

Summary
Mucopolysaccharidoses (MPS) are a group of lysosomal storage diseases caused by mutations in lysosomal enzymes involved in degradation of glycosaminoglycans (GAGs). Patients with MPS grow poorly and become physically disabled due to systemic bone disease. While many of the major skeletal effects in mouse models for MPS have been described, no detailed analysis that compares GAGs levels and characteristics of bone by micro-CT has been done.
The aims of this study were to assess severity of bone dysplasia among four MPS mouse models (MPS I, IIIA, IVA and VII), to determine the relationship between severity of bone dysplasia and serum keratan sulfate (KS) and heparan sulfate (HS) levels in those models, and to explore the mechanism of KS elevation in MPS I, IIIA, and VII mouse models.
Clinically, MPS VII mice had the most severe bone pathology; however, MPS I and IVA mice also showed skeletal pathology. MPS I and VII mice showed severe bone dysplasia, higher bone mineral density, narrowed spinal canal, and shorter sclerotic bones by micro-CT and radiographs. Serum KS and HS levels were elevated in MPS I, IIIA, and VII mice. Severity of skeletal disease displayed by micro-CT, radiographs and histopathology correlated with the level of KS elevation. We showed that elevated HS levels in MPS mouse models could inhibit N-acetylgalactosamine-6-sulfate sulfatase enzyme.
These studies suggest that KS could be released from chondrocytes affected by accumulation of other GAGs and that KS could be useful as a biomarker for severity of bone dysplasia in MPS disorders.
Introduction
Mucopolysaccharidoses (MPS) are a group of lysosomal storage diseases caused by deficiencies of lysosomal enzymes required for the stepwise degradation of glycosaminoglycans (GAGs). Each type of MPS is associated with a specific enzyme deficiency that results in accumulation of a specific group of GAG(s) in cellular lysosomes throughout the body, their release into the bloodstream, and excretion in urine. Elevated levels of some combination of the GAGs, keratan sulfate (KS), heparan sulfate (HS), dermatan sulfate (DS), chondroitin sulfate (CS), and hyaluronan are commonly found in the blood and urine of MPS patients. The clinical manifestations of MPS are progressive and vary widely both among and within specific types of MPS. The most common clinical manifestations include skeletal deformities (dysostosis multiplex), coarse facial features, mental retardation, organomegaly, short stature, and corneal opacity (Neufeld et al. 2001; Aldenhoven et al. 2009). Most clinical phenotypes in MPS patients have been well studied and characterized. Recently, mouse models of MPS have been found naturally or have been generated using targeted mutagenesis. These models have been used to develop and evaluate the effectiveness of therapies including enzyme replacement therapy, hematopoietic stem cell transplantation, gene therapy, and substrate reduction therapy for multiple types of MPS (Fraldi et al. 2007; Haskins 2007; Grubb et al. 2008; Herati et al. 2008; Jung et al. 2010; Tomatsu et al. 2010). Thorough characterization of these mouse models provides critical information about the similarities and differences between the mouse model phenotypes and the corresponding human disease.
Recent studies showed severe skeletal abnormalities in MPS I (Hurler syndrome) and MPS VII (Sly syndrome) mouse models and milder skeletal abnormalities in MPS IIIA (Sanfilippo syndrome) and MPS IVA (Morquio type A syndrome) mouse models (Clarke et al. 1997; Bhaumik et al. 1999; Bhattacharyya et al. 2001; Tomatsu et al. 2005a; Sly et al. 2001). Skeletal abnormalities found in MPS I mice include a flattened facial profile; thickening of digits; anterior flaring of ribs; and thickening of facial bones, ribs, and fibulae (Clarke et al. 1997). Similar abnormalities have been found in MPS VII mice, including facial dysmorphism, growth retardation, shortened extremities, sclerosis of the calvarium, sclerotic long bones, broad zygomatic arches, narrow rib cage, and blunted nose (Sly et al. 2001). Although skeletal defects in MPS IIIA mouse models are milder, they include thickened calvarium, vertebral deformation, and cartilaginous matrix proliferation into the spinal canal (Bhaumik et al. 1999; Bhattacharyya et al. 2001). While severe skeletal abnormalities are seen in patients with MPS IVA, a radiographic analysis of mixed background MPS IVA mouse models at 4 months of age did not show any obvious abnormalities in long bones, thorax, or calvaria (Tomatsu et al. 2003; Tomatsu et al. 2005a; Tomatsu et al. 2007).
Analysis of skeletal abnormalities was also carried out on a mouse model for Hunter syndrome (MPS II). Radiographs showed sclerosis and enlargement of skull bones in 4-week-old mice and appendicular bone enlargement in 10- to 13-week-old mice. Nearly all bones were affected in mice 39–40 weeks of age. Micro-CT scans of the tarsotibial joints at age 48 weeks showed severe calcification of the calcaneous tendon and periosteal bone formation on the lateral aspect of the distal tibia. Dual energy X-ray absorptiometry showed slightly increased whole-body bone mineral density (BMD) in the model mice compared with wild-type (WT) control mice. However, BMD of individual bones was not measured. Computed tomography was used to measure bone cross-sectional area and showed that the cross-sectional area was larger in the model mice than in WT mice (Garcia et al. 2007). Although the major skeletal abnormalities in mouse models have been described, skeletal pathology and bone density have not been well characterized, and no detailed analysis has compared the bone dysplasia among different MPS mouse models.
Until recently, each type of MPS was thought to be characterized by accumulation of a specific group of GAGs in tissues and specific cell types. Thus, KS was thought to be accumulated only in MPS IV patients, since the enzyme deficiency in MPS IV is involved in the degradation pathway of KS. However, it has been shown recently that KS is also elevated in the serum of patients with MPS I, II, III, VI, and VII (Tomatsu et al. 2005b). The mechanism of this secondary KS elevation is not known.
In this study, we performed a detailed characterization of the skeletal dysplasia in mouse models for MPS types I, IIIA, IVA, and VII using micro-CT, radiographs, and histopathology. In addition, we measured the serum HS and KS levels of MPS I, IIIA, IVA, and VII to evaluate the relation between severity of bone dysplasia and specific GAG levels. We also performed an enzyme inhibition assay with the goal of explaining the secondary KS elevation described in MPS patients.
