

Abstract
Varicella zoster virus (VZV) is one of the human herpesviruses. To date, over 40 complete VZV genomes have been sequenced and analyzed. The VZV genome contains around 125,000 base pairs including 70 open reading frames (ORFs). Enumeration of single nucleotide polymorphisms (SNPs) has determined that the following ORFs are the most variable (in descending order): 62, 22, 29, 28, 37, 21, 54, 31, 1 and 55. ORF 62 is the major immediate early regulatory VZV gene. Further SNP analysis across the entire genome has led to the observation that VZV strains can be broadly grouped into clades within a phylogenetic tree. VZV strains collected in Singapore provided important sequence data for construction of the phylogenetic tree. Currently 5 VZV clades are recognized; they have been designated clades 1 through 5. Clades 1 and 3 include European/North American strains; clade 2 includes Asian strains, especially from Japan; and clade 5 includes strains from India. Clade 4 includes some strains from Europe, but its geographic origins need further documentation.. Within clade 1, five variant viruses have been isolated with a missense mutation in the gE (ORF 68) glycoprotein; these strains have an altered increased cell spread phenotype. Bioinformatics analyses of the attenuated vaccine strains have also been performed, with a subsequent discovery of a stop-codon SNP in ORFO as a likely attenuation determinant. Taken together, these VZV bioinformatics analyses have provided enormous insights into VZV phylogenetics as well as VZV SNPs associated with attenuation.
1. VZV, a brief introduction
Varicella zoster virus (VZV) is present in all peoples around the world. VZV is one of the nine human herpes viruses. VZV causes the childhood illness called varicella or chickenpox(Weller, 1983). Following primary infection, the virus enters a latent state in the dorsal root ganglia along the spinal column. Upon reactivation in late adulthood, VZV emerges from the sensory nerves to cause the dermatomal exanthem known as herpes zoster (zona) or shingles. Even in the most remote populations, VZV antibodies have been detected in serum samples(Black et al., 1974). An excellent example is Amazonia. In the late 1960’s, serological investigations were carried out among an isolated tribe living in Amazonia, in order to determine the prevalence of VZV. Around 80% of the tribe had VZV antibodies when surveyed by a complement fixation (CF) test. This test has a relatively low sensitivity, so the true seroprevalence is undoubtedly higher than 80%, perhaps approaching 100%. The Amazon tribes are the remnants of a human migration across the Bering land bridge from Asia 10–15 kya. The ancestral groups of humans continued their migrations southward along the Western coasts of North America and Central America, until reaching Amazonia within South America.
The fact that VZV can persist in isolated populations is dependent on the property of latency and reactivation as herpes zoster(Hope-Simpson, 1965). In other words, when an older adult has the disease herpes zoster, that individual will transmit VZV to most individuals in the same family group who have never had VZV infection. The newly infected children and adults will have the disease varicella, after which the virus will enter a latent state within their dorsal root ganglia. The cycle will repeat itself in another 20–40 years, when one of the previously infected people will reactivate a latent VZV infection and develop herpes zoster. Thus, through the dual diseases of varicella and herpes zoster, VZV has persisted since anatomically modern humans first left Africa 90–130 thousand years ago (kya) and entered Arabia(Grose, 2012). Thereafter, VZV was carried along with humankind during subsequent migrations into Asia and Europe. In this review, we will assess the genetic variation of VZV within different human populations, with an emphasis on the importance of an early study of SNPs found in VZV strains from Singapore(Chow et al., 1993).
2. Discovery of VZV SNPs
The fact that SNPs could define geographic differences between VZV strains was first discovered in 2001(Faga et al., 2001). Prior to that time, the general opinion was that VZV strains around the world were so similar that they could not be easily distinguished. In a 2001 exploratory analysis of VZV diversity, VZV strains were collected from around the United States. In addition, the Japanese VZV vaccine strain Oka was included(Takahashi, 2004). The VZV genome consists of nearly 125,000 bp separated into 70 open reading frames; the ORFs include 9 glycoprotein genes(Davison and Scott, 1986; Storlie et al., 2008). For our analysis, we sequenced 5 of the 9 glycoprotein genes as well as the major immediate early gene called ORF62(Ruyechan, 2010).
ORF62 was selected because it is one of the largest VZV genes and also because it is the major regulator of VZV transcription (Fig. 1). When the sequences were compared, we were surprised at the number of SNPs that were detectable. Furthermore, the SNPs were not randomly distributed; rather they segregated into distinct groups. Based on their SNP profiles, the majority of the VZV strains separated into 2 groups. However, what was unusual is that the SNPs in one of the VZV strains collected in Los Angeles segregated with the Japanese vaccine strain, rather than the other strains from the USA. Since Los Angeles is a city on the West Coast of the USA with a large Asian population, the last result strongly suggested an Asian origin for one Los Angeles VZV strain. This strain, called LAX1, has now been completely sequenced and is indeed a Japanese (or Asian) strain (Peters et al, 2012).
