

Abstract
Dilated cardiomyopathy (DCM) is a disease characterized by dilation of the ventricular chambers and reduced contractile function. We examined the contractile performance of chemically-skinned ventricular strips from two heterozygous murine models of DCM-causing missense mutations of myosin, F764L/+ and S532P/+, in an α-myosin heavy chain (MyHC) background. In Ca2+-activated skinned myocardial strips, the maximum developed tension in F764L/+ was only ~50% that of litter-mate controls (+/+). The F764L/+ also exhibited significantly reduced rigor stiffness, loaded shortening velocity and power output. Corresponding indices for S532P/+ strips were not different from controls. Manipulation of MgATP concentration in conjunction with measures of viscoelasticity, which provides estimates of myosin detachment rate 2πc, allowed us to probe the molecular basis of changes in crossbridge kinetics that occur with the myosin mutations. By examining the response of detachment rate to varying MgATP we found the rate of MgADP release was unaffected by the myosin mutations. However, MgATP binding rate was higher in the DCM groups compared to controls (422±109 mM−1.s−1 in F764L/+, 483±74 mM−1.s−1 in S532P/+ and 303± 18 mM−1.s−1 in +/+). In addition, the rate constant of force development, 2πb, was significantly higher in DCM groups compared to controls (at 5 mM MgATP: 36.9±4.9 s−1 in F764L/+, 32.9±4.5 s−1 in S532P/+ and 18.2±1.7 s−1 in +/+). These results suggest that elevated rates of force development and MgATP binding are features of cardiac myofilament function that underlie the development of DCM.
Keywords: hypertrophic cardiomyopathy, myocardium, detachment rate, time-on, sinusoidal analysis
1. INTRODUCTION
Dilated cardiomyopathy (DCM) is a disease characterized by ventricular dilation and impaired contraction. Hereditary forms of DCM have been associated with mutations in a number of sarcomeric proteins, including cardiac β-myosin heavy chain (β-MyHC), myosin binding protein-C, actin, troponin-T, myosin regulatory light chain and others [1–4]. This study focuses on examining the mechanical and kinetic properties of cardiac myofilaments bearing one of two DCM β-MyHC mutations associated with early-onset ventricular dilation in humans: S532P and F764L [5, 6]. Residue S532 is in a highly conserved α-helix in the lower 50-kDa domain of the myosin head that binds strongly with actin [7]. Residue F764 is in the converter region that transmits structural changes that arise in the nucleotide binding site to the myosin lever arm [7].
Schmitt et al. [8] engineered these point mutations into cardiac α-MyHC in two murine models and examined the effect of these mutations on cardiac morphology and function in homozygous and heterozygous mice. At the whole heart level, the homozygous mice exhibit morphological and functional characteristics of profound DCM. The heterozygous mice exhibited DCM to a lesser degree. Contractile function of myocytes isolated from the heterozygotes was also depressed. These findings in the heterozygotes demonstrated that cardiac morphology and function in these mutant mice are consistent with the human phenotype.
Myosin harvested from the homozygous S532P and F764L mouse hearts exhibit reduced actin filament velocities and reduced actin-activated myosin ATPases measured in standard in vitro motility and biochemical assays [8, 9]. Notably, in nearly unloaded laser trap experiments carried out at low MgATP concentration (10 μM), the time that myosin was attached to actin (ton) was prolonged in the S532P but unchanged in the F764L compared to their values in wild type controls [8]. These data suggested that DCM mutations reduce performance of myosin at the molecular level, which contrasts against the enhanced performance observed with point mutations leading to familial hypertrophic cardiomyopathy (FHC) [6, 9].
In the current study we asked whether the distinct phenotypes exhibited in isolated myosin from S532P and F764L homozygous mice were also present in the context of an intact myofilament lattice in skinned strips of heterozygous mice. We found that these mice developed DCM as they aged as indicated by reduced contractile function at the whole heart level and increased β-MyHC content. We asked further whether changes in characteristic function of these myosin in younger heterozygous mice could underlie the development of DCM [8]. To answer this question, we used small length perturbation analysis of the myocardial skinned strip to examine myosin crossbridge detachment at various MgATP concentrations and to deduce rates of MgADP release and MgATP binding. While MgADP release rate with these mutations appears dictated by myosin isoform content, MgATP binding rate was higher in the myocardium bearing the DCM mutations and could not be explained by the presence of β-MyHC. Another parameter, the rate of force development (2πb), was also higher in the DCM mutant myosin compared to controls independent of β-MyHC. These enhanced rates can only be attributed to characteristics of the DCM mutations, which reside in very different structural regions of the myosin molecule, and point to common functional properties of myofilament function that underlie the development of DCM.
2. METHODS
2.1 Mouse Models
All procedures were reviewed and approved by the Institutional Animal Care and Use Committees of The University of Vermont College of Medical and The Harvard Medical School. Two lines of heterozygous transgenic mice, one containing the F764L myosin mutation (F764L/+) and one containing the S532P myosin mutation (S532P/+), were generated as previously described [8]. Functional data from these DCM mice were compared against age-matched litter-mate controls (+/+).
2.2 Left Ventricular Mechanics
Hearts from five F764L/+, five S532P/+, and nine +/+ control mice were used for isolated heart studies. Mice used for these experiments were 40–60 weeks old. The isovolumic mouse heart preparation has been described in detail elsewhere [10]. Briefly, after anesthesia and mechanical ventilation the heart was excised, the aorta was cannulated and perfused with oxygenated Krebs buffer (2.5 mmol/L Ca2+, 35°C to 37°C, pH 7.35 to 7.45). A custom-made balloon mounted on a catheter was placed in the LV via the mitral orifice. A micromanometer catheter was introduced just above the mitral orifice via a side port. Pacing electrodes were attached to the LV and heart rate maintained at 240 beats per minute. The heart was then placed in a chamber maintained at 35°C to 37°C. Coronary perfusion pressure was controlled by a pressurized arterial reservoir at ~100 mm Hg. After a 30-minute stabilization period, total LV volume was set to 31 and then 35 μL using a manual micrometer syringe driver. LV pressure was measured under steady-state conditions at each volume. For analysis of contractile function we selected LV volumes common to all hearts that were as close as possible to 31 and 35 μL and compared LV developed pressure (peak systolic pressure minus end-diastolic pressure) at each level. These volumes were selected because they encompass the physiologic range in normal mice [10].