Materials and Methods
Materials
Mouse models
We used four different mouse models (MPS I, MPS IIIA, MPS IVA, and MPS VII) to assess bone dysplasia in this study. The enzyme deficiency of each mouse model is as follows: α-L-iduronidase (IDUA) for MPS I, heparan-N-sulfatase for MPS IIIA, N-acetylgalactosamine-6-sulfate sulfatase (GALNS) for MPS IVA, and β-glucuronidase (GUSB) for MPS VII. All MPS mouse models used in this study have been generated using a recombinant technique. Each model mouse was backcrossed onto C57BL/6J to create the congenic background. All experiments were conducted with the highest standards of humane animal care approved by the local committee at Saint Louis University
MPS I knockout mouse
MPS I knockout model mice older than 4 weeks have facial characteristics similar to MPS I patients and dysostosis multiplex including thickened ribs, zygomatic arches, long bones and skull (Clarke et al. 1997). The mouse model was purchased from Jackson Laboratory (Bar Harbor, ME: catalogue number: 004068).
MPS IIIA knockout mouse
No differences have been observed between MPS IIIA knockout mice and normal mice before 6–7 months of age. After 6–7 months, MPS IIIA mice appear less active; their coats are scruffy; and they have a hunched posture, distended bladder, hepatosplenomegaly, corneal opacity, vertebral deformation, and thickened calvarium (Bhaumik et al. 1999; Bhattacharyya et al. 2001). The mouse model was purchased from Jackson Laboratory (Bar Harbor, ME: catalogue number: 003780).
MPS IVA tolerant mouse
This mouse was developed because MPS IVA knockout mice (Tomatsu et al. 2003) developed immune responses to infusions of human GALNS enzyme and gene therapy. In addition to a disrupted GALNS gene, this mouse has a transgene expressing an inactive form of the human GALNS enzyme, which confers immune tolerance to administered human GALNS. This mouse has a similar phenotype to the initial MPS IVA mouse model, except that the mouse has increased GAG accumulation in lysosomes (Tomatsu et al. 2005a).
MPS VII tolerant mouse
A tolerant mouse model for MPS VII was developed from the original Birkenmeir β-glucuronidase-deficient mouse (gusmps/mps) (Birkenmier et al. 1989) and has been used for evaluation of the effectiveness of a variety of experimental treatments. This mouse has characteristics very similar to humans with MPS VII including a shortened face and facial dysmorphism, growth retardation, deafness, shortened lifespan, and behavioral deficits (Sly et al. 2001).
GAG Assays
Whole blood was collected from mice and centrifuged to isolate the serum. The High Sensitive KS ELISA Kit and HS ELISA Kit were produced and kindly provided by Seikagaku Biobusiness Corporation (Tokyo, Japan) and were used to measure serum HS and KS levels. Absorbance readings were taken at 450 nm using an EL800 automated microplate reader (Bio-Tek Instruments, Inc.; Winooski, VT), and HS and KS concentrations were calculated using a calibration curve.
Micro-CT Analysis and Radiography
Mice were euthanized using CO2. At dissection, leg bones and spines were placed in 95% ethanol. A micro-CT scan was performed on each bone using a Scanco μCT40 system (Scanco Medical; Brüttisellen, Switzerland) according to manufacturer’s instructions. Scans were focused on the cervical vertebrae and the knee joint. A three dimensional reconstruction of each bone was made, and BMD was calculated using the micro-CT scan data from mouse knees (Nazarian et al. 2008). The mean and standard deviation of the BMD for each mouse model and age were calculated and compared with controls using unpaired Student’s t-tests. Radiographs were also taken for each leg, spine, and ribs and compared.
Legs were measured using plain radiographs for each mouse. Mice older than 10 weeks were measured, and the mean length measurement and standard deviation are reported for each mouse model.
Pathology
The knee joint tissues were immersion-fixed in 4% paraformaldehyde/2% glutaraldehyde in PBS, postfixed in osmium tetroxide, and embedded in Spurr’s resin. For evaluation of lysosomal storage by light microscopy, toluidine blue-stained 0.5-μm-thick sections were examined.
Results
Appearance
MPS VII mice had the most severe clinical phenotype. MPS VII mice were not distinguishable phenotypically from WT littermates at birth but could be identified easily by the time of weaning. MPS VII mice had shortened faces and were slightly smaller. As they aged, their growth retardation, lack of fat tissue, shortened extremities, and facial dysmorphism with a blunted nose became more prominent. MPS I mice had the second-most severe skeletal abnormalities. By four weeks of age, homozygous null mice developed a flattened facial profile and a thickening of digits. MPS IIIA mice did not show any abnormal phenotype in their extremities. MPS IVA mice did not show any abnormal clinical phenotype on the mixed background as described previously (Tomatsu et al. 2005a). However, after backcrossing onto the C57/B6 strain, MPS IVA-affected mice had an upward angled calcaneus at age 6–7 months (Fig. 1).
Figure 1.
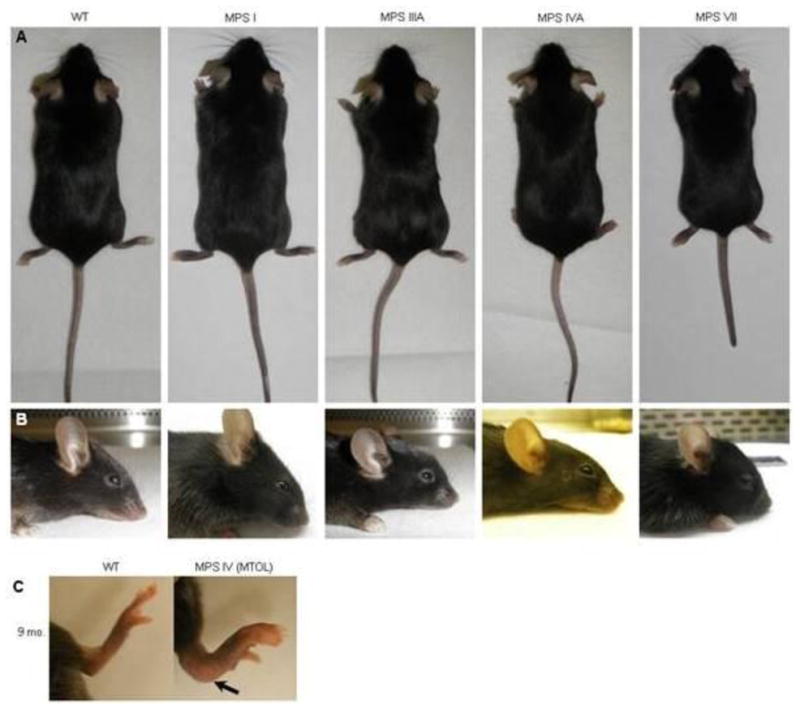
Clinical photographs of MPS I, IIIA, IVA, and VII mice.