Figure 1. Representation of the domains within the VZV immediate early regulatory protein IE62.

VZV ORF62 contains a high number of SNPs, as shown in Table 1. Therefore, this ORF has played an important role in early phylogenetic analysis of VZV strains (Faga et al, 2001). Two SNPs within ORF62 that may be involved in the VZV attenuation genotype are shown in bold type in the center of the figure.
3. Identification of VZV clades in Singapore
Based on the above observation about Asian strains, we collected additional strains from Singapore and analyzed the same 6 genes(Wagenaar et al., 2003). This second analysis was even more informative than the first analysis. The studies in Singapore were particularly informative because the isolates clearly segregated into 2 clusters (Fig. 2). As can be seen by analysis of the Fig. 2, the SNPs within the glycoprotein genes, as well as the regulatory gene ORF62, were easily separated into one group that resembled the Japanese strain Oka while the other group of Singapore VZV strains did not. In addition to SNPs, there was one interesting insertion, namely, one-half of the Singapore strains had a distinctive 27atg28 insertion within the gL glycoprotein (ORF60) gene, similar to that found in Japanese VZV strains. At this point, we proposed a phylogenetic tree with four clades designated A, B, C and D (Fig. 3). Clades A and D represented the two clades previously found in the USA, while clade B included the Japanese strain Oka. Clade C represented the other cluster of Singapore VZV strains. However, there have been surprises as further strains were sequenced after we performed the Singapore study. For example, in 2003 we proposed that the 2 clusters discovered in Singapore represented distinct Asian clades. However, we have subsequently identified a VZV strain in the eastern Maritime Provinces of Canada (strain 8) that is closely related to one of the 2 Singapore clades (the non-Oka clade)(Peters et al., 2006). The SNPs in Canada strain 8 suggest that the clade C in Singapore may represent European VZV strains introduced into Singapore over the last 400 years, with successive incursions by Portuguese, Dutch, and British colonists.
Figure 2. SNPs within the two clusters of Singapore VZV strains.

VZV strains were collected from school children with varicella in Singapore (Chow et al, 1993; Wagenaar et al, 2003). Three of the sequenced VZV ORFs from these strains are illustrated in the figure. One cluster of Singapore VZV strains segregates with the Japanese VZV Oka clade, originally called clade B and now renamed clade 2. The other Singapore cluster resembles clade C viruses, now renamed clade 4. Black bars indicate nonsynonymous SNPs and white bars indicate synonymous SNPs.
Figure 3. The five VZV clades.
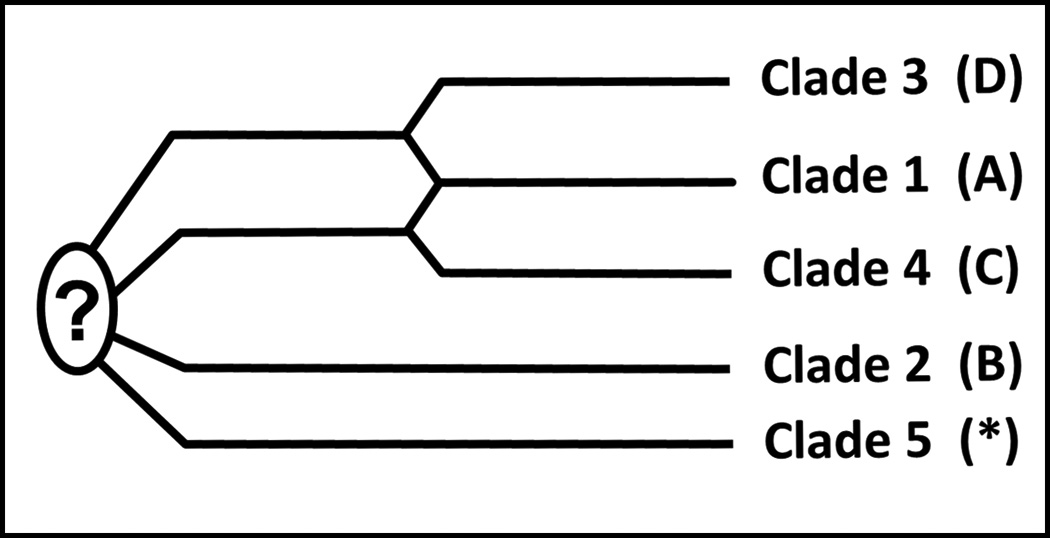
In an initial analysis of 18 completely sequenced VZV genomes, only four clades were discovered (Peters et al, 2006). These four clades were designated A, B, C and D. Subsequently a fifth clade was found to include VZV strains from India. The clades were subsequently re-designated by the numbers 1 through 5 at a VZV nomenclature meeting held in 2008 (Breuer et al, 2010). A common ancestor to the five clades has not yet been isolated and sequenced.
