

Abstract
This Letter describes the further optimization of an MLPCN probe molecule (ML137) through the introduction of 5- and 6- membered spirocycles in place of the isatin ketone. Interestingly divergent structure-activity relationships, when compared to earlier M1 PAMs, are presented. These novel spirocycles possess improved efficacy relative to ML137, while also maintaining high selectivity for the human and rat muscarinic M1 receptor subtype.
Keywords: Muscarinic acetylcholine receptor 1, M1, Spirocyclic, Positive allosteric modulator (PAM), ML137, VU0413162
The tremendous potential for truly selective muscarinic acetylcholine (ACh) ligands in a variety of disease states is well appreciated; in reality, however, this potential has been difficult to achieve due to a historical reliance on targeting the highly conserved orthosteric ACh binding site shared by the five muscarinic ACh receptor subtypes (mAChRs, M1 through M5).1 As such, many of our current beliefs pertaining to the potential utility of muscarinic ligands in central nervous system (CNS) disorders has come from the phenotypes of M1–5 knock out (KO) mice,2 CNS receptor localization and the promising clinical outcomes observed with the muscarinic agonist xanomeline in both Alzheimer’s disease and schizophrenia patients.3,4 Unfortunately, xanomeline’s limited muscarinic subtype selectivity has given rise to unacceptable levels of adverse events consistent with peripheral activation of the M3 receptor. Although switching focus to allosteric agonists is a credible tactic for circumventing the selectivity issues associated with engaging the orthosteric site, a fair number of allosteric agonists have recently been shown to bind in a bitopic sense to the mAChRs, bringing the orthosteric site issues back into unwelcome consideration.5,6 Another tactic for achieving high mAChR subtype selectivity has been the development of positive allosteric modulators (PAMs), compounds with selectivity profiles that benefit from binding solely at sites distinct from the ACh binding site where amino acid homology between the muscarinic receptors has the potential to be reduced. This approach has been validated through the development of numerous, highly selective PAMs for M1,7 M4,8 and M5.9
Recently, we described our initial success at replacing the isatin portion of ML137 and were able to arrive at a variety of novel structures partially summarized by compounds 1 and 2 in Figure 1.10 Reduction or modification of the ketone carbonyl in ML137 served as the starting point for these benzo-fused bicyclic replacements. Alternatively, it might be envisioned that this carbonyl could be partially hydrated when water is available (in vitro/ in vivo) or even preferentially present as the hydrate when bound to the allosteric site on the hM1 receptor. This idea formed the rationale for the preparation of the spirocyclic ketal 3 (Figure 1) as a discrete and stable entity. When tested against the human M1 (hM1) receptor for functional activity, 3 provided a hM1 PAM EC50 of 2.0 µM with an AChmax response of 62%, showing reasonable potency and efficacy, respectively. Although not as potent (EC50) as its parent ML137 (hM1 EC50 = 0.60 µM, AChmax = 45 ± 3 %, pEC50 = 6.22 ± 0.02),11 compound 3 produced improved efficacy (% AChmax) at the highest concentration tested of 30 µM. This novel spirocycle was the impetus for exploring a wider range of diverse spirocycles at this location. Some of these spirocycles were available from commercial sources and were alkylated with the appropriate benzyl chloride as described previously.10 However, the majority of subsequent analogs were prepared according to Schemes 1, 2 and 3.
Figure 1.

Prior replacements for the isatin in ML137 and a rationale for the preparation of the spirocyclic ketal 3.
Scheme 1.

Reagent and conditions: (a) K2CO3, MeCN; (b) PdCl2(dppf)•DCM, Cs2CO3, THF/H2O, 160 °C, 10 min; (c) TFA or HCl, 0 °C to rt.
Scheme 2.

Reagents and conditions: (a) tert-butyl sulfinamide, Ti(OiPr)4, 0 °C to rt, DCM, 84% yield; (b) THF, −78 °C to rt, 72% yield; (c) HCl in dioxane (4 M), 0 °C to rt; (d) NaHB(OAc)3, DCE, rt, 58% yield, 2 steps; (e) (CH2O)n, CO2H2, H2O, 100 °C, 2 h, 70% yield; (f) PdCl2(dppf)•DCM, Cs2CO3, THF/H2O, 160 °C, 10 min, 40% yield.