A. Full body pictures
B. Side view of head
C. Leg of 9-month-old MPS IVA mouse. Arrow shows abnormal curvature of ankle compared with WT mice
Radiography
Radiographic analysis of the long bones from MPS VII mice showed skeletal dysplasia similar to that seen in humans with MPS VII. Radiographs comparing the lower extremities of mutant and normal mice are presented in Figure 2. The long bones were shortened, broad, and sclerotic at 9 months of age when compared with WT mice. The ribcage was narrow with short and thickened ribs. The sternal ends of the ribs showed decreased radiodensity on plain radiographs. The cervical vertebrae showed severely increased radiodensity when compared with WT mice (Fig. 2B). Bone lengths for WT, MPS VII and MPS I mice are presented in Fig. 3. MPS VII mice had significantly shortened tibia and feet and an increased mid-femur diameter in comparison with WT mice. MPS VII mice appeared to have the most severe skeletal pathology.
Figure 2.
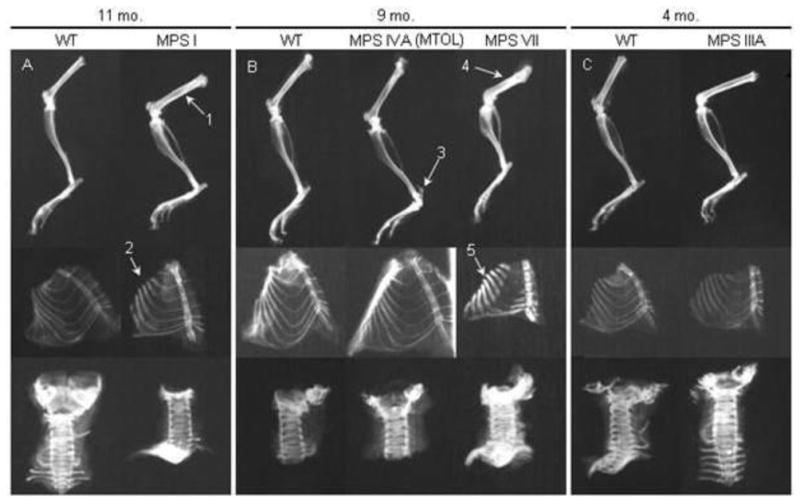
Radiographs of age- and sex-matched MPS I, IIIA, IVA, and VII mouse legs, ribcages, and cervical vertebrae. (A) 11 month-old MPS I and control mice. Arrow 1 identifies the sclerotic femur in an MPS I mouse. Arrow 2 identifies the sclerotic “oar-like” ribs. (B) 9-month-old MPS IVA and MPS VII mice with control mice. Arrow 3 shows abnormal calcaneus angled upwards. Arrow 4 identifies severely shortened and sclerotic femur of MPS VII mice. Arrow 5 shows short, thickened ribs, with lack of calcification on the medial aspect of the ribs of MPS VII mice. (C) 4-month-old MPS IIIA mouse bones with control mice. MPS IIIA mouse leg and spine show increased radiodensity.
Figure 3.
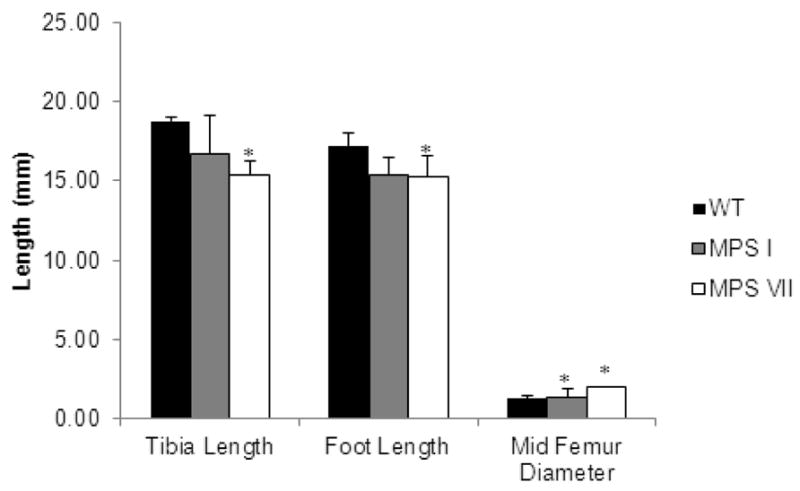
Leg measurements of WT, MPS I, and VII mice. Measurements of the length of the tibia and foot from the posterior calcaneus to the distal end of the distal phalanges, and the diameter of the mid-femur of mice over 12 weeks of age. Error bars are +/− 1 SD. P-values less than 0.05 in comparison with WT mice are represented by an asterisk. WT mice, n=5. MPS I, n=3. MPS VII, n=4.
In MPS I model mice, the femur and tibia were sclerotic on plain radiographs at age 11 months (Fig. 2A). Ribs were enlarged and oar-like with increased density, while the cervical vertebrae appeared normal when compared with WT mice. MPS I mice had a significantly increased mid-femur diameter when compared with WT mice (Fig. 3). The tibia and foot lengths of MPS I mice were decreased compared with those of WT mice; however, the difference was not as profound as in MPS VII mice.
Radiographs of MPS IIIA mouse legs showed increased radiodensity at age 16–19 weeks, while ribs and spine did not show any abnormalities at 16–19 weeks when compared with WT mice (Fig. 2C). MPS IVA mice had normal legs, ribs, and spine compared with WT mice; however, a calcaneus abnormality in MPS IVA mice was present on a radiograph and clinically in which the posterior aspect of the calcaneus was angled upwards (Fig. 2B – arrow 3).