Overall, the VZV genetic analyses on the Singapore strains were important because we verified that VZV strains could be segregated into geographic groupings based on SNPs. Secondly, we decided to call these geographic clusters of VZV strains as clades. Among the strains from three continents, the single most important observation was a separation between clades in Europe/ North America and clades in the Asian countries of Singapore and Japan. Other VZV research groups were also publishing similar observations about the genetic differences found in VZV strains isolated in different geographic regions (Barrett-Muir et al., 2003; Norberg et al., 2006). The latter observations led to the hypothesis that the clades represented the coevolution of VZV with humankind(Grose, 2012).
Through the property of latency and reactivation, VZV was carried along with human populations as they migrated out of east Africa across the Strait of Bab el Mandeb around 90–130 kya (Grose, 2012). An earlier assumption that the out-of-Africa migration occurred as recently as 60 kya appears to be been an error in determining the correct human sequence mutation rate (Gibbons, 2012). From Arabia, anatomically modern humans migrated eastward into India, then out of India along the coastal regions of southeast Asia into China, then out of China into Japan. A second great human migration traveled northward out of Arabia into central Asia, then out of central Asia westward into Europe, then eventually out of Europe into North America and South America. Throughout these migrations of humankind, VZV traveled along hidden within the dorsal root ganglia, only occasionally emerging to cause herpes zoster. Since herpes zoster is contagious, travelers accompanying the index cases would themselves have contracted the primary infection varicella, had they not been previously infected at an earlier date. In this manner, throughout the world, VZV has survived even in relatively isolated human populations, such as the tribes in Amazonia (Black et al, 1974).
4. Clades based on complete sequencing of VZV genomes
To further investigate the actual variability of the entire VZV genome and to catalogue the location and number of genetic differences, 11 new complete genomic sequences were compared with those previously in the public domain (18 complete genomes in total). The VZV sequencing was performed at the Canadian National Microbiology Laboratory in Winnipeg, Manitoba, Canada(Peters et al., 2006; Tyler et al., 2007). The analysis revealed that while VZV is relatively stable genetically, it does possess a certain degree of variability. For example, the five reiteration regions, the origin of replication region and the intergenic homopolymer regions were particularly variable between VZV strains. In addition, the terminal viral sequences were found to vary within and between strains specifically at the 3' end of the genome. Enumeration of SNPs within the 18 complete VZV genomes identified a total of 557 variable sites, 451 of which were found in coding regions and resulted in 187 different amino acid substitutions. The ORFs with the most SNPs are listed in Table 1; note in particular the position of ORF62. When we completed the analyses, we discovered that the SNPs within the previously mentioned 6 genes were adequate to distinguish the clusters of VZV strains and segregate the strains into 4 clades, designated A, B, C and D.
Table 1.
VZV ORFs with the most SNPs
| Total | Coding | Length |
|---|---|---|
| 62 | 62 | 1 |
| 22 | 22 | 62 |
| 29 | 28 | 60 |
| 28 | 31 | 0 |
| 37 | 33 | 58 |
| 21 | 37 | 35 |
| 54 | 68 | 36 |
| 31 | 54 | 56 |
| 1 | 21 | 65 |
| 55 | 48 | 15 |
Note: The total column lists the ORFs with the most SNPs. The coding column indicates the ORFs with the most nonsynonymous SNPs. The length column indicates the ORFs with most SNPs per 100 nucleotides.
In 2008, the groups who had published in the field of VZV genomics met in London to compare sequence data and establish a common nomenclature. The principal investigators included Drs. J. Breuer (UK), C. Grose (USA), P. Norberg (Sweden), G. Tipples (Canada)and D. Schmid (USA). When all complete genomic sequences were analyzed, an extensive SNP analysis separated the strains into 5 clades (Fig. 3). The genomic database and conclusions of this meeting were published in 2010(Breuer et al., 2010). Four of the five clades were the same as previously identified, beginning in 2001. One new clade was defined among VZV strains subsequently found to originate from India(Zell et al., 2012). The clades were enumerated from 1 to 5, based on the order in which the first complete sequence within each clade was published. Thus the first VZV sequence to be published in 1986, called VZV Dumas, was designated clade 1; strain MSP was included as a second reference strain for clade 1(Davison and Scott, 1986; Grose et al., 2004). The first Japanese VZV sequence to be published, that for the vaccine and parental Oka strains, were designated clade 2(Gomi et al., 2002). The three additional clades included the following reference sequences: clade 3 were HJO and 03–500, clade 4 were DR and 8, and clade 5 was Ca123(Breuer et al., 2010; Norberg et al., 2006; Peters et al., 2006). The geographic locations for the 5 VZV clades are shown in the Fig. 4.
Figure 4. The geographical locations of the five VZV clades.