Scheme 3.

Reagents and conditions: (a) allyltrimethylsilane, SnCl4, DCM, 0 °C, 5 h, 60% yield; (b) TBAF, THF, 60 °C, 18 h, 28% yield; (c) 15, PdCl2(dppf)•DCM, Cs2CO3, THF/H2O, 160 °C, 10 min, 75% yield.
Scheme 1 shows the alkylation of commercially available Boc-protected spirocyclic amines 4 with benzylic bromides 5 employing potassium carbonate in acetonitrile to provide aryl bromides 6. Compound 6 can then undergo Suzuki couplings with a variety of boronic acids/esters, such as 7, to form the N-Boc protected biaryl compounds 8. The N-Boc protecting group can then be removed with a strong acid (TFA or anhydrous HCl) to give the secondary amine 9. Amine 9 was an invaluable intermediate poised to engage in a wide variety of well-precedented reactions with a near limitless number of electrophiles.
Scheme 2 depicts the construction of an isomeric spiropyrrolidine analog, which was not commercially available, starting from the known, non-selective M1,3,5 PAM VU0119498.9a Condensation of this isatin with tert-butyl sulfonamide promoted by titanium (IV) tetraisopropoxide in DCM was followed, in a separate step, by the addition of Grignard 10 to yield 11. In this manner, both the nitrogen and the carbon chain required to form the spiropyrrolidine were efficiently introduced. Removal of both protecting groups from 11 with HCl in dioxane gave the aminoaldehyde 12 (alternatively in a protonated/cyclized version, not shown) which readily underwent reductive cyclization to form the spiropyrrolidine 13. The secondary amine of 13 was reductively methylated with formaldehyde in an aqueous mixture of formic acid at elevated temperatures to give the tertiary amine 14. Suzuki coupling with 15 provided the fully elaborated spiropyrrolidine 18c (See Table 1 for structure). The analogous spirotetrahydrofuran was prepared starting from the same isatin (Scheme 3). Tin (IV) chloride promoted the formal dipolar cycloaddition of allyltrimethylsilane to the ketone in VU0119498 giving the spirocycle 16 in a single step and in good yield (60%).12 Heating 16 with TBAF in THF yielded the unadorned spirotetrahydrofuran 17, which was efficiently converted to the desired biaryl 18b under our standard Suzuki coupling conditions.
Table 1.
Structures and activities of analogs 3, 18a–h showing spirocyclic alternatives to the isatin core.
 | |||||
|---|---|---|---|---|---|
| Compd | X | hM1 EC50 (µM)a |
AChmax (%)a,b |
pEC50a | |
| 3 | 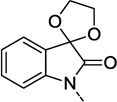 |
F | 2.0 | 62 ± 2 | 5.70 ± 0.11 |
| 18a |  |
F | >10 | 43 ± 2 | |
| 18b |  |
H | >10 | 43 ± 3 | |
| 18c |  |
H | >10 | 53 ± 4 | |
| 18d | 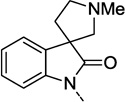 |
F | 3.2 | 65 ± 3 | 5.49 ± 0.10 |
| 18e |  |
F | - | ||
| 18f | 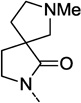 |
F | >10 | 28 ± 3 | |
| 18g |  |
F | 3.8 | 49 ± 4 | 5.42 ± 0.06 |
| 18h |  |
F | >10 | 29 ± 2 | |
Values represent the mean ± standard error of the means of at least three independent determinations performed in triplicate. "-" indicates an inactive compound showing no PAM activity up to the highest concentration tested (30 µM).
When hM1 EC50 is reported as >10, this value represents the percent maximal ACh response when the test compound was present at its highest concentration (30 µM).