Micro-CT examination
Micro-CT scans of MPS I and VII mice showed abnormal periosteal bone formation in the knee joint (Fig. 4) and thickened posterior vertebral arches of cervical vertebrae 1 and 2 (Fig. 5). Cross sectional images also show thickened periosteum in the bones of the knee joint (Fig. 4) and spine (cervical vertebrae 1 and 2) (Fig. 5) in MPS I and VII mice compared with those of WT mice. These abnormal findings in bone were more severe in MPS VII mice than in MPS I mice. In addition, cervical spinal stenosis was observed in both MPS I and VII mice. Again, the narrowing of the spinal canal was more prominent in MPS VII mice (Fig. 5). Micro-CT scans and cross-sectional images of knee bones and cervical spine from MPS IIIA and MPS IVA mice did not show any obvious abnormalities when compared with WT mice (Figs. 4 and 5).
Figure 4.
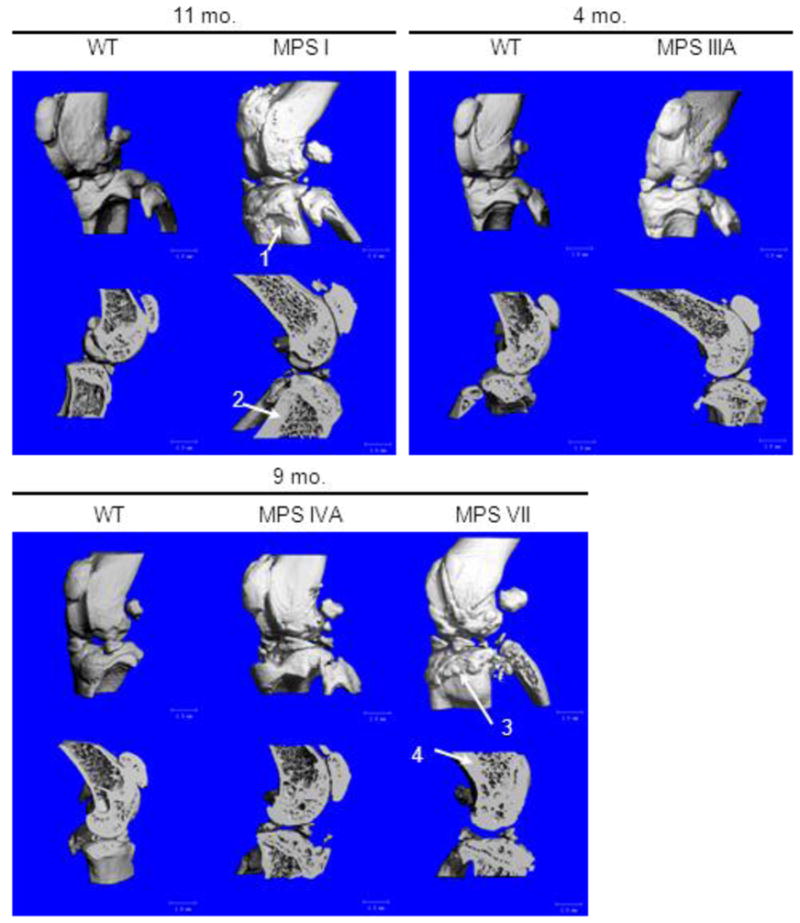
Three dimensional micro-CT reconstructions of knee joints in MPS I, IIIA, IVA, and VII mice. Cross-sections are sagittal through the midline of the knee joint. Arrow 1 shows abnormal periosteal bone formation on the tibia of an MPS I mouse. Arrow 2 shows an area of increased bone thickness in an MPS I mouse compared to the WT control mouse. Arrow 3 shows severe abnormal periosteal bone formation on the tibia of an MPS VII mouse. Arrow 4 shows abnormally thickened bone in an MPS VII mouse compared with the bone thickness in an age-matched WT mouse.
Figure 5.

Three dimensional micro-CT reconstructions of cervical vertebrae 1 and 2 in MPS I, IIIA, IVA, and VII mice. Cross sections are sagittal sections in the midline. Arrow 1 shows thickened vertebral arches in MPS I. Arrow 2 shows thickened bone in vertebrae of an MPS I mouse. Arrow 3 shows thickened vertebral arches in an MPS VII mouse. Arrow 4 shows thickened bone in the vertebral arches of an MPS VII mouse.
The micro-CT studies of knee bones also yielded BMD measurements. The mean BMD measurements were as follows: WT: 496.24 ± 38.75 mgHA/cc, MPS I: 480.63 ± 28.57 mgHA/cc, MPS IIIA: 563.18 ± 54.20 mgHA/cc, MPS IVA: 500.73 ± 22.25 mgHA/cc, and MPS VII: 567.92 ± 57.59 mgHA/cc. The mean BMD of WT mice increased until approximately 10 weeks of age and plateaued. Although the differences between WT and age-matched MPS I, IIIA, and IVA mouse BMDs were not statistically significant, the BMD of MPS VII females older than 2 months was 18% higher than that for age-matched male MPS VII and WT mice. In 4- to 6-week old MPS I and VII mice, the BMD was lower than for WT mice. The trends shown in the mean BMD values from micro-CT mirrored the radiodensity trends seen in the plain radiographic results.
Spine micro-CT studies were used to measure the dimensions of the spinal canal in WT, MPS I, and MPS VII mice (Fig. 6). All measurements of spinal-canal dimensions in MPS VII and MPS I mice were significantly reduced when compared with WT mice. Spinal stenosis was more severe in MPS VII mice compared with MPS I mice; however, differences in measurements were not significant.
Figure 6.
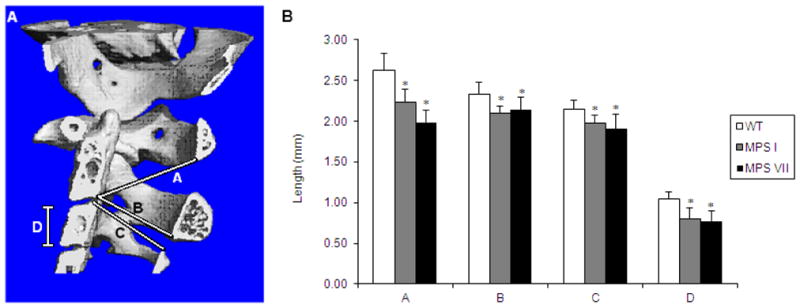
Measurements of dimensions in cervical vertebrae of WT, MPS I, and MPS VII mice.