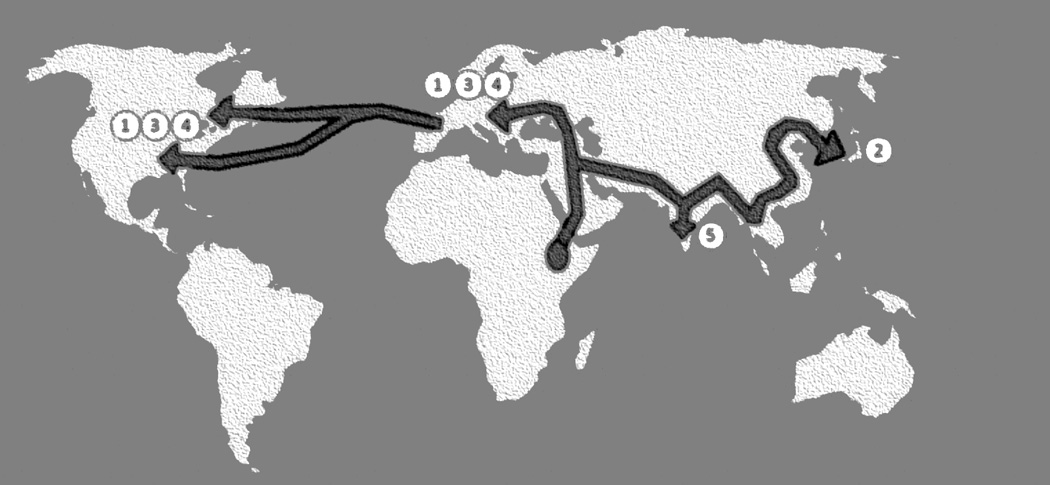
Clades 1 and 3 are European/North American VZV strains. Clade 2 includes Asian VZV strains, especially from Japan. Clade 5 includes VZV strains from India. Clade 4 appears to include European strains, but analyses of more strains are required.
Another separate clade analysis of the VZV SNP data available in 2008 was performed in 2009(McGeoch, 2009). For this analysis, 494 SNPs were selected that were relatively evenly distributed across the genome. Of the 494 SNPs, 281 were considered singleton SNPs, because they were found in only one strain and therefore not ideal for establishing linages. Thus 213 non-singleton SNPs were further analyzed. This re-inspection demonstrated that they segregated into the previously defined 5 clades. The incidence of SNPs was slightly higher in the short region, especially the short repeats. The short repeat region is also notable because its G+C content is 59%, versus 44% for the unique long region and 43% for the unique short region of the VZV genome. Additional analysis was then performed to define the relationship between the 5 clades, in other words, are any of the current 5 clades derived from the 4 other clades. Of great interest, the alleles in clade 1 match alleles in clade 3 and to a somewhat lesser extent alleles in clade 4. In other words, clade 1 appears to have descended from an ancestral recombination event between clades 3 and 4 (Fig. 3). The other two clades, namely clades 2 and 5, both possess lineages of great depth with as yet no identified common ancestor (Fig. 3).
Yet another analysis of VZV SNPs was performed in 2012, by a group which sequenced an additional 21 complete VZV genomes(Zell et al., 2012). The new strains were collected in Germany: 19 strains were collected from patients and 2 strains were known to be derivatives of the Oka Japanese strain. In their SNP analysis, 7 strains belonged to clade 1, 5 strains to clade 3, and 5 strains to clade 5; the 2 Japanese strains belonged to clade 2, as predicted. Two strains did not fit into established criteria for any clade, although their data suggest that the criteria for clade 4 may need to be readjusted. But a major conclusion is that the VZV clade designations have been confirmed for clades 1, 2, 3, and 5. As previously defined, clades 1 and 3 represent European strains, clade 2 represents Japanese strains, and clade 5 Indian strains. These authors estimated an evolutionary rate for the VZV thymidine kinase/polymerase genes of 3.9×10E-9 substitutions per site per year. The same group also estimated that VZV radiation in modern humankind began around 110 kya, correlating closely with the previously described out-of-Africa model of VZV evolution and phylogeography (Grose, 2012). Furthermore, based on the out-of-Africa model, further clades likely will be discovered, especially in VZV strains isolated from peoples living in remote geographical regions.
5. Clade 1 viruses with new phenotypes
The viruses in the above clades invariably have the same phenotype when grown in cultured cells. There is however, one exception to the above general rule. Certain clade 1 viruses have a phenotype distinguishable from all other VZV strains. The first of these variant strains was described in 1998(Santos et al., 1998). The virus was isolated from a boy with leukemia who was admitted to a hospital after contracting disseminated varicella. The infection was successfully treated with acyclovir, but the isolate could not be identified as VZV by a commercial diagnostic rapid immunostaining kit. Because of this failure, the isolate was sent to a VZV research laboratory for further characterization. The source of confusion was identified, namely, the VZV strain had undergone a missense mutation in the gE (ORF 68) protein, which is the target of the monoclonal antibody in the VZV diagnostic kit. The missense mutation led to a change in amino acid from aspartic acid to asparagine, which in turn disrupted a B-cell epitope harbored near the N-terminus of the gE glycoprotein. The strain was called VZV-MSP, because it was isolated in Minneapolis-St. Paul, Minnesota.