2.3 Solutions for Skinned Strips
Chemicals and reagents were obtained from Sigma Corp. (St. Louis, MO) and concentrations are expressed in mmol/L unless noted otherwise. Concentrations were calculated using an ionic equilibrium program [11]. Relaxing solution: pCa 8.0, 20 N,N-bis(2-hydroxyethyl)-2-aminoethanesulfonic acid (BES), 5 ethylene glycol tetraacetic acid (EGTA), 5 MgATP, 1 Mg2, 35 phosphocreatine (PCr), 300 U/mL creatine kinase (CK), 1 dithiothreitol (DTT), ionic strength 200 mEq adjusted with sodium methane sulfate, and pH 7.0. Activation solution: same as relaxing with pCa 4.0. Rigor solution: same as relaxing with pCa 4.8 and without MgATP, PCr and CK. Skinning solution: same as relaxing without PCr and CK and with 10 μg/mL leupeptin, 1% Triton-X100 wt/vol and subsequently 50% glycerol wt/vol. Storage solution: same as skinning without Triton-X100.
2.4 Skinned Strips
Myofilament experiments were performed using mice aged 20–30 wks. LV myocardial skinned strips were studied as described previously at 17°C and 2.2 μm sarcomere length [12]. Resting, or minimum tension (Tmin) was measured at pCa 7 and maximum calcium-activated tension (Tmax) was measured at pCa 4.8. Maximum developed tension (Tdev) was calculated as Tmax–Tmin. MgATP concentrations ranging from 5 to <0.05 mM were applied at pCa 4.8 by exchanging volumes of rigor solution. In a separate set of strips, Pi concentrations ranging from ~0 to 4 mM were applied at pCa 4.8 by exchanging volumes of solution containing 8 mM Pi.
While two control groups (n=6 each) were examined eight months apart to coincide with the examination of the two DCM groups at 20–30 wks of age, the functional data from the control groups were not different from each other and were therefore pooled to provide one control group.
2.5 Tension-Velocity Relationship
Various mechanical loads expressed as fractions of maximum absolute tension (Tmax) were imposed by feedback control of muscle length. Force was maintained over a 400 ms, and velocity was calculated as the slope of length change at the earliest steady-state force, ~50 ms after feedback initiation. Tension-velocity (T-V) relationships were fit to a hyperbolic Hill equation normalized to Tmax:
| (Equation 1) |
where T′ = T/Tmax, a′= a/Tmax, and a and b are the parameters of the non-normalized hyperbolic Hill equation. The physiological characteristics maximum unloaded shortening velocity (Vmax), velocity at maximum power (Vopt), tension at maximum power (T′opt) and maximum power production (Pmax) were calculated from a′ and b, as follows [13]:
2.6 Length perturbation analysis and crossbridge kinetics
At each ionic concentration, sinusoidal length perturbations of amplitude 0.125% strip length were applied (0.125–250 Hz) as described previously [14]. The elastic and viscous moduli were measured from the in-phase and out-of-phase portions of the tension response to length perturbation. The following mathematical expression was fitted to the complex modulus using non-linear least squares analysis:
| (Equation 2) |
where A, B and C are magnitudes expressed in kN/m2, k = a unitless exponent, b and c are characteristic frequencies expressed in Hz, ω = angular frequency in units of s−1 of the length perturbation equal to 2π×frequency of perturbation, and i = −11/2 [14].
Parameters A and k reflect the viscoelastic properties of various structural elements of the muscle strip, including a Ca2+-dependent portion ascribed to the myosin heads attached to actin at any given time [15]. Coefficients B and C are proportional to both the unitary stiffness of the crossbridge and the number of force-generating myosin heads attach to actin at any given time [16]. The apparent rate constants 2πb and 2πc reflect respectively the rates of myosin crossbridge formation and detachment [16–18]. The rate 2πb measured in the frequency domain is equivalent to the rate of developed force in response to a step length change and is akin to kdf of Phase 3 in stretch activation experiments [14, 17]. The rate 2πc is the equivalent of the myosin crossbridge detachment rate and its reciprocal is equal to ton, the average time myosin is attached to actin [14]. This interpretation of 2πc as the myosin crossbridge detachment rate is conceptually similar to that proposed by Campbell et al. [19].
2.7 MgATP-dependent Myosin Enzyme Kinetics
We assume that the total myosin crossbridge lifetime, ton, is represented adequately as the sum of two time periods: time to release MgADP and time to bind MgATP as explained in Tyska and Warshaw [20]. Noting the myosin crossbridge detachment rate, 2πc, is the reciprocal of the mean ton, we can represent the myosin crossbridge detachment rate in terms of its dependence upon MgATP concentration as follows:
| (Equation 3) |
where k−ADP = rate of MgADP release to which the detachment rate 2πc asymptotically approaches (s−1), k+ATP = rate of MgATP binding per MgATP concentration (M−1.s−1), and the ratio (k−ADP/k+ATP) = concentration of MgATP producing half k−ADP.
2.8 Analysis
Statistical analyses were performed using SPSS 20.0 (IBM, Chicago, IL). Non-linear least-squares fitting was performed using Sigma Plot 8.0 (SPSS, Chicago, IL). Results for two muscle strips from each heart were averaged to provide a single measure for that heart. All data are reported as mean ± SEM unless indicated. Student-Newman-Keuls (S-N-K) was used to test for differences among three populations, two DCM groups and one +/+ control group. Significant differences between a DCM mutant and control are reported at the P<0.05 and P<0.01 levels. When in support of other statistically significant findings, P-values less than 0.1 are also reported to minimize the chance of reporting a false negative (Type II error) [21].
3. RESULTS
3.1 Left ventricular mechanics
Table 1 summarizes the morphometric and hemodynamic data for the mouse hearts used in the whole heart studies. Cardiac mass per body weight and β-MyHC content were greater in the F764L/+ compared to +/+ aged 40–60 wks. Although we did not find a statistically higher cardiac mass per body weight (P=0.348) or β-MyHC content (P=0.105) in the S532P/+ compared to +/+, values for these variables reported in Table 1 are consistent with a subtle cardiomyopathy in the S532P/+ reported at 48 wks [8].