Table 1 presents a survey of spirocycles and their functional PAM activity at hM1. Compounds 18a and 18f showed an interesting divergence in the SAR between these spirocycles and our earlier series of non-spiro isatin replacement analogs in that deletion of the amide carbonyl (18a) or deletion of the phenyl ring (18f) did not abolish PAM activity, as was previously observed.10 Although an EC50 could not be determined for these two compounds, a measureable positive allosteric modulation of ACh’s activity was observed (AChmax > 20%) and presumably indicates beneficial interactions between the receptor and portions of the new spirocycle ring.13 Relative to 3, the tetrahydrofuran 18b and N-methyl pyrrolidine 18c lost potency but retained a measurable level of hM1 PAM efficacy. This loss in potency is most likely not related to the absence of a fluorine on the phenyl ring (vide infra, compare 19d with 20i from Tables 2 and 3, respectively). The isomeric N-methyl spiropyrrolidine 18d displayed measurable potency with a hM1 EC50 of 3.2 µM. However, the desmethyl analog 18e lacked any PAM activity at concentrations up to 30 µM. While the N-methyl spiropiperidine 18g was about 2-fold less potent than the lead spirocycle 3, for this structure, removal of the N-methyl group did not abolish all the PAM activity for 18h.
Table 2.
Structures and activities of spiropyrrolidine analogs 19a–t.
 | |||||
|---|---|---|---|---|---|
| Compd | *a | R | hM1 EC50 (µM)b |
AChmax (%)b,c |
pEC50b |
| 19a | A | Me | 2.9 | 67 ± 3 | 5.54 ± 0.06 |
| 19b | B | Me | 2.9 | 50 ± 3 | 5.54 ± 0.07 |
| 19c | ± | c-butyl | >10 | 48 ± 3 | |
| 19d | ± | c-pentyl | 3.1 | 57 ± 3 | 5.51 ± 0.02 |
| 19e | ± | c-hexyl | 5.2 | 60 ± 2 | 5.29 ± 0.06 |
| 19f | ± | c-heptyl | 7.5 | 50 ± 3 | 5.13 ± 0.05 |
| 19g | A | c-pentyl | 1.6 | 69 ± 1 | 5.80 ± 0.03 |
| 19h | B | c-pentyl | >10 | 33 ± 3 | |
| 19i | ± | i-Pr | >10 | 30 ± 2 | |
| 19j | ± | Et | 3.2 | 59 ± 2 | 5.49 ± 0.03 |
| 19k | ± | CH2CH2F | 4.1 | 60 ± 3 | 5.39 ± 0.02 |
| 19l | ± | CH2CHF2 | >10 | 52 ± 3 | |
| 19m | ± | CH2CF3 | - | ||
| 19n | ± | Boc | - | ||
| 19o | ± | Ac | >10 | 41 ± 2 | |
| 19p | ± | (C=O)c-butyl | 6.3 | 45 ± 3 | 5.20 ± 0.07 |
| 19q | ± | (C=O)c-hexyl | 5.0 | 50 ± 4 | 5.30 ± 0.05 |
| 19r | ± | SO2Me | >10 | 41 ± 1 | |
| 19s | ± | Ph | - | ||
| 19t | ± | 2-pyrimidyl | 2.5 | 64 ± 1 | 5.61 ± 0.07 |
Absolute stereochemistry is unknown, but each resolved enantiomer displayed an ee of >95% as determined by analytical chiral HPLC
Values represent the mean ± standard error of the means of at least three independent determinations performed in triplicate. "-" indicates an inactive compound showing no PAM activity up to the highest concentration tested (30 µM).
When hM1 EC50 is reported as >10, this value represents the percent maximal ACh response when the test compound was present at its highest concentration (30 µM).
Table 3.
Structure and activities of analogs 20a–i.
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Compd | X | Y | Z | R3 | R5 | hM1 EC50 (µM)a |
AChmax (%)a,b |
pEC50a |
| 20a | F | H | H | Me | H | 3.0 | 41 ± 4 | 5.52 ± 0.02 |
| 20b | F | H | H | CF3 | H | - | ||
| 20c | F | H | H | H | Me | - | ||
| 20d | F | H | H | Me | Me | - | ||
| 20e | H | F | H | H | H | 2.0 | 28 ± 3 | 5.70 ± 0.09 |
| 20f | F | H | F | H | H | 2.6 | 66 ± 2 | 5.58 ± 0.07 |
| 20g | F | F | H | H | H | >10 | 43 ± 2 | |
| 20h | H | F | F | H | H | 4.1 | 51 ± 3 | 5.39 ± 0.05 |
| 20i | H | H | H | H | H | 1.8 | 44 ± 4 | 5.74 ± 0.05 |
Values represent the mean ± standard error of the means of at least three independent determinations performed in triplicate. "-" indicates an inactive compound showing no PAM activity up to the highest concentration tested (30 µM).