A. Location of measurements is shown in Fig. 6A.
Line A: From posterior, inferior aspect of the body of cervical vertebrae 2 to the inferior aspect of the arch of cervical vertebrae 1.
Line B: From posterior, inferior aspect of the body of cervical vertebrae 2 to the inferior aspect of the arch of cervical vertebrae 2 (diameter of spinal canal of cervical vertebrae 2).
Line C: From the posterior, superior aspect of the body of cervical vertebrae 3 to the superior aspect of the arch of cervical vertebrae 3 (diameter or spinal canal of cervical vertebrae 3).
Line D: Height of the body of cervical vertebrae 3.
B. Measurements of Lines A–D were made using micro-CT software. Error bars are +/− 1 SD. P-values less than 0.05 compared with WT mice values are represented by an asterisk. Sample sizes are: WT, n=8; MPS I, n=3; MPS VII, n=4 for Line A and B and n=5 for Line C and D.
Pathology in growth plate region
MPS VII
Glycosaminoglycan storage in bone at 16 weeks of age was marked, with lysosomal distention in osteoblasts and osteocytes lining the cortical and trabecular bone, in chondrocytes, and in the sinus lining cells in the bone marrow (Fig. 7). The column structure through all layers of the growth plate was markedly disorganized, and all chondrocytes were prominently ballooned with vacuoles. The bone and joints had altered architecture with synovial thickening and vacuolated cells in the synovium. All articular cartilage cells showed marked distention with a thicker articular cartilage layer. The cells in the periosteum also had marked vacuolar distension.
Figure 7.

Histopathologic changes of the proximal growth plates (tibia) and articular cartilage area (tibia) at 16 weeks of age. Tissue was stained with toluidine blue. The vacuoles in epiphyseal cartilage layer are more prevalent in order of MPS VII > MPS IVA > MPS I > MPS IIIA mice while the vacuoles in articular cartilage region are more in order of MPS VII > MPS I > MPS IVA > MPS IIIA mice. The perimeters of growth plate and articular cartilage regions are more irregular in MPS VII > MPS I > MPS IVA > MPS IIIA mice.
MPS I
Figure 7 shows the histopathologic changes of the proximal tibial and distal femoral growth plates at 16 weeks of age. Figure 7 represents polarized light microscopy of tibial cortical bone at 16 weeks of age. The light microscopic views reveal a loss of the parallel order of the bone matrix with loss of the concentric arrangement of lamellae or haversian system formation. The cortex is thickened in affected mice. The osteocytes have clearly increased cytoplasmic volumes.
MPS IIIA
At age 16 weeks, MPS IIIA mice exhibited a small amount of storage in articular and epiphyseal cartilage cells (Fig. 7). The cartilage layer at the growth plate was regular, and the column structure was normal. The femur of MPS IIIA mice did not show obvious storage materials in osteoblasts lining the cortical bone and osteocytes. The sinus lining cells in bone marrow also contained a small amount of storage.
MPS IVA
At age 16 weeks, the epiphyseal growth plate of MPS IVA mice was irregular, and chondrocytes with lysosomal distension were obvious (Fig. 7). The femur of a 16-week-old MPS IVA tolerant mouse showed osteoblasts lining the cortical bone and osteocytes within the bone with slight storage material. The sinus lining cells in bone marrow also contained a small amount of storage. A 16-week-old MPS IVA tolerant mouse showed the cells in the periosteum and the articular cartilage cells which also contained storage material.
KS Assay
Measurement of mouse serum KS levels (Fig. 8) by High Sensitive ELISA assay showed significantly increased serum KS levels in MPS VII (p < 0.0001), MPS I (p < 0.0001), and MPS IIIA (p < 0.005) when compared with WT mice. The serum KS in MPS VII mice was the most increased, while MPS I and IIIA mice showed a smaller increase. MPS IVA mice did not have elevated serum KS levels in comparison to WT mice. Keratan sulfate levels increased with age in all mouse models.
Figure 8.
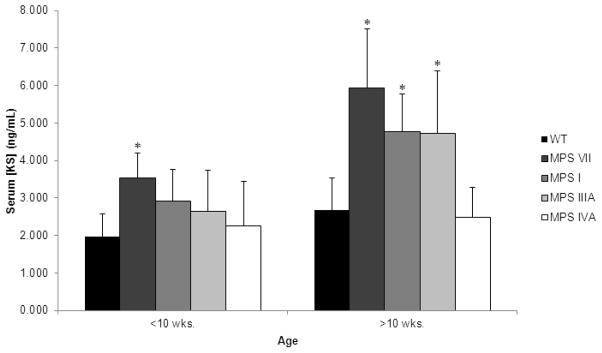
Mouse serum concentration of KS. Error bars are +/− 1 SD. P-value of less than 0.05 compared with serum KS level in age-matched WT mice is represented with an asterisk. Sample sizes are as follows: Less than 10 weeks of age: WT, n=15; MPS VII, n=5; MPS I, n=2; MPS IIIA, n=7; MPS IVA, n=5. Greater than 10 weeks of age: WT, n=24; MPS VII, n=9; MPS I, n=9; MPS IIIA, n=3; MPS IVA, n=12.
HS Assay
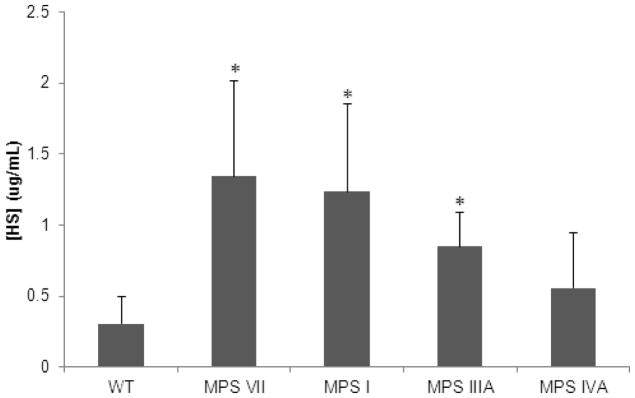
Serum HS levels in 5-week-old MPS I, IIIA, and VII mice were significantly (p < 0.05) elevated when compared with the serum HS levels in 5-week-old WT mice. Serum HS levels in 5-week-old MPS IVA mouse models were not significantly elevated when compared with 5-week-old WT mice (Fig. 9). HS levels increased with age (data not reported).