Table 1.
Characteristics of mice aged 40–60 weeks used in LV studies and hemodynamics.
| F764L/+ (n=5) | S532P/+ (n=5) | +/+ (n=9) | |
|---|---|---|---|
| Body mass (g) | 30.1±1.4 | 31.1±2.4 | 34.5±1.9 |
| LV/Body (mg/g) | 8.4±1.1 *† | 6.0±0.8 | 5.1±0.3 |
| RV+LV/Body (mg/g) | 10.1±1.4 *† | 7.5±1.0 | 6.3±0.4 |
| β-MyHC (% of total) | 45±7 * | 29±5 | 17±3 |
|
| |||
| Diastolic Pressure (mmHg) | |||
| LV volume 31 μL | 4.6±1.9 | 11.6±3.3 | 9.4±3.3 |
| LV volume 35 μL | 20.1±5.5 | 22.2±3.1 | 14.8±5.6 |
| Developed Pressure (mmHg) | |||
| LV volume 31 μL | 30.4±4.4 * | 44.8±5.1 * | 63.9 ±6.1 |
| LV volume 35 μL | 47.2±8.4 * | 60.0±7.0 # | 83.2 ±7.6 |
= different from +/+ controls at P<0.05 by S-N-K.
= different from S532P/+ at P<0.05 by S-N-K.
P=0.065 compared to +/+ controls.
n = number of hearts. For MyHC, n=5 for each group.
At 31 μL balloon volume, diastolic wall stress (estimated by Laplace’s law as diastolic pressure × chamber radius/wall thickness and using measured pressures, set volumes and previously reported wall thickness [8]) is likely lower in the F764L/+ compared to the S532P/+ and controls; consequently, the preload in the F764L/+ hearts is also lower and the lower developed pressure recorded in the F764L/+ may be underestimated as a result. Nevertheless, the preloads for the S532P/+ and +/+ controls are comparable at 31 μL volume, and we report a depressed contractile function in the S532P/+ (Table 1) consistent with the development of a cardiomyopathy in the S532P/+.
At 35 μL balloon volume, diastolic wall stress is on the order of 22 mmHg for the DCM groups and about 16 mmHg for the controls. Under these experimental conditions, one would expect the higher preloads in the DCM groups to result in a higher developed pressure compared to controls [10]. However, instead we observed lower developed pressure in both DCM groups. Based on this result, i.e., opposite to that predicted had preload bias been a factor, we conclude that hearts bearing the DCM myosin exhibit significantly depressed contractile function compared to +/+ controls.
In summary, the DCM heterozygous mice demonstrated reduced LV systolic performance and signs of increased cardiac mass and β-MyHC content consistent with the development of DCM as reported previously in these heterozygotes of comparable age [8].
3.2 Skinned Strip Mechanics
Morphometric data of younger mice used in the skinned strip experiments are presented in Table 2. At 20–30 wks of age LV mass normalized to body mass was greater in the F764L/+ mice compared to their +/+ controls. Except for a lower RV mass in the S532P/+, there were no other detectable morphometric differences between S532P/+ and +/+ controls. These findings are consistent with the earlier onset of cardiac remodeling in the F764L/+ compared to S532P/+ described previously [8].
Table 2.
Characteristics of mice aged 20–30 weeks used in myocardial skinned strip experiments.
| F764L/+ (n=6) | S532P/+ (n=6) | +/+ (n=12) | |
|---|---|---|---|
| Body mass (g) | 26.8±0.8 * | 25.4±0.8 * | 31.4±1.1 |
| LV mass (mg) | 120±6 | 100±2 | 114±4 |
| RV mass (mg) | 51±9 | 29±1 * | 35±1 |
| LV/Body (mg/g) | 4.49±0.24 ** | 3.96±0.18 | 3.65±0.08 |
| LV+RV/Body (mg/g) | 6.45±0.54 ** | 5.11±0.24 | 4.80±0.10 |
| β-MyHC (% of total) | 33±7 * | 9±3 | 15±2 |
= different from +/+ controls at P<0.05,
P<0.01 by S-N-K.
n = number of hearts. For MyHC, n= 5 F764L/+, 5 S532P/+ and 10 +/+.
The F764L/+ demonstrated significantly higher (~25–40%) content of β-MyHC compared to controls (Figure 1A and Table 2). There was no significant differential incorporation of β-MyHC in the S532P/+ mice compared to controls. The presence of the β-MyHC, which demonstrates slower kinetics and longer ton compared to α-MyHC [12, 14, 20, 22, 23], must be kept in mind when interpreting the kinetic results from the F764L/+.
Figure 1.

One heart from each population is shown to illustrate the relative α- vs β-MyHC isoform content in these heterozygous mice compared to controls. A. In older mice aged 40–60 wks, a significant proportion of β-MyHC, a hallmark of developing cardiomyopathy, was found in the F764L/+ compared to S532P/+ and +/+ controls. B. In younger mice aged 20–30 wks, β-MyHC content was also higher in F764L/+ compared to S532P/+ and +/+ controls. C. Normalized isometric tension vs pCa demonstrated a maximum calcium activated condition at pCa 4.8, 17°C and 2.2 μm sarcomere length used throughout the study. n = number of hearts, two strips per heart.
3.3 Tension-pCa relationships
Maximum Tdev in F764L/+ was only ~50% of controls, while Tdev in the S532P/+ was similar to controls (Table 3). The normalized developed tension-pCa relationships revealed that calcium activation at pCa 6.0 was lower in the DCM mutants compared to controls, but sensitivity to calcium, pCa50, was not different among the three groups (5.84±0.03 for F764L/+, 5.84±0.02 for S532P/+ and 5.88±0.02 for +/+). As illustrated in Figure 1C, maximum calcium activation was achieved with pCa 4.8, which was the activation concentration used for the remaining examinations.
Table 3.