When hM1 EC50 is reported as >10, this value represents the percent maximal ACh response when the test compound was present at its highest concentration (30 µM).
Because compound 18d showed measurable potency, with the potential for a twofold potency increase upon chiral resolution, and was readily available for rapid analog preparation, this spiropyrrolidine was chosen for the additional modifications shown in Table 2. HPLC chiral column chromatography resolution of 18d provided the individual enantiomers (> 95% ee) 19a and 19b which surprisingly shared the identical potency and only slightly different efficacy, favoring enantiomer 19a. A series of N-cycloalkyl groups were explored (19c–f), showing the cyclopentyl ring to be the preferred size, which exhibited an equivalent potency to the N-methyl version (18d). The increase in alkyl group size from R = Me to R = c-pentyl may help to explain why there was now an enantioselective preference for one enantiomer (19g) over the other (19h), both with unknown absolute configurations. Given the range of activities seen with the various cycloalkyl groups, it was surprising to see such a large decline in activity upon going to the isopropyl analog 19i. The N-ethyl analog 19j shared the same potency as the N-methyl analog, but as fluorines were introduced onto the ethyl terminus, potency decreased (19k–m). Whether this trend in potency resulted from decreasingly basic pyrrolidine nitrogens, an increase in steric size, or a combination of both factors was not rigorously explored. Carbonate, acyl and sulfonyl groups were also attached to this nitrogen (19n–r) but resulted only in compounds with less functional activity than the parent analog 18d. Interestingly, the N-phenyl pyrrolidine 19s was prepared and found to be inactive at the hM1 receptor, whereas the 2-pyrimidine analog 19t produced low micro-molar activity. Table 2 serves to illustrate the type of shallow SAR frequently observed with allosteric ligands, where even minor changes often result in few gains, and more frequently, significant losses in functional activity.
Having explored the spirocycle region of these new M1 PAMs, we decided to re-examine the biaryl region in the context of the N-cyclopentyl spiropyrrolidine 19d. The results appearing in Table 3 indicated that the 3-position of the pyrazole could be substituted with a methyl group, leaving PAM activity virtually unaffected (20a). However, if a larger and more electron withdrawing trifluoromethyl group was introduced at the same location, all PAM activity was absent. Methyl substitution at the 5-position eliminated PAM activity (20c) and simultaneous methylations at both the 3- and 5- positions provided an inactive compound (20d). Although not shown in Table 3, compounds not methylated on the N-1 position of the pyrazole were devoid of PAM activity. Fluorination patterns on the central ring provided relatively flat SAR. The slightly preferred location for a single fluorine atom was that found in 20e. Of the difluorides tested, compound 20f was equipotent with the best monofluoride, while the other difluorides brought significantly reduced activity. Unexpectedly, compound 20i demonstrated that an absence of fluorination on the central phenyl was potentially superior with respect to potency. This fluorine SAR is very different from the SAR elucidated during the development of ML137,7b and exemplifies the potential pitfalls associated with trying to transfer established SAR from one series into even a closely related PAM series.