The phenotype of the variant VZV strain is characterized by a faster cell-to-cell spread with greater syncytial formation, when grown in cultured cells(Santos et al., 2000). When examined in the severe combined immunodeficient (SCID) mouse model of VZV pathogenesis, VZV infection of the human skin xenograft was markedly more severe than after a typical wild type VZV infection, a sign of increased virulence. Since discovery of the VZV-MSP strain in Minnesota in 1998, 4 additional VZV gE variant strains have been described. A Canadian case involved an elderly man with extensive zoster(Tipples et al., 2002). Two similar variant strains were discovered in a repository of VZV strains collected by the Karolinska University Hospital in Stockholm, Sweden, from patients who had been treated in their hospital for severe infection(Wirgart et al., 2006). The fifth case was a child in Rome, who contracted a fatal case of varicella(Natoli et al., 2006). When the above reports are reviewed, these cases appear to represent more severe varicella or zoster infections. Yet the clade 1 variant viruses do not appear to have spread widely since several investigations have failed to identify the VZV gE mutated virus within community outbreaks of varicella.
6. Attenuation SNP within ORF0 of varicella vaccine
One of the goals of VZV researchers over the past decade has been to identity the SNPs which are responsible for the attenuation phenotype of the varicella vaccine virus. When the entire sequence of the vaccine Oka strain (vOka) and its parental strain were published in 2002, there was an early attempt to define the determinants of attenuation (Gomi et al, 2002). Based on numerous SNPs within ORF62, the immediate early protein IE62 was a prime candidate. Yet, successive analyses have failed to attribute attenuation to a single SNP within IE62 (Yamanishi, 2008; Zerboni et al., 2005).
A consensus has emerged that a combination of SNPs, probably including one or two within ORF62, are responsible for attenuation. To define those other SNPs, we decided to take a bioinformatics approach. In a past experiment, we had demonstrated that the laboratory strain called VZV Ellen was highly attenuated when grown in the SCID mouse model of VZV pathogenesis. To further investigate the nature of attenuation, we completed next-generation sequencing of the VZV Ellen genome(Peters et al., 2012). VZV Ellen was a particularly good candidate because Ellen does not belong to the same clade as the vaccine strain Oka; Oka is a member of clade 2 while Ellen belongs to clade 3. After a bioinformatics analysis, we noted that Ellen shared relatively few SNPs with vOka. But one shared SNP in ORF0, sometimes called ORF S/L, was both unexpected and interesting. An earlier report had already found the transition SNP (TGA into CGA) in the vOka ORF0 gene, which eliminates a stop-codon and thereby extends the ORF0 protein by 92 amino acids, until reaching a second stop codon within ORF1 (Fig. 5). Thus the mutated vOka ORF0 is longer than the wild type ORF0 found in parental Oka. Of note, the attenuated Ellen strain had an identical stopcodon mutation in ORF0 (Table 2). When we performed further bioinformatics analyses, we estimated the likelihood that 2 geographically unrelated VZV strains would have an identical stop-codon mutation within the same ORF as 1×10E-8, obviously very unlikely. Instead, a more likely explanation is that the stop-codon SNP is a determinant of attenuation. However, we postulate that there are other as yet undetermined attenuation SNPs within the vOka genome. For example, vOka and Ellen strains share two ORF62 coding SNPs shown in Fig. 1. Further analyses of recombinant VZV viruses containing these SNPs will be required to assign with certainty the role of each SNP in the attenuation genotype.
Figure 5. Diagram of wild type and mutated ORF0 proteins.

The mutated ORF0 protein is a candidate for an attenuation determinant in the varicella vaccine. Mutated ORF0 is the result of a stop-codon mutation in wild type ORF0 that leads to an extension of the wild type protein from 129 to 221 amino acids in length.
Table 2.