Parameters of maximum calcium-activated tension and tension-velocity relationships at 17°C and 2.2 μm sarcomere length.
| F764L/+ (n=6) | S532P/+ (n=6) | +/+ (n=12) | |
|---|---|---|---|
| Tmax (mN.mm−2) | 16.74±2.00 ** | 27.66±1.70 | 27.76±1.21 |
| Tdev (mN.mm−2) | 14.65±1.92 ** | 25.15±1.47 | 25.14±1.15 |
| Vmax (10−3 ML/s) | 246±27 | 272±28 | 265±17 |
| Vopt (10−3 ML/s) | 89±10 | 111±8 | 109±5 |
| Topt/Tmax (%) | 36.3±7.7 * | 41.5±1.8 | 41.7±1.2 |
| Pmax (10−3 ML/s × T/Tmax) | 32.5±3.7 * | 45.7±2.7 | 44.9±1.9 |
Differences from controls reported at *P<0.05, **P<0.01 by S-N-K. n= number of hearts, two strips per heart.
3.4 Force-velocity relationships
Myocardial shortening velocities in the F764L/+ were statistically depressed at loads greater than 0.4 × maximum tension (Tmax) compared to controls (Figure 2A). Mechanical power output was similarly reduced in the F764L/+ (Figure 2B). Maximum unloaded velocity (Vmax) and velocity of maximum power production (Vopt) were not different between the DCM mutant groups and their respective controls (Table 3). Maximum normalized power (Pmax) and the tension at which Pmax is reached (Topt/Tmax) were significantly lower in the F764L/+ versus controls (Table 3). In contrast, no force-velocity parameters were different in the S532P/+ compared to controls (Table 3).
Figure 2.
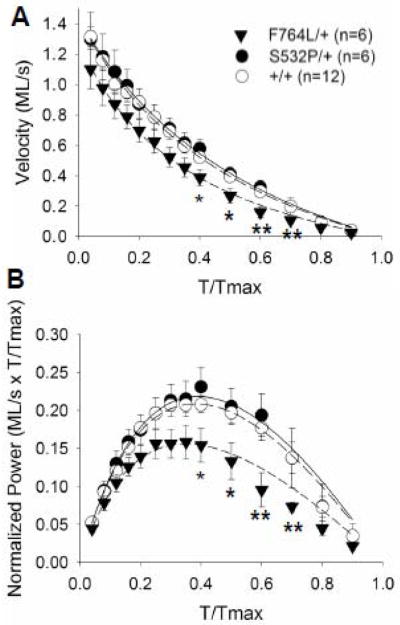
Velocity- and power-tension relationships. A. Shortening velocity in the F764L/+ myocardial strips was significantly lower at several loads compared to +/+, while maximum velocity near unloaded conditions was similar between F764L/+ and +/+. There were no differences between S532P/+ and +/+. B. Power normalized to maximum isometric tension was also significantly lower in F764L/+ at specific loads, but was similar between S532P/+ and controls. Curves represent fit of Equation 1 to mean values. *P<0.05, **P<0.01 compared to +/+ controls by S-N-K. n = number of hearts, two strips per heart.
3.5 Length perturbation analysis and crossbridge stiffness
Calcium-activated F764L/+ myocardial strips displayed lower elastic and viscous moduli at frequencies greater than 1.6 Hz compared to +/+ controls (Figures 3A and 3B). Under rigor conditions with no MgATP, the elastic and viscous moduli were reduced by ~40% across the entire frequency range in the F764L/+ compared to +/+ controls (Figures 3C and 3D). Previous work suggests that the converter domain, where the F764L mutation resides, is responsible to a high degree for crossbridge stiffness [24, 25]. Our data suggest that the F764L mutation leads to a more compliant converter domain and myosin crossbridge, which in turn is responsible for the reduced developed tension and reduced elastic and viscous moduli under activated conditions.
Figure 3.

Elastic and viscous moduli measured at maximum calcium activation and rigor conditions. A. At maximum calcium activation and 5 mM MgATP, elastic modulus of the F764L/+ was significantly lower compared to +/+ over all frequencies examined. Elastic modulus of the S532P/+ was reduced for some low frequencies (inset). B. Viscous modulus was likewise lower in the F764L/+ for frequencies greater than 2 Hz. C and D. Under rigor conditions, both elastic and viscous moduli in the F764L/+ were reduced to about 50% compared to +/+. These data indicate that the F764L mutation, which resides in the myosin converter domain, significantly reduces the stiffness of the strongly bound myosin crossbridge. Values for elastic and viscous moduli at any frequency were similar between S532P/+ and controls. *P<0.05 for F764L/+ compared to controls. †P<0.05 for S532P/+ compared to controls. n = number of hearts, two strips per heart.
The elastic moduli of the S532P/+ strips was reduced at several frequencies between 1–15 Hz compared to +/+ controls (Figure 3A, inset). The elastic and viscous moduli under rigor conditions were not different between S532P/+ and +/+ (Figures 3C and 3D), indicating no change in crossbridge compliance with the S532P mutation.
3.6 Effects of MgATP on mutation-related kinetic differences
Figure 4 illustrates the influence of varying MgATP concentration on model parameters observed from low (0.1 mM) to saturating (5 mM) MgATP. The parameter A, which reflects the magnitude of myocardial visco-elastic stiffness, was significantly reduced in the F764L/+ compared to controls over all MgATP examined. Parameter A was similarly reduced in S532P/+ for concentrations of MgATP above 0.4 mM (Figure 4A). Parameter k indicates the degree of viscous quality (as k approaches 1) vs elastic quality (as k approaches 0) of the muscle viscoelasticity. The value of k was not affected in the F764L/+, but was elevated in the S532P/+ (Figure 4B). These data suggest that under activated conditions both DCM mutations effectively reduced overall myocardial stiffness and that an additional effect of the S532P mutation was to promote a more viscous quality to the overall stiffness.
Figure 4.