Even after a considerable amount of effort was invested to explore a great deal of structural diversity, no improvements in potency were forthcoming relative to the lead compound ML137. However, as shown in Table 4, encouraging improvements in efficacy were realized in the arena of hM1 ACh fold-shift values. Our initial foray into spirocycles, ketal 3, demonstrated an order of magnitude increase in its ability to shift the ACh concentration-response relative to ML137. Perhaps the most interesting pair of compounds in Table 4 was the pair of enantiomers 19a and 19b. Although both compounds share the same potency of 2.9 µM (hM1 EC50), the enantiomer with a slightly better efficacy (19a: AChmax = 67% versus 19b: AChmax = 50%) is roughly 3-times better at enhancing the potency of ACh at the hM1 receptor. This can be seen graphically in Figure 2 by comparing 19a (open inverted triangles) and 19b (open circles). Returning to Table 4, in the context of the N-ethyl spiropyrrolidines 19j and 19k, the potency trend relative to the number of fluorines present was not strictly recapitulated in their abilities to produce an ACh fold-shift. For example, although 19j was slightly more potent than 19k, with both sharing the same AChmax, 19k elicited a higher fold-shift of the full ACh concentration-response curve (CRC). For the final compound in Table 4, 19t, it was encouraging to see that such a structurally diverse analog could also show such robust efficacy.
Table 4.
ACh fold-shifts for selected hM1 PAMs.
| Compd | hM1 ACh fold-shifta |
|---|---|
| ML137 | 2.7 ± 0.2 |
| 3 | 27 ± 3 |
| 19a | 21 ± 2 |
| 19b | 7.2 ± 0.2 |
| 19g | 28 ± 2 |
| 19j | 8.3 ± 0.8 |
| 19k | 13.5 ± 0.7 |
| 19l | 5.9 ± 0.5 |
| 19t | 17.4 ± 0.9 |
Leftward shift of an ACh CRC in the presence of 30 µM compound relative to the ACh CRC control. Values represent the mean ± standard error of the means of at least three independent determinations.
Figure 2.

Fold-shift CRCs for the hM1 PAMs: ML137 (VU0366369-1), 3 (VU0413162-1), 19a (VU0450158-1), 19b (VU0450257-1).
To round out the characterization of these novel spirocyclic M1 PAMs, we profiled 3 and 18d across all five human and all five rat muscarinic acetylcholine receptors. Up to the highest concentrations tested (30 µM) both compounds were completely selective for the M1 receptors. A representative example of these results is shown in Figure 3 for compound 3 (VU0413162-1) against the human receptors.
Figure 3.

Muscarinic selectivity for 3 (VU0413162-1).
In summary, a number of isatin replacements containing various spirocycles have been explored and these novel structures have been confirmed as M1 PAMs. Although flat SAR was observed with respect to potency, significant improvements in efficacy were achieved for a range of structures. Very high receptor subtype selectivity for 3 and 18d was observed across the spectrum of muscarinic subtypes (M2 through M5). Further optimization within any of these scaffolds holds great promise for the development of highly active and selective M1 PAMs and will be reported in due course.
Acknowledgments
The authors thank the NIH (U54MH084659) and NIMH (1RO1MH082867) for support of our M1 program. Vanderbilt University is a Specialized Chemistry Center within the MLPCN, and all ML# probes are freely available upon request.14
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Dencker D, Thomsen M, Wörtwein G, Weikop P, Cui Y, Jeon J, Wess J, Fink-Jensen A. ACS Chem. Neurosci. 2012;3:80–89. doi: 10.1021/cn200110q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wess J, Eglen RM, Gautam D. Nat. Rev. Drug Discov. 2007;6:721–733. doi: 10.1038/nrd2379. [DOI] [PubMed] [Google Scholar]