Figure 9.

Mouse serum concentration of HS in 5-week-old mice. Error bars are +/− 1 SD. P-value of less than 0.05 compared with serum HS level in age-matched WT mice is represented with an asterisk. Sample sizes are as follows: WT, n=4; MPS VII, n=4; MPS I, n=2; MPS IIIA, n=3; MPS IVA, n=4.
GALNS Inhibition Assay
The GALNS inhibition assays were performed to determine the effect of varying GAG concentrations on GALNS activity. CS and DS were shown to have no effect on GALNS activity. GALNS activity was 100% in up to 80 μg/ml CS. GALNS activity also remained at 100% in the presence of DS concentrations up to 20 μg/ml. However, GALNS activity was shown to decrease in the presence of increasing concentrations of HS. At 1 μg/ml HS, GALNS activity was decreased to 81%; at 2 μg/ml, it was decreased to 79%; at 5 μg/ml, it was decreased to 57%; at 20 μg/ml, it was decreased to 20%; and at 40 μg/ml it was decreased to 5% of normal activity (Fig. 10).
Figure 10.
GALNS inhibition assay results. Graphs represent GALNS activity in the presence of increasing concentrations of: A) CS, B) DS, and C) HS. Values represent percent of GALNS activity with 0 ug/ml concentrations of CS, DS, or HS.
Discussion
In this study, we demonstrated the following: 1) the severity of bone dysplasia in MPS mouse models is as follows: MPS VII > MPS I > MPS IIIA ≥ MPS IVA, 2) elevated serum KS and HS levels in MPS I, IIIA, and VII mice that correlated with severity of bone lesions found in micro-CT and pathohistology, 3) a correlation between secondary elevation of KS levels in MPS I, IIIA, and VII mice with high HS levels, and 4) inhibition of GALNS activity by concentrations of HS found in MPS I, IIIA, and VII mouse models.
To our knowledge, this is the first comparison of bone lesions in MPS I, IIIA, IVA, and VII mice using micro-CT. Micro-CT scans revealed severe thickening of the periosteum of vertebrae and bones in the knee joint in MPS VII mice. Thickened vertebral bone resulted in a narrowing of the spinal canal. In the knee joint bones, the thickening produced a rough articular surface. Both plain radiographs and BMD measurements by micro-CT showed increased BMD in female MPS VII mice when compared with age-matched WT mice. Bone dysplasia in MPS I mice was similar to that described in MPS VII, but it was not as severe.
Evaluation of bone from MPS IIIA model mice by micro-CT did not reveal any structural abnormalities compared with WT mice. However, plain radiographs and micro-CT scans showed elevated BMD in two of three 4-month-old MPS IIIA male mice when compared with controls. Micro-CT scans of MPS IVA mice also showed no obvious abnormalities compared with WT mice. In addition, BMD also appears to be normal in MPS IVA mice. An abnormality of the calcaneus bone was noted on radiographs of 9-month-old MPS IVA mice, and the same abnormality was clinically apparent in four other mice at 6–7 months of age. This is the first description of a clinical finding in bone in MPS IVA mice, although other pathological findings in cartilage and connective tissues have been described (Tomatsu et al. 2005a). A similar abnormality in the calcaneus bone was reported in the MPS II mouse model (Garcia et al. 2007). The pathogenesis of this localized abnormality is of great interest.
A KS assay showed elevated KS in MPS I, IIIA, and VII mice compared with WT mice; the highest levels of serum KS were found in MPS VII mice, followed by MPS I mice. This finding was unexpected, since MPS IVA is the only form of MPS known to have a deficiency of an enzyme required for the degradation of KS. However, similarly elevated blood KS levels were identified in human patients with MPS I, II, III, VI, and VII (Tomatsu et al. 2005b), and the elevation of KS in these mouse models has been termed secondary elevation of KS. Studies reported here allow us to propose the mechanism. In this study we found that HS was significantly elevated in MPS I, IIIA, and VII mice when compared with WT mice. Since GALNS is the enzyme that degrades KS, we performed a GALNS enzyme inhibition assay to measure the inhibitory effects of CS, DS, and HS on GALNS activity. This assay showed that only HS (and neither CS nor DS) inhibited the activity of the GALNS enzyme. This result supports the hypothesis that the secondary elevation of KS levels seen in MPS I, IIIA, and VII patients could be caused by inhibition of the GALNS enzyme by elevated levels of HS. This hypothesis is further supported by the fact that the levels of HS found to significantly inhibit GALNS activity in vitro correlate with the serum levels of HS found in MPS I, IIIA, and VII patients (Tomatsu et al. 2005b) and mice (this study).
Chondroitin sulfate and KS are the major GAGs in articular and growth plate cartilage where bone grows (Klüppel et al. 2005; Rani et al. 1999). Dermatan sulfate and HS, degraded by both GUSB and IDUA responsible for MPS VII and MPS I, are minor components of the GAGs in this cartilage region (Kodama et al. 1997; Ernst et al. 1996; Grimaud et al. 2002). Skeletal disease is reported to be mild in MPS IIIA (Stewart et al. 1992) and MPS IIIB (Sims et al. 2005) mice which accumulate HS. Bone pathology in MPS VII mice is more severe than in MPS I mice. One potential reason for the difference is that MPS VII accumulates chondroitin 4-sulfate (C4S) in cartilage (Herati et al. 2008). MPS VI mice with deficiency of N-acetylgalactosamine 4-sulfatase (Heinrich et al. 1998), critical for degradation of C4S, also have a severe skeletal disease.
Skeletal disease is severe in human MPS IVA patients who cannot degrade chondroitin 6-sulfate (C6S) and KS because of deficiency of GALNS. However, MPS IVA mice have minimal skeletal disease (Tomatsu et al. 2005a; this manuscript) even though the storage materials are evident in cartilage cells. This was likely recently explained by the fact that C6S constitutes a minor component of cartilage (Oberle et al. 2006; Gaffen et al. 1995) and KS synthesis in mice is substantially limited when compared with human. Taken together, data from MPS mouse models are consistent with the hypothesis that accumulation of C4S is responsible for more severe chondrodysplasia in MPS VI and MPS VII than other MPS mouse models (Herati et al. 2008).