Model parameters over varying MgATP concentrations. A and B. Parameters A and k, which reflect the passive viscoelastic stiffness characteristics of the muscle strips, demonstrated significantly lower stiffness, A, in the DCM mutants. This can be most easily seen in the raw data of Figures 3A and 3C as lower values for the elastic moduli in both mutants. The higher value for k in the S532L/+ indicates a higher viscous rather than elastic quality in the myocardial viscoelastic stiffness. C. The magnitude B was not different among the groups. D. The rate parameter 2πb, which reflects the rate of force development after quick stretch, was significantly higher in the DCM mutants compared to respective controls at higher MgATP concentrations. E. Magnitude C was elevated in the S532P/+ and, given no change in rigor stiffness with this mutation (Figure 3D), indicates a higher myosin crossbridge duty ratio. F. The rate parameter 2πc, which reflects the myosin crossbridge detachment rate, was significantly lower in the F764L/+ compared to +/+ at high MgATP, and significantly higher in the S532P/+ at 0.8 mM MgATP. *P<0.05 and **P<0.01 for F764L/+ compared to controls, †P<0.05 for S532P/+ compared to controls.
Parameter B, which reflects the number and stiffness of crossbridges undergoing work-producing isomerization [16, 17], was not different among the groups (Figure 4C). Parameter C reflects the product of the number and stiffness of crossbridges in the post-power stroke state [14] and was higher in the S532P/+ at MgATP greater than 1 mM (Figure 4E). Considering that crossbridge stiffness is not affected in the S532P/+, the higher value for magnitude C would suggest a greater recruitment, or duty ratio, in the S532P/+.
Parameter 2πb was markedly higher in the myocardial strips bearing DCM mutant myosin compared to controls (Figure 4D) and will be interpreted in further detail in the Discussion section. Parameter 2πc was significantly lower in the F764L/+ compared to controls at MgATP greater than 1 mM (Figure 4F). These data reflect a longer myosin crossbridge lifetime, ton, in the F764L/+ compared to +/+ at saturating MgATP (Figure 5A). Values for 2πc were significantly higher in the S532L/+ compared to +/+ at intermediate concentrations 0.4–0.8 mM MgATP, but were not different at more saturating concentrations of MgATP. The higher sensitivity of the S532P/+ to MgATP at these sub-saturating concentrations of MgATP is more apparent when the detachment rates are normalized as shown in Figure 5B.
Figure 5.

Estimated myosin crossbridge lifetime, ton, and normalized detachment rate over varying MgATP concentrations. A. Myosin crossbridge ton is reduced with increasing MgATP concentrations. Crossbridge ton at saturating MgATP was longer in the F764L/+ indicative of the significant incorporation of β-MyHC. B. Myosin detachment was normalized to the average detachment rate between 2 and 5 mM MgATP. The detachment rates of the myocardium bearing the DCM mutants reach saturation values at lower MgATP concentrations compared to controls and is reflected in their higher values for k+ATP and MgATP50. C. The rate parameter 2πb was significantly higher in the DCM mutants compared to respective controls, but 2πb was not differentially sensitive to Pi concentration due to incorporation of DCM mutant myosin. *P<0.05 for F764L/+ compared to controls, †P<0.05 for S532P/+ compared to controls.
3.7 MgATP-dependent kinetics
Myosin detachment rates and their sensitivity to MgATP, illustrated in Figure 4F, were obtained from fits of equation 4 to these data. The rate of MgADP release, k−ADP, was significantly lower in F764L/+ compared to +/+, but was not different between S532P/+ and +/+ (Table 4). Values for k−ADP in the S532P/+ and both sets of +/+ controls were similar to those values reported previously for 100% α-MyHC of the same mouse strain, 129/SvEv [12]. Using values for k−ADP from our previous work [12] and assuming a linear combination of 33% β-MyHC at 24 s−1 and 67% α-MyHC at 111 s−1, k−ADP for the F764L/+ would be predicted to be 82 s−1, which is within the standard error found in the present study (Table 4). Therefore, the lower value for k−ADP we report here in the F764L/+ can be explained by the presence of ~33% β-MyHC, although we cannot completely discount the possibility that k−ADP might be lower in the F764L mutant α-MyHC compared to wild-type α-MyHC at saturating MgATP.
Table 4.
Variables of myosin enzyme kinetics model applied to myosin detachment rate 2πc vs MgATP relationship at 17°C and 2.2 μm sarcomere length. MgATP50 = (k−ADP/k+ATP).
| F764L/+ (n=6) | S532P/+ (n=6) | +/+ (n=12) | |
|---|---|---|---|
| k−ADP (s−1) | 80.9±9.1 ** | 118.7±9.5 | 111.7±4.4 |
| k+ATP (mM−1.s−1) | 422±109 # | 483±74 * | 303±18 |
| t−ADP (ms) | 13.2±1.8 * | 8.7±0.6 | 9.1±0.5 |
| MgATP50 (μM) | 204±31 ** | 276±45 * | 377±18 |
Different from +/+ controls at *P<0.05, **P<0.01 by S-N-K, #P=0.069. n = number of hearts, two strips per heart.
The MgATP binding rate, k+ATP, was higher in both DCM mutants compared to controls (Table 4). This result is also reflected in the significantly lower MgATP50. We might have expected a lower k+ATP in the F764L/+ given the incorporation of 33% β-MyHC, which displays a k+ATP about half that of α-MyHC [12]. The higher k+ATP measured for the F764L/+ must therefore be due to the incorporation of F764L myosin and not due to β-MyHC.
3.8 Pi-dependent kinetics
The parameter 2πb has been interpreted as being proportional to Pi release and rebinding rates [16, 17]. The markedly higher values of 2πb in both DCM mutants suggests the mutant myosin possesses greater sensitivity and susceptibility to detachment after Pi rebinding. We added Pi to the activating solution to test the hypothesis that the elevated 2πb was due to higher rates of Pi release and/or rebinding in the myocardium bearing the DCM myosin. The elastic and viscous moduli demonstrated frequency shifts to higher frequency with added Pi. These frequency shifts in the moduli were visually similar among all groups examined here (see Supplemental Material).
As Pi was added from 0 to 4 mM, 2πb remained high in the DCM compared to controls (Figure 5C). The Pi-sensitivities of other model parameters are provided in the Supplemental Material. A repeated-measures ANOVA was applied to 2πb over the varying Pi concentrations using a between-subject variable genotype defined as DCM or control. There were significant main effects for 2πb due to genotype and Pi concentration, but there was no significant genotype × Pi interaction and therefore no suggestion of a differential sensitivity to Pi between the DCM mutants and controls.