SNPs shared by the attenuated varicella strains and VZV Ellen Strain
| ORF | SNP | Residue | Ellen | 32/P72 | pOKA | vOKA | VarilRix | VariVax |
|---|---|---|---|---|---|---|---|---|
| ORF0 | T/C | Y72 | Y72H | |||||
| ORF0 | T/C | *130 | *130R | *130R | *130R | *130R | ||
| ORF6 | C/T | A1053 | A1053T | |||||
| ORF8 | A/G | D44 | syn | |||||
| ORF11 | G/A | A130 | A130T | |||||
| ORF19 | T/C | L404 | syn | |||||
| ORF21 | C/T | I191 | syn | |||||
| ORF27 | G/A | Q261 | syn | |||||
| ORF33 | C/A | P383 | syn | |||||
| ORF33 | T/C | H291 | H291R | |||||
| ORF36 | A/G | T26 | syn | |||||
| ORF48 | C/T | T172 | T172I | |||||
| ORF54 | T/C | V210 | syn | |||||
| ORF55 | T/C | R57 | syn | |||||
| ORF56 | A/G | A83 | syn | |||||
| ORF57 | A/G | C66 | C66R | |||||
| ORF59 | T/C | Q257 | Q257R | |||||
| ORF59 | A/G | H157 | syn | |||||
| ORF61 | C/T | A6 | A6T | |||||
| ORF62 | T/C | I1260 | I1260V | I1260V | I1260V | I1260V | ||
| ORF62 | A/C | S1254 | S1254A | |||||
| ORF62 | T/C | E1243 | E1243G | |||||
| ORF62 | T/C | S1241 | S1241G | |||||
| ORF62 | T/C | Q1215 | Q1215R | |||||
| ORF62 | T/C | A1093 | syn | |||||
| ORF62 | T/C | Q1072 | Q1072R | |||||
| ORF62 | T/C | Q1057 | Q1057R | |||||
| ORF62 | T/C | K1045 | K1045E | |||||
| ORF62 | A/G | L1028 | syn | |||||
| ORF62 | A/G | L963 | syn | |||||
| ORF62 | T/C | R958 | R958G | R958G | R958G | R958G | R958G | |
| ORF62 | T/C | Q768 | Q768R | Q768Q/R | ||||
| ORF62 | T/C | S628 | S628G | S628G | S628G | S628G | S628G | |
| ORF62 | T/C | N195 | N195D | |||||
| ORF62 | T/C | Q190 | syn | |||||
| ORF62 | A/G | V172 | syn |
NOTE: Strain VZV Ellen is an attenuated strain. Strain 32/p72 is high passage 72 of a laboratory strain VZV 32. The parental strain of varicella vaccine is called pOka and the attenuated vaccine strain is called vOka. Varivax and Varilvax are the names of the commercial varicella vaccines.
Abbrreviation syn = synonymous SNP.
7. Summary of VZV bioinformatics
VZV bioinformatics has been extremely revealing(Grose, 2006). In one sense, the VZV genome is sufficiently stable that it can be analyzed in a similar manner to the human genome. By delineation of just a few alleles, geographic origins can be assigned to a VZV strain with reasonable certainty. To date, five VZV clades have been delineated (Breuer et al, 2010). Yet, the common ancestor to the five clades remains to be isolated. Based on the likely out-of-Africa explanation for the origin of human VZV throughout the world, isolated populations within Africa may still harbor ancestral VZV strains. Furthermore, ancient VZV strains may still reside within isolated human populations living in other remote geographic regions of South America or Australia.
VZV bioinformatics has also provided insight into the genetic explanation for attenuation of the varicella vaccine strain vOka, also called Varivax (Merck) or VarilRix(Glaxo). By knowing the clade-specific SNPs and eliminating them, SNPs specific for attenuation in the varicella vaccine viruses can be deduced. For example, the stop-codon SNP in ORF0 appears to be a determinant of attenuation in varicella vaccine(Peters et al., 2012). The same ORF0 SNP is found in a live attenuated varicella vaccine produced independently of vOka in Korea and named Suduvax(Kim et al., 2011). Thus, these extensive SNP analyses have provided and will continue to provide valuable knowledge that is required for future production of second generation varicella and zoster vaccines.
Finally, it is noteworthy that genomic analyses of the closely related alphaherpesvirus herpes simplex virus type 1 (HSV-1) have found strikingly differing results. When the complete HSV-1 genomic sequences of 10 clinical HSV-1 strains and 2 highly passaged laboratory strains called F and 17 were subjected to phylogenetic analysis, the investigators discovered that all 12 strains appeared to be mosaic recombinant viruses (Norberg et al, 2011). In fact, the frequent recombination events in the various HSV-1 genomes of the 12 strains disallowed any classification into distinct clades. Based on these data, the vast majority of HSV-1 strains circulating throughout the world are likely to be recombinant viruses. The authors provide an hypothesis for this difference between HSV-1 and VZV genomics, namely, that the number of replication cycles per year is much greater for HSV-1, which frequently reactivates from latency while VZV may reactivate only once in a human host’s lifetime (Norberg et al, 2011). This same explanation had been proposed earlier to explain the apparent relative stability of the VZV genome over the millennia (Faga et al, 2001; Grose, 2012).