Our findings support the hypothesis that severity of bone dysplasia found in MPS mouse models correlates with serum KS levels. While the origin of KS elevation in MPS mouse models is still not clear, we have described a mechanism which may contribute to the KS elevation that we have described in MPS VII, I and IIIA mice. The elevated levels of KS in MPS VII, MPS I, and MPS IIIA mice and limited elevation of KS in MPS IVA mice also suggests that serum KS elevation may be caused by release of KS from chondrocytes which are damaged by accumulation of other GAGs. In the MPS IVA mouse model, severe bone dysplasia is not present because of limited GAG accumulation in chondrocytes and therefore KS would not be released from affected chondrocytes. Consequently, KS elevation in blood is limited despite the absence of functional GALNS enzyme. Further study will be required to characterize the etiology of the secondary KS elevation that we have described in MPS VII, I, and IIIA mouse models. In addition, characterization of KS in the different MPS mouse models may help to clarify the mechanism of secondary KS elevation. The previous and our studies in different species indicate that accumulation of CS and/or KS are important for bone disease in human MPS patients.
In conclusion, this work demonstrates the utility of micro-CT analysis to characterize bone dysplasia in murine models of MPS I, IIIA, IVA, and VII. In addition, it provides evidence for a correlation between the severity of bone dysplasia (micro-CT and bone pathology) and serum KS levels. An understanding of the mechanism of KS elevation in MPS will help to clarify the role of accumulation of KS and other GAGs in causing bone dysplasia.
Acknowledgments
Funding: Supported by grants from Austrian Research Society for Mucopolysaccharidoses and Related Diseases, and International Morquio Organization, and National Institutes of Health grant GM34182.
Supported by grants from Austrian Research Society for Mucopolysaccharidoses and Related Diseases and Carol Ann Foundation (International Morquio Organization). W.S.S. and J.H.G. were also supported by National Institutes of Health grant GM34182. The content of the article has not been influenced by the sponsors.
KS and HS kits were kindly provided by Seikagaku Corporation, Japan.
Footnotes
Competing interest statement: There is no potential conflict of interest, real or perceived, in this manuscript for all authors.
Ethics approval: not applicable
Patient consent statement: not applicabl
IACUC approval: approved. The protocol number is 1484 at Saint Louis University.
Contributions of specific authors:
Daniel Rowan: he planned and conducted the experiments of CT, assay, and pathology and reported the work on this project under supervision of Dr. Tomatsu.
Shunji Tomatsu: As PI of this project, he designed and planned the experiments with all collaborators and supervised and reviewed all work, responsible for the experiment and manuscript.
Jeffrey H. Grubb: He planned and provided mouse colonies and reviewed the manuscript.
Adriana M. Montańo: She supported planning of the experiment and reviewed the manuscript.
William S. Sly: He designed and planned the experiment with Dr. Tomatsu and reviewed the manuscript.
Guarantor: Dr. Tomatsu will serve as the guarantor for this manuscript. He accepts full responsibility for the work and/or conduct of the study, had access to the data, and controlled the decision to publish.
References
- Aldenhoven M, Sakkers RJB, Boelens J, de Koning TJ, Wulffraat NM. Musculoskeletal manifestations of lysosomal storage disorders. Ann Rheum Dis. 2009;68:1659–1665. doi: 10.1136/ard.2008.095315. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya R, Gliddon B, Beccari T, Hopwood JJ, Stanley P. A novel missense mutation in lysosomal sulfamidase is the basis of MPS III A in a spontaneous mouse mutant. Glycobiology. 2001;11:99–103. doi: 10.1093/glycob/11.1.99. [DOI] [PubMed] [Google Scholar]
- Bhaumik M, Muller VJ, Rozaklis T, et al. A mouse model for mucopolysaccharidosis type III A (Sanfilippo syndrome) Glycobiology. 1999;9:1389–1396. doi: 10.1093/glycob/9.12.1389. [DOI] [PubMed] [Google Scholar]
- Birkenmier EH, Davisson MT, Beamer WG, et al. Murine mucopolysaccharidosis type VII. Characterization of a mouse with beta-glucuronidase deficiency. J Clin Invest. 1989;83:1258–1266. doi: 10.1172/JCI114010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke LA, Russell CS, Pownall S, et al. Murine mucopolysaccharidosis type I: targeted disruption of the murine α-L-iduronidase gene. Hum Mol Genet. 1997;6:503–511. doi: 10.1093/hmg/6.4.503. [DOI] [PubMed] [Google Scholar]
- Ernst M, Oates A, Dunn AR. Gp130-mediated signal transduction in embryonic stem cells involves activation of Jak and Ras/mitogen-activated protein kinase pathways. J Biol Chem. 1996;271:30136–30143. doi: 10.1074/jbc.271.47.30136. [DOI] [PubMed] [Google Scholar]
- Fraldi A, Hemsley K, Crawley A, et al. Functional correction of CNS lesions in an MPS-IIIA mouse model by intracerebral AAV-mediated delivery of sulfamidase and SUMF1 genes. Hum Mol Genet. 2007;16:2693–2702. doi: 10.1093/hmg/ddm223. [DOI] [PubMed] [Google Scholar]
- Gaffen JD, Gleave SJ, Crossman MV, Bayliss MT, Mason RM. Articular cartilage proteoglycans in osteoarthritic STR/Ort mice. Osteoarthritis Cartilage. 1995;3:95–104. doi: 10.1016/s1063-4584(05)80042-1. [DOI] [PubMed] [Google Scholar]
- Garcia AR, Pan J, Lamsa JC, Muenzer J. The characterization of a murine model of mucopolysaccharidosis II (Hunter syndrome) J Inherit Metab Dis. 2007;30:924–934. doi: 10.1007/s10545-007-0641-8. [DOI] [PubMed] [Google Scholar]