4. DISCUSSION
The most significant findings of this study were the following. (i) LV developed pressure was significantly lower in both DCM groups compared to controls and more so in F764L/+ mice than in S532P/+. This indicates a site-specific differential effect of the point mutations on myofilament function in the working heart that roughly correlated with the extent of DCM as the mice aged. (ii) In younger mice of the same genotypes, the maximum developed tension achieved in Ca2+-activated skinned strips from the F764L/+ was about half that of controls. There were similarly lower velocities and power at higher applied loads in the F764L/+. Corresponding indices in S532P/+ strips were not different from controls. (iii) Ca2+-activated myofilament stiffness was significantly lower in both DCM mutants compared to controls, although myofilament stiffness under rigor conditions was lower only in F764L/+ and not in S532P/+. (iv) The rate of force development (2πb) was higher in F764L/+ and S532P/+ strips compared to controls at saturating MgATP and independent of Pi. And (v) MgATP-dependency of the myosin detachment rate (2πc) demonstrated a higher MgATP binding rate in both F764L/+ and S532P/+. Points iii, iv and v indicate alterations in myofilament function that underlie developing DCM and at least in the case of the S532P/+ precede discernable alterations in ventricular morphology indicative of DCM.
4.1 Effect of mutations on molecular vs myocardial function
We observed a reduction in contractile function for the F764L/+ ventricle and skinned myocardium compared to S532P/+ and +/+ as reported previously [8]. Our findings, however, are not consistent with results at the molecular level, where actin sliding velocities were reduced in both F764L and S532P mutant myosins and force generation was reduced in the S532P [9]. Furthermore, our finding a higher MgATP binding rate, k+ATP, for the DCM myocardial strips suggests a shorter myosin crossbridge lifetime, ton, in the DCM myosin at low MgATP concentrations; however, ton at 10 μM MgATP was not shorter in the DCM myosin but reported longer in the S532P [8]. These discrepancies between molecular-level and myocardial-level function may be due in part to differences in assay geometry and in post-translational modifications. Our preparations maintain an intact myofilament lattice structure, which imparts internal stresses not present in the isolated molecule assays [12]. Differential post translational modifications may also arise due to different stages of development of the cardiomyopathy in the F764L/+ and S532P/+ mice used here compared to the homozygotes used in the isolated molecule assays or to differences in sample preparation procedures between assays. Any one of these differences could theoretically account for discrepancies between the isolated DCM mutant myosin and the skinned DCM myocardial strips. How exactly these differences may be responsible for the different functional results is unclear. We believe the reduced function in these DCM mutant myosin observed at the molecular level may arise through mechanisms that dominate myosin function outside the context of the sarcomere and myofilament lattice [6, 12].
4.2 Molecular mechanisms of DCM development
The prolonged ton in F764L/+ can be explained by the partial switch in myosin heavy chain isoform expression from fast α-MyHC to slow β-MyHC with ensuing reduction in MgADP release rate [12, 14, 20, 22, 23]. Values for the rates 2πb and k+ATP for β-MyHC were previously reported to be ~50% those for α-MyHC [12]; thus β-MyHC in the F764L/+ cannot account for the higher 2πb and k+ATP we report, and we conclude the F764L mutant myosin must be responsible for the elevated rates. A reduced activated stiffness would also not be expected from the presence of β-MyHC in the F764L/+ [12]. Similarly, the S532P mutant myosin, which is the only other myosin besides the wildtype α-MyHC in the S532P/+ myocardium, must underlie the higher 2πb and k+ATP and reduced activated stiffness observed in the S532P/+. These results suggest that an enhanced rate of force development, 2πb, higher rate of MgATP binding and reduced Ca2+-activated stiffness are common functional characteristics of these two myosin mutations in the context of an intact myofilament lattice and may ultimately underlie or predict the development of DCM.
How can two point mutations, one located in the converter region (F764L) and the other in the actin-binding domain (S532P), result in a similar elevation in 2πb and k+ATP and reduction in Ca2+-activated stiffness? And how can these functional characteristics lead to DCM? We address these questions below.
4.2.1 Higher rate of force development, 2πb, and reduced Ca2+-activated stiffness
The rate parameter 2πb reflects the rate of force development similar to that observed in phase 3 of a quick stretch experiment as demonstrated previously [14, 17]. This rate of force development was significantly higher in both DCM mutants compared to controls over several concentrations of MgATP (Figure 4D) and, as measured using quick stretch, was similarly elevated in another mouse model of DCM caused by the absence of myosin binding protein-C [26] and in two mouse models of R92 mutations in troponin-T causing FHC [27].
The parameter 2πb has been interpreted as being proportional to the sum of the transition rates between the weakly-bound and strongly-bound myosin crossbridge states, i.e., between the non-force generating and force generating states of myosin [16–18]. With this interpretation, the DCM myosins would be inherently more likely to undergo a forward-going or reverse-going power stroke [16, 17]. If the DCM mutations lead to a higher probability of power stroke reversal independent of Pi, as our data suggest, this mechanism could conceivably account for a reduced Ca2+-activated stiffness and a reduced resistance to myocardial lengthening, which in turn could account for a higher myocardial susceptibility to stretch and eventual dilation under normal myocardial stresses. The reduced crossbridge stiffness due to the more compliant F764L mutated converter domain would conceivably further contribute to the reduced Ca2+-activated stiffness observed in the F764L/+ and the further susceptibility to stretch by myocardial stresses.
4.2.2 Higher rate of MgATP binding, k+ATP
We propose that k+ATP is dictated in part by the degree of mechanical compression on the side of the myosin molecule harboring the nucleotide binding site. In the case of the F764L mutated α-MyHC, which leads to a highly compliant converter domain, the degree of nucleotide binding site compression would be low relative to that in a wildtype α-MyHC. The reduced compression of the pocket results in a higher probability of MgATP binding at the nucleotide binding site and thus a higher k+ATP. A similar phenomenon occurs when an assistive load is applied to isolated myosin and the probability of MgATP binding is enhanced [28]. In the case of the S532P, we speculate that the point mutation leads to a similarly less compressed nucleotide binding site by a different means, perhaps by affecting the stiffness of the region localized to the nucleotide binding pocket or of the actin binding domain.