Acknowledgements
The corresponding author thanks Julian Tang for the invitation to present a talk on VZV Evolution and Phylogenetics at the Workshop on Phylogenetics of Infectious Diseases—with a Focus on DNA Viruses, sponsored by the Institute for Mathematical Sciences at the National University of Singapore in October 2011. Research by C. Grose is supported by NIH grant AI89716.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Barrett-Muir W, Scott FT, Aaby P, John J, Matondo P, Chaudhry QL, Siqueira M, Poulsen A, Yaminishi K, Breuer J. Genetic variation of varicella-zoster virus: evidence for geographical separation of strains. J Med Virol. 2003;70(Suppl 1):S42–S47. doi: 10.1002/jmv.10319. [DOI] [PubMed] [Google Scholar]
- Black FL, Hierholzer WJ, Pinheiro F, Evans AS, Woodall JP, Opton EM, Emmons JE, West BS, Edsall G, Downs WG, Wallace GD. Evidence for persistence of infectious agents in isolated human populations. Amer J Epidemiol. 1974;100:230–250. doi: 10.1093/oxfordjournals.aje.a112032. [DOI] [PubMed] [Google Scholar]
- Breuer J, Grose C, Norberg P, Tipples G, Schmid DS. A proposal for a common nomenclature for viral clades that form the species varicella-zoster virus: summary of VZV Nomenclature Meeting 2008, Barts and the London School of Medicine and Dentistry, 24–25 July 2008. J Gen Virol. 2010;91:821–828. doi: 10.1099/vir.0.017814-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow VT, Wan SS, Doraisingham S, Ling AE. Comparative analysis of the restriction endonuclease profiles of the Dumas and Singapore strains of varicella-zoster virus. J Med Virol. 1993;40:339–342. doi: 10.1002/jmv.1890400415. [DOI] [PubMed] [Google Scholar]
- Davison AJ, Scott JE. The complete DNA sequence of varicella-zoster virus. J Gen Virol. 1986;67:1759–1816. doi: 10.1099/0022-1317-67-9-1759. [DOI] [PubMed] [Google Scholar]
- Faga B, Maury W, Bruckner DA, Grose C. Identification and mapping of single nucleotide polymorphisms in the varicella-zoster virus genome. Virology. 2001;280:1–6. doi: 10.1006/viro.2000.0775. [DOI] [PubMed] [Google Scholar]
- Gibbons A. Turning back the clock: Slowing the pace of prehistory. Science. 2012;338:189–191. doi: 10.1126/science.338.6104.189. [DOI] [PubMed] [Google Scholar]
- Gomi Y, Sunamachi H, Mori Y, Nagaike K, Takahashi M, Yamanishi K. Comparison of the complete DNA sequences of the Oka varicella vaccine and its parental virus. J Virol. 2002;76:11447–11459. doi: 10.1128/JVI.76.22.11447-11459.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grose C. Varicella-zoster virus: out of Africa and into the research laboratory. Herpes. 2006;12:88–92. [PubMed] [Google Scholar]
- Grose C. Pangaea and the out-of-Africa model of varicella-zoster virus evolution and phylogeography. J Virol. 2012;86:9558–9565. doi: 10.1128/JVI.00357-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grose C, Tyler S, Peters G, Hiebert J, Stephens GM, Ruyechan WT, Jackson W, Storlie J, Tipples GA. Complete DNA sequence analyses of the first two varicella-zoster virus glycoprotein E (D150N) mutant viruses found in North America: evolution of genotypes with an accelerated cell spread phenotype. J Virol. 2004;78:6799–6807. doi: 10.1128/JVI.78.13.6799-6807.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hope-Simpson RE. The nature of herpes zoster: a long-term study and a new hypothesis. Proc R Soc Med. 1965;58:9–20. [PMC free article] [PubMed] [Google Scholar]
- Kim JI, Jung GS, Kim YY, Ji GY, Kim HS, Wang WD, Park HS, Park SY, Kim GH, Kwon SN, Lee KM, Ahn JH, Yoon Y, Lee CH. Sequencing and characterization of Varicella-Zoster virus vaccine strain SuduVax. Virol J. 2011;8:547. doi: 10.1186/1743-422X-8-547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeoch DJ. Lineages of varicella-zoster virus. J Gen Virol. 2009;90:963–969. doi: 10.1099/vir.0.007658-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natoli S, Ciotti M, Paba P, Testore GP, Palmieri G, Orlandi A, Sabato AF, Leonardis F. A novel mutation of varicella-zoster virus associated to fatal hepatitis. J Clin Virol. 2006;37:72–74. doi: 10.1016/j.jcv.2006.06.004. [DOI] [PubMed] [Google Scholar]
