scholar]
- 2.Barr AJ. Protein tyrosine phosphatases as drug targets: strategies and challenges of inhibitor development. Future Med Chem. 2010;2:1563–1576. doi: 10.4155/fmc.10.241. [DOI] [PubMed] [Google Scholar]
- 3.Blaskovich MA. Drug discovery and protein tyrosine phosphatases. Curr Med Chem. 2009;16:2095–2176. doi: 10.2174/092986709788612693. [DOI] [PubMed] [Google Scholar]
- 4.Wang WQ, Sun JP, Zhang ZY. An overview of the protein tyrosine phosphatase superfamily. Curr Top Med Chem. 2003;3:739–748. doi: 10.2174/1568026033452302. [DOI] [PubMed] [Google Scholar]
- 5.Tabernero L, Aricescu AR, Jones EY, Szedlacsek SE. Protein tyrosine phosphatases: structure-function relationships. FEBS J. 2008;275:867–882. doi: 10.1111/j.1742-4658.2008.06251.x. [DOI] [PubMed] [Google Scholar]
- 6.Brugge JS, Jarosik G, Andersen J, Queral-Lustig A, Fedor-Chaiken M, Broach JR. Expression of rous sarcoma virus transforming protein pp60 v-src in Saccharomyces cerevisiae cells. Mol Cell Biol. 1987;7:2180–2187. doi: 10.1128/mcb.7.6.2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kornbluth S, Jove R, Hanafusa H. Characterization of avian and viral p60src proteins expressed in yeast. Proc Natl Acad Sci USA. 1987;84:4455–4459. doi: 10.1073/pnas.84.13.4455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Florio M, Wilson LK, Trager JB, Thorner J, Martin GS. Aberrant protein-phosphorylation at tyrosine is responsible for the growth-inhibitory action of pp60(v-Src) expressed in the yeast Saccharomyces cerevisiae. Mol Biol Cell. 1994;5:283–296. doi: 10.1091/mbc.5.3.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Montalibet J, Kennedy BP. Using yeast to screen for inhibitors of protein tyrosine phosphatase 1B. Biochem Pharmacol. 2004;68:1807–1814. doi: 10.1016/j.bcp.2004.06.024. [DOI] [PubMed] [Google Scholar]
- 10.Montalibet J, Skorey K, McKay D, Scapin G, Asante-Appiah E, Kennedy BP. Residues distant from the active site influence protein-tyrosine phosphatase 1B inhibitor binding. J Biol Chem. 2006;281:5258–5266. doi: 10.1074/jbc.M511546200. [DOI] [PubMed] [Google Scholar]
- 11.Hemsley A, Arnheim N, Toney MD, Cortopassi G, Galas DJ. A simple method for site-directed mutagenesis using the polymerase chain reaction. Nucleic Acids Res. 1989;17:6545–6551. doi: 10.1093/nar/17.16.6545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gietz RD, Woods RA. Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Methods Enzymol. 2002;350:87–96. doi: 10.1016/s0076-6879(02)50957-5. [DOI] [PubMed] [Google Scholar]
- 13.Kushnirov VV. Rapid and reliable protein extraction from yeast. Yeast. 2000;16:857–860. doi: 10.1002/1097-0061(20000630)16:9<857::AID-YEA561>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 14.Andersen JN, Mortensen OH, Peters GH, Drake PG, Iversen LF, Olsen OH, Jansen PG, Andersen HS, Tonks NK, Moller NP. Structural and evolutionary relationships among protein tyrosine phosphatase domains. Mol Cell Biol. 2001;21:7117–7136. doi: 10.1128/MCB.21.21.7117-7136.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Elchebly M, Payette P, Michaliszyn E, Cromlish W, Collins S, Loy AL, Normandin D, Cheng A, Himms-Hagen J, Chan CC, Ramachandran C, Gresser MJ, Tremblay ML, Kennedy BP. Increased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatase-1B gene. Science. 1999;283:1544–1548. doi: 10.1126/science.283.5407.1544. [DOI] [PubMed] [Google Scholar]
- 16.You-Ten KE, Muise ES, Itie A, Michaliszyn E, Wagner J, Jothy S, Lapp WS, Tremblay ML. Impaired bone marrow microenvironment and immune function in T cell protein tyrosine phosphatase-deficient mice. J Exp Med. 1997;186:683–693. doi: 10.1084/jem.186.5.683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chan G, Kalaitzidis D, Neel BG. The tyrosine phosphatase Shp2 (PTPN11) in cancer. Cancer Metast Rev. 2008;27:179–192. doi: 10.1007/s10555-008-9126-y. [DOI] [PubMed] [Google Scholar]
- 18.Davidson D, Shi X, Zhong MC, Rhee I, Veillette A. The phosphatase PTP-PEST promotes secondary T cell responses by dephosphorylating the protein tyrosine kinase Pyk2. Immunity. 2010;33:167–180. doi: 10.1016/j.immuni.2010.08.001. [DOI] [PubMed] [Google Scholar]
- 19.Côté JF, Chung PL, Théberge JF, Hallé M, Spencer S, Lasky LA, Tremblay ML. PSTPIP is a substrate of PTP-PEST and serves as a scaffold guiding PTP-PEST toward a specific dephosphorylation of WASP. J Biol Chem. 2002;277:2973–2986. doi: 10.1074/jbc.M106428200. [DOI] [PubMed] [Google Scholar]
- 20.Vang T, Miletic AV, Arimura Y, Tautz L, Rickert RC, Mustelin T. Protein tyrosine phosphatases in autoimmunity. Ann Rev Imm. 2008;26:29–55. doi: 10.1146/annurev.immunol.26.021607.090418. [DOI] [PubMed] [Google Scholar]
- 21.Vang T, Miletic AV, Bottini N, Mustelin T. Protein tyrosine phosphatase PTPN22 in human autoimmunity. Autoimmunity. 2007;40:453–461. doi: 10.1080/08916930701464897. [DOI] [PubMed] [Google Scholar]
- 22.Hou S, Suresh PS, Qi X, Lepp A, Mirza SP, Chen G. p38γ Mitogen-activated protein kinase signals through phosphorylating Its phosphatase PTPH1 in regulating Ras protein oncogenesis and stress response. J Biol Chem. 2012;287:27895–27905. doi: 10.1074/jbc.M111.335794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang Z, Shen D, Parsons DW, Bardelli A, Sager J, Szabo S, Ptak J, Silliman N, Peters BA, van der Heijden MS, Parmigiani G, Yan H, Wang TL, Riggins G, Powell SM, Willson JK, Markowitz S, Kinzler KW, Vogelstein B, Velculescu VE. Mutational analysis of the tyrosine phosphatome in colorectal cancers. Science. 2004;304:1164–1166. doi: 10.1126/science.1096096. [DOI] [PubMed] [Google Scholar]
- 24.Pallen CJ. Protein tyrosine phosphatase alpha (PTPalpha): a Src family kinase activator and mediator of multiple biological effects. Curr Top Med Chem. 2003;3:821–835. doi: 10.2174/1568026033452320. [DOI] [PubMed] [Google Scholar]
- 25.Blanchetot C, Chagnon M, Dubé N, Hallé M, Tremblay ML. Substrate-trapping techniques in the identification of cellular PTP targets. Methods. 2005;35:44–53. doi: 10.1016/j.ymeth.2004.07.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.