- Grimaud E, Blanchard F, Charrier C, Gouin F, Redini F, Heymann D. Leukaemia inhibitory factor (lif) is expressed in hypertrophic chondrocytes and vascular sprouts during osteogenesis. Cytokine. 2002;20:224–230. doi: 10.1006/cyto.2002.2002. [DOI] [PubMed] [Google Scholar]
- Grubb JH, Vogler C, Tan Y, Shah GN, MacRae AF, Sly WS. Infused Fc-tagged β-glucuronidase crosses the placenta and produces clearance of storage in utero in mucopolysaccharidosis VII mice. Proc Natl Acad Sci U S A. 2008;105:8375–8380. doi: 10.1073/pnas.0803715105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haskins ME. Animal models for mucopolysaccharidosis disorders and their clinical relevance. Acta Paediatr. 2007;96:56–62. doi: 10.1111/j.1651-2227.2007.00211.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinrich PC, Behrmann I, Müller-Newen G, Schaper F, Graeve L. Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem J. 1998;334:297–314. doi: 10.1042/bj3340297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herati RS, Ma X, Tittiger M, Ohlemiller KK, Kovacs A, Ponder KP. Improved retroviral vector design results in sustained expression after adult gene therapy in mucopolysaccharidosis I mice. J Gene Med. 2008;10:972–982. doi: 10.1002/jgm.1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung SC, Park ES, Choi EN, Kim CH, Kim SJ, Jin DK. Characterization of a novel mucopolysaccharidosis type II model mouse and recombinant AAV2/8 vector-mediated gene therapy. Mol Cells. 2010;30:13–18. doi: 10.1007/s10059-010-0083-2. [DOI] [PubMed] [Google Scholar]
- Klüppel M, Wight TN, Chan C, Hinek A, Wrana JL. Maintenance of chondroitin sulfation balance by chondroitin-4-sulfotransferase 1 is required for chondrocyte development and growth factor signaling during cartilage morphogenesis. Development. 2005;132:3989–4003. doi: 10.1242/dev.01948. [DOI] [PubMed] [Google Scholar]
- Kodama H, Fukuda K, Pan J, et al. Leukemia inhibitory factor, a potent cardiac hypertrophic cytokine, activates the JAK/STAT pathway in rat cardiomyocytes. Circ Res. 1997;81:656–663. doi: 10.1161/01.res.81.5.656. [DOI] [PubMed] [Google Scholar]
- Masu Y, Wolf E, Holtmann B, Sendtner M, Brem G, Thoenen H. Disruption of the CNTF gene results in motor neuron degeneration. Nature. 1993;365:27–32. doi: 10.1038/365027a0. [DOI] [PubMed] [Google Scholar]
- Nazarian A, Snyder BD, Zurakowski D, Muller R. Quantitative micro-computed tomotraphy: A non-invasive method to assess equivalent bone mineral density. Bone. 2008;43:302–311. doi: 10.1016/j.bone.2008.04.009. [DOI] [PubMed] [Google Scholar]
- Neufeld EF, Muenzer J. The mucopolysaccharidosis. In: Scriver CR, Beaudet al, Sly WS, Valle D, Childs B, Kinzler KW, Vogelstein B, editors. The Metabolic and Molecular Bases of Inherited Disease. 8. New York: McGraw-Hill; 2001. pp. 3421–3452. [Google Scholar]
- Oberle S, Schober A, Meyer V, et al. Loss of leukemia inhibitory factor receptor beta or cardiotrophin-1 causes similar deficits in preganglionic sympathetic neurons and adrenal medulla. J Neurosci. 2006;26:1823–1832. doi: 10.1523/JNEUROSCI.4127-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rani MR, Leaman DW, Han Y, et al. Catalytically active TYK2 is essential for interferon-beta-mediated phosphorylation of STAT3 and interferonalpha receptor-1 (IFNAR-1) but not for activation of phosphoinositol 3-kinase. J Biol Chem. 1999;274:32507–32511. doi: 10.1074/jbc.274.45.32507. [DOI] [PubMed] [Google Scholar]
- Sims NA, Jenkins BJ, Nakamura A, et al. Interleukin-11 receptor signaling is required for normal bone remodeling. J Bone Miner Res. 2005;20:1093–1102. doi: 10.1359/JBMR.050209. [DOI] [PubMed] [Google Scholar]
- Sly WS, Vogler C, Grubb JH, et al. Active site mutant transgene confers tolerance to human β-glucuronidase without affecting the phenotype of MPS VII mice. Proc Natl Acad Sci U S A. 2001;98:2205–2210. doi: 10.1073/pnas.051623698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart CL, Kaspar P, Brunet LJ, Bhatt H, Gadi I, Köntgen F, Abbondanzo SJ. Blastocyst implantation depends on maternal expression of leukaemia inhibitory factor. Nature. 1992;359:76–79. doi: 10.1038/359076a0. [DOI] [PubMed] [Google Scholar]
- Tomatsu S, Orii KO, Vogler C, et al. Mouse model of N-acetylgalactosamine-6-sulfate sulfatase deficiency (Galns−/−) produced by targeted disruption of the gene defective in Morquio A disease. Hum Mol Genet. 2003;12:3349–3358. doi: 10.1093/hmg/ddg366. [DOI] [PubMed] [Google Scholar]
- Tomatsu S, Gutierrez M, Nishioka T, et al. Development of MPS IVA mouse. Genet. 2005a;14:3321–3335. doi: 10.1093/hmg/ddi364. [DOI] [PubMed] [Google Scholar]
- Tomatsu S, Okamura K, Maeda H, et al. Keratan sulphate levels in mucopolysaccharidosis and mucolipidoses. J Inherit Metab Dis. 2005b;28:187–202. doi: 10.1007/s10545-005-5673-3. [DOI] [PubMed] [Google Scholar]
- Tomatsu S, Vogler C, Montano AM, et al. Murine model (Galnstm(C76S)slu) of MPS IVA with missense mutation at the active site cystine conserved among sulfatase proteins. Mol Genet Metab. 2007;91:251–258. doi: 10.1016/j.ymgme.2007.02.009. [DOI] [PubMed] [Google Scholar]
- Tomatsu S, Montano AM, Dung VC, et al. Enhancement of drug delivery: enzyme-replacement therapy for murine Morquio A syndrome. Mol Ther. 2010;18:1094–1102. doi: 10.1038/mt.2010.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ware CB, Horowitz MC, Renshaw BR, et al. Targeted disruption of the low-affinity leukemia inhibitory factor receptor gene causes placental, skeletal, neural and metabolic defects and results in perinatal death. Development. 1995;121:1283–1299. doi: 10.1242/dev.121.5.1283. [DOI] [PubMed] [Google Scholar]