- Norberg P, Liljeqvist JA, Bergstrom T, Sammons S, Schmid DS, Loparev VN. Complete-genome phylogenetic approach to varicella-zoster virus evolution: genetic divergence and evidence for recombination. J Virol. 2006;80:9569–9576. doi: 10.1128/JVI.00835-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norberg P, Tyler S, Severini A, Whitley R, Liljeqvist J, Bergstrom T. A genome wide comparative evolutionary analysis of herpes simplex virus type 1 and varicella zoster virus. PLoS One. 2011;6:e22527. doi: 10.1371/journal.pone.0022527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters GA, Tyler SD, Carpenter JE, Jackson W, Mori Y, Arvin AM, Grose C. The attenuated genotype of varicella-zoster virus includes an ORF0 transitional stop-codon mutation. J Virol. 2012;86:10695–10703. doi: 10.1128/JVI.01067-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters GA, Tyler SD, Grose C, Severini A, Gray MJ, Upton C, Tipples GA. A full-genome phylogenetic analysis of varicella-zoster virus reveals a novel origin of replication-based genotyping scheme and evidence of recombination between major circulating clades. J Virol. 2006;80:9850–9860. doi: 10.1128/JVI.00715-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruyechan WT. Roles of cellular transcription factors in VZV replication. Curr Topics Microbiol Immunol. 2010;342:43–65. doi: 10.1007/82_2010_42. [DOI] [PubMed] [Google Scholar]
- Santos RA, Hatfield CC, Cole NL, Padilla JA, Moffat JF, Arvin AM, Ruyechan WT, Hay J, Grose C. Varicella-zoster virus gE escape mutant VZV-MSP exhibits an accelerated cell-to-cell spread phenotype in both infected cell cultures and SCID-hu mice. Virology. 2000;275:306–317. doi: 10.1006/viro.2000.0507. [DOI] [PubMed] [Google Scholar]
- Santos RA, Padilla JA, Hatfield C, Grose C. Antigenic variation of varicella zoster virus Fc receptor gE: loss of a major B cell epitope in the ectodomain. Virology. 1998;249:21–31. doi: 10.1006/viro.1998.9313. [DOI] [PubMed] [Google Scholar]
- Storlie J, Maresova L, Jackson W, Grose C. Comparative analyses of the 9 glycoprotein genes found in wild-type and vaccine strains of varicella-zoster virus. J Infect Dis. 2008;197(Suppl 2):S49–S53. doi: 10.1086/522127. [DOI] [PubMed] [Google Scholar]
- Takahashi M. Effectiveness of live varicella vaccine. Expert Opin Biol Ther. 2004;4:199–216. doi: 10.1517/14712598.4.2.199. [DOI] [PubMed] [Google Scholar]
- Tipples GA, Stephens GM, Sherlock C, Bowler M, Hoy B, Cook D, Grose C. New variant of varicella-zoster virus. Emerg Infect Dis. 2002;8:1504–1505. doi: 10.3201/eid0812.020118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyler SD, Peters GA, Grose C, Severini A, Gray MJ, Upton C, Tipples GA. Genomic cartography of varicella-zoster virus: a complete genome-based analysis of strain variability with implications for attenuation and phenotypic differences. Virology. 2007;359:447–458. doi: 10.1016/j.virol.2006.09.037. [DOI] [PubMed] [Google Scholar]
- Wagenaar TR, Chow VT, Buranathai C, Thawatsupha P, Grose C. The out of Africa model of varicella-zoster virus evolution: single nucleotide polymorphisms and private alleles distinguish Asian clades from European/North American clades. Vaccine. 2003;21:1072–1081. doi: 10.1016/s0264-410x(02)00559-5. [DOI] [PubMed] [Google Scholar]
- Weller TH. Varicella and herpes zoster. Changing concepts of the natural history, control, and importance of a not-so-benign virus. New Engl J Med. 1983;309:1434–1440. doi: 10.1056/NEJM198312083092306. [DOI] [PubMed] [Google Scholar]
- Wirgart BZ, Estrada V, Jackson W, Linde A, Grose C. A novel varicella-zoster virus gE mutation discovered in two Swedish isolates. J Clin Virol. 2006;37:134–136. doi: 10.1016/j.jcv.2006.06.007. [DOI] [PubMed] [Google Scholar]
- Yamanishi K. Molecular analysis of the Oka vaccine strain of varicella-zoster virus. J Infect Dis. 2008;197(Suppl 2):S45–S48. doi: 10.1086/522122. [DOI] [PubMed] [Google Scholar]
- Zell R, Taudien S, Pfaff F, Wutzler P, Platzer M, Sauerbrei A. Sequencing of 21 varicella-zoster virus genomes reveals two novel genotypes and evidence of recombination. J Virol. 2012;86:1608–1622. doi: 10.1128/JVI.06233-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerboni L, Hinchliffe S, Sommer MH, Ito H, Besser J, Stamatis S, Cheng J, Distefano D, Kraiouchkine N, Shaw A, Arvin AM. Analysis of varicella zoster virus attenuation by evaluation of chimeric parent Oka/vaccine Oka recombinant viruses in skin xenografts in the SCIDhu mouse model. Virology. 2005;332:337–346. doi: 10.1016/j.virol.2004.10.047. [DOI] [PubMed] [Google Scholar]