Because physiological MgATP content is thought to be at saturating concentrations ~10 mM [29], it is unlikely that an elevated MgATP binding rate would play a role in the development of DCM. If, however, MgATP levels were in fact abnormally low (e.g., <1 mM) as occurs with heart failure [29], the higher k+ATP could shorten myosin crossbridge lifetimes compared to non-DCM myocardium under similar conditions, a kinetic alteration that could contribute to the observed reduction in myocardial stiffness. As there is as yet no basis for postulating such low MgATP levels, it is more likely that the higher k+ATP is a concomitant side-effect or byproduct of the molecular mechanisms responsible for the higher rate of power stroke reversal reflected in 2πb, and that the higher k+ATP may be a marker or even a predictor of DCM but not a direct contributor to DCM development.
4.3 Limitations
The quantitative analysis and interpretation of our results rely upon the application of model Equation 2, which describes the measured viscoelastic characteristics of a myocardial skinned strip. We have demonstrated previously that this model equation fits very well the raw data collected from mouse myocardium, and that the specific mathematical expression given in the C-term of Equation 2 is expected to arise from the enzymatic activity of myosin in striated muscle [14]. We present in the Supplemental Material a demonstration of the fits over varying MgATP concentrations and also the residuals of these fits. The plots of the residuals demonstrate that some features of the raw data are slightly, yet systematically misrepresented by the model equation and that more work is necessary to fully account for the viscoelastic properties of striated muscle that arise from myosin crossbridge formation. Nevertheless, the representation of the C-term at the highest frequencies appears to be well founded, and in this frequency range >3 Hz the residuals are never more than 5% of the magnitude of the complex modulus. We believe the corresponding calculation of crossbridge ton yields valid, even if imprecise, values for making comparisons between groups.
We are also limited by our not knowing the exact content of mutated α-MyHC in the DCM mouse hearts. One might assume that somewhere between ~30–70% endogenous α-MyHC carries the mutation in the DCM mice, and it would be difficult to estimate the influence of the DCM myosin on myofilament function. Nevertheless, we would expect a more pronounced phenotype and even higher values for 2πb and k+ATP in myocardium from homozygotes compared to those measured here in heterozygotes.
4.4 Conclusion
We report that an enhanced rate of force development, 2πb, higher rate of MgATP binding, k+ATP, and reduced Ca2+-activated stiffness are common functional characteristics of the two DCM mutations in mouse α-MyHC examined here at the myocardial level. Whether these two DCM mutations in human β-MyHC would result in the same function effects is not known at this time, although an enhanced rate of force development has been reported with a FHC-causing mutation in troponin-T in a mouse β-MyHC background [27]. We speculate that the DCM-related functional characteristics reported here reflect the underlying cause (or are concomitant with the underlying cause) of DCM.
Supplementary Material
Highlights.
F764L and S532P point mutations were engineered into mouse cardiac myosin.
Dilated cardiomyopathy developed in heterozygous F764L/+ and S532P/+ mice.
Myocardial strips exhibited higher rates of force development and MgATP binding.
These alterations in myofilament function preceded development of DCM.
Acknowledgments
This study was funded by NIH grant P01 HL59408.
Footnotes
DISCLOSURES
None declared.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Watkins H, Rosenzweig A, Hwang DS, Levi T, McKenna W, Seidman CE, et al. Characteristics and prognostic implications of myosin missense mutations in familial hypertrophic cardiomyopathy. N Engl J Med. 1992;326(17):1108–14. doi: 10.1056/NEJM199204233261703. [DOI] [PubMed] [Google Scholar]
- 2.Towbin JA. The role of cytoskeletal proteins in cardiomyopathies. Current opinion in cell biology. 1998;10(1):131–9. doi: 10.1016/s0955-0674(98)80096-3. [DOI] [PubMed] [Google Scholar]
- 3.Seidman JG, Seidman C. The genetic basis for cardiomyopathy: from mutation identification to mechanistic paradigms. Cell. 2001;104(4):557–67. doi: 10.1016/s0092-8674(01)00242-2. [DOI] [PubMed] [Google Scholar]
- 4.Daehmlow S, Erdmann J, Knueppel T, Gille C, Froemmel C, Hummel M, et al. Novel mutations in sarcomeric protein genes in dilated cardiomyopathy. Biochemical and biophysical research communications. 2002;298(1):116–20. doi: 10.1016/s0006-291x(02)02374-4. [DOI] [PubMed] [Google Scholar]
- 5.Kamisago M, Sharma SD, DePalma SR, Solomon S, Sharma P, McDonough B, et al. Mutations in sarcomere protein genes as a cause of dilated cardiomyopathy. N Engl J Med. 2000;343(23):1688–96. doi: 10.1056/NEJM200012073432304. [DOI] [PubMed] [Google Scholar]
- 6.Moore JR, Leinwand L, Warshaw DM. Understanding cardiomyopathy phenotypes based on the functional impact of mutations in the myosin motor. Circ Res. 2012;111(3):375–85. doi: 10.1161/CIRCRESAHA.110.223842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rayment I, Rypniewski WR, Schmidt-Base K, Smith R, Tomchick DR, Benning MM, et al. Three-dimensional structure of myosin subfragment-1: a molecular motor. Science. 1993;261(5117):50–8. doi: 10.1126/science.8316857. [DOI] [PubMed] [Google Scholar]
- 8.Schmitt JP, Debold EP, Ahmad F, Armstrong A, Frederico A, Conner DA, et al. Cardiac myosin missense mutations cause dilated cardiomyopathy in mouse models and depress molecular motor function. Proc Natl Acad Sci U S A. 2006;103(39):14525–30. doi: 10.1073/pnas.0606383103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Debold EP, Schmitt JP, Moore JR, Patlak JB, Beck SE, Seidman JG, et al. Hypertrophic and dilated cardiomyopathy mutations differentially affect the molecular force generation of mouse {alpha}-cardiac myosin in the laser trap assay. Am J Physiol Heart Circ Physiol. 2007 doi: 10.1152/ajpheart.00128.2007. [DOI] [PubMed] [Google Scholar]
- 10.Kameyama T, Chen Z, Bell SP, Fabian J, LeWinter MM. Mechanoenergetic studies in isolated mouse hearts. Am J Physiol. 1998;274(1 Pt 2):H366–74. doi: 10.1152/ajpheart.1998.274.1.H366. [DOI] [PubMed] [Google Scholar]
- 11.Godt RE, Lindley BD. Influence of temperature upon contractile activation and isometric force production in mechanically skinned muscle fibers of the frog. J Gen Physiol. 1982;80(2):279–97. doi: 10.1085/jgp.80.2.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Y, Tanner BC, Lombardo AT, Tremble SM, Maughan DW, Vanburen P, et al. Cardiac myosin isoforms exhibit differential rates of MgADP release and MgATP binding detected by myocardial viscoelasticity. J Mol Cell Cardiol. 2013;54(1):1–8. doi: 10.1016/j.yjmcc.2012.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McDonald KS, Wolff MR, Moss RL. Force-velocity and power-load curves in rat skinned cardiac myocytes. The Journal of physiology. 1998;511 ( Pt 2):519–31. doi: 10.1111/j.1469-7793.1998.519bh.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Palmer BM, Suzuki T, Wang Y, Barnes WD, Miller MS, Maughan DW. Two-state model of acto-myosin attachment-detachment predicts C-process of sinusoidal analysis. Biophysical journal. 2007;93(3):760–9. doi: 10.1529/biophysj.106.101626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mulieri LA, Barnes W, Leavitt BJ, Ittleman FP, LeWinter MM, Alpert NR, et al. Alterations of myocardial dynamic stiffness implicating abnormal crossbridge function in human mitral regurgitation heart failure. Circ Res. 2002;90(1):66–72. doi: 10.1161/hh0102.103221. [DOI] [PubMed] [Google Scholar]
- 16.Kawai M, Saeki Y, Zhao Y. Crossbridge scheme and the kinetic constants of elementary steps deduced from chemically skinned papillary and trabecular muscles of the ferret. Circ Res. 1993;73(1):35–50. doi: 10.1161/01.res.73.1.35. [DOI] [PubMed] [Google Scholar]
- 17.Kawai M, Brandt PW. Sinusoidal analysis: a high resolution method for correlating biochemical reactions with physiological processes in activated skeletal muscles of rabbit, frog and crayfish. J Muscle Res Cell Motil. 1980;1(3):279–303. doi: 10.1007/BF00711932. [DOI] [PubMed] [Google Scholar]
- 18.Kawai M, Zhao Y, Halvorson HR. Elementary steps of contraction probed by sinusoidal analysis technique in rabbit psoas fibers. Advances in experimental medicine and biology. 1993;332:567–77. doi: 10.1007/978-1-4615-2872-2_51. discussion 77–80. [DOI] [PubMed] [Google Scholar]
- 19.Campbell KB, Chandra M, Kirkpatrick RD, Slinker BK, Hunter WC. Interpreting cardiac muscle force-length dynamics using a novel functional model. Am J Physiol Heart Circ Physiol. 2004;286(4):H1535–45. doi: 10.1152/ajpheart.01029.2003. [DOI] [PubMed] [Google Scholar]
- 20.Tyska MJ, Warshaw DM. The myosin power stroke. Cell Motil Cytoskeleton. 2002;51(1):1–15. doi: 10.1002/cm.10014. [DOI] [PubMed] [Google Scholar]
- 21.Williams JL, Hathaway CA, Kloster KL, Layne BH. Low power, type II errors, and other statistical problems in recent cardiovascular research. Am J Physiol. 1997;273(1 Pt 2):H487–93. doi: 10.1152/ajpheart.1997.273.1.H487. [DOI] [PubMed] [Google Scholar]
- 22.Sugiura S, Kobayakawa N, Fujita H, Yamashita H, Momomura S, Chaen S, et al. Comparison of unitary displacements and forces between 2 cardiac myosin isoforms by the optical trap technique: molecular basis for cardiac adaptation. Circ Res. 1998;82(10):1029–34. doi: 10.1161/01.res.82.10.1029. [DOI] [PubMed] [Google Scholar]
- 23.VanBuren P, Harris DE, Alpert NR, Warshaw DM. Cardiac V1 and V3 myosins differ in their hydrolytic and mechanical activities in vitro. Circ Res. 1995;77(2):439–44. doi: 10.1161/01.res.77.2.439. [DOI] [PubMed] [Google Scholar]
- 24.Kohler J, Winkler G, Schulte I, Scholz T, McKenna W, Brenner B, et al. Mutation of the myosin converter domain alters cross-bridge elasticity. Proc Natl Acad Sci U S A. 2002;99(6):3557–62. doi: 10.1073/pnas.062415899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Seebohm B, Matinmehr F, Kohler J, Francino A, Navarro-Lopez F, Perrot A, et al. Cardiomyopathy mutations reveal variable region of myosin converter as major element of cross-bridge compliance. Biophysical journal. 2009;97(3):806–24. doi: 10.1016/j.bpj.2009.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stelzer JE, Dunning SB, Moss RL. Ablation of cardiac myosin-binding protein-C accelerates stretch activation in murine skinned myocardium. Circ Res. 2006;98(9):1212–8. doi: 10.1161/01.RES.0000219863.94390.ce. [DOI] [PubMed] [Google Scholar]
- 27.Ford SJ, Mamidi R, Jimenez J, Tardiff JC, Chandra M. Effects of R92 mutations in mouse cardiac troponin T are influenced by changes in myosin heavy chain isoform. J Mol Cell Cardiol. 2012;53(4):542–51. doi: 10.1016/j.yjmcc.2012.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kad NM, Patlak JB, Fagnant PM, Trybus KM, Warshaw DM. Mutation of a conserved glycine in the SH1-SH2 helix affects the load-dependent kinetics of myosin. Biophysical journal. 2007;92(5):1623–31. doi: 10.1529/biophysj.106.097618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ingwall JS, Weiss RG. Is the failing heart energy starved? On using chemical energy to support cardiac function. Circ Res. 2004;95(2):135–45. doi: 10.1161/01.RES.0000137170.41939.d9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.