- 3.Bymaster FP, Whitesitt CA, Shannon HE, DeLapp N, Ward JS, Calligaro DO, Shipley LA, Buelke-Sam JL, Bodick NC, Farde L, Sheardown MJ, Olesen PH, Hansen KT, Suzdak PD, Swedberg MDB, Sauerberg P, Mitch CH. Drug Dev. Res. 1997;40:158–170. [Google Scholar]
- 4.Shekhar A, Potter WZ, Lightfoot J, Lienemann J, Dube S, Mallinckrodt C, Bymaster FP, McKinzie DL, Lelder CC. Am. J. Psychiatry. 2008;165:1033–1039. doi: 10.1176/appi.ajp.2008.06091591. [DOI] [PubMed] [Google Scholar]
- 5.Melancon BJ, Gogliotti RD, Tarr JC, Saleh SA, Chauder BA, Lebois EP, Cho HP, Utley TJ, Sheffler DJ, Bridges TM, Morrison RD, Daniels JS, Niswender CM, Conn PJ, Lindsley CW, Wood MR. Bioorg. Med. Chem. Lett. 2012;22:3467–3472. doi: 10.1016/j.bmcl.2012.03.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Digby GJ, Utley TJ, Lamsal A, Sevel C, Sheffler DJ, Lebois EP, Bridges TM, Wood MR, Niswender CM, Lindsley CW, Conn PJ. ACS Chem. Neurosci. 2012 doi: 10.1021/cn300103e. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Ma L, Seager MA, Wittmann M, Jacobson M, Bickel D, Burno M, Jones K, Graufelds VK, Xu G, Pearson M, McCampbell A, Gaspar R, Shughrue P, Danziger A, Regan C, Flick R, Pascarella D, Garson S, Doran S, Kreatsoulas C, Veng L, Lindsley CW, Shipe W, Kuduk S, Sur C, Kinney G, Seabrook GR, Ray WJ. Proc. Natl. Acad. Sci. USA. 2009;106:15950–15955. doi: 10.1073/pnas.0900903106. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Bridges TM, Kennedy JP, Noetzel MJ, Breininger ML, Gentry PR, Conn PJ, Lindsley CW. Bioorg. Med. Chem. Lett. 2010;20:1972–1975. doi: 10.1016/j.bmcl.2010.01.109. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Reid PR, Bridges TM, Sheffler DJ, Cho HP, Lewis LM, Days E, Daniels JS, Jones CK, Niswender CM, Weaver CD, Conn PJ, Lindsley CW, Wood MR. Bioorg. Med. Chem. Lett. 2011;21:2697–2701. doi: 10.1016/j.bmcl.2010.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Tarr JC, Turlington ML, Reid PR, Utley TJ, Sheffler DJ, Cho HP, Klar R, Pancani T, Klein MT, Bridges TM, Morrison RD, Blobaum AL, Xiang Z, Daniels JS, Niswender CM, Conn PJ, Wood MR, Lindsley CW. ACS Chem. Neurosci. 2012 doi: 10.1021/cn300068s. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Brady AE, Jones CK, Bridges TM, Kennedy JP, Thompson AD, Heiman JU, Breininger ML, Gentry PR, Yin H, Jadhav SB, Shirey JK, Conn PJ, Lindsley CW. J. Pharmacol. Exp. Ther. 2008;327:941–953. doi: 10.1124/jpet.108.140350. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kennedy JP, Bridges TM, Gentry PR, Brogan JT, Kane AS, Jones CK, Brady AE, Shirey JK, Conn PJ, Lindsley CW. ChemMedChem. 2009;4:1600–1607. doi: 10.1002/cmdc.200900231. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Salovich JM, Vinson PN, Sheffler DJ, Lamsal A, Utley TJ, Blobaum AL, Bridges TM, Le U, Jones CK, Wood MR, Daniels JS, Conn PJ, Niswender CM, Lindsley CW, Hopkins CR. Bioorg. Med. Chem. Lett. 2012;22:5084–5088. doi: 10.1016/j.bmcl.2012.05.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Bridges TM, Marlo JE, Niswender CM, Jones CK, Jadhav SB, Gentry PR, Plumley HC, Weaver CD, Conn PJ, Lindsley CW. J. Med. Chem. 2009;52:3445–3448. doi: 10.1021/jm900286j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Bridges TM, Kennedy JP, Cho HP, Breininger ML, Gentry PR, Hopkins CR, Conn PJ, Lindsley CW. Bioorg. Med. Chem. Lett. 2010;20:558–562. doi: 10.1016/j.bmcl.2009.11.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Melancon BJ, Poslusney MS, Gentry PR, Tarr JC, Sheffler DJ, Mattmann ME, Bridges TM, Utley TJ, Daniels JS, Niswender CM, Conn PJ, Lindsley CW, Wood MR. Bioorg. Med. Chem. Lett. doi: 10.1016/j.bmcl.2012.11.092. Accepted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Although the M1 PAM EC50 has been previously reported as 0.83 µM with a %AChmax = 65% (Ref. 7b), the inherent variability associated with functional assays supports that the most recent EC50 = 0.60 µM for ML137 is equally valid and can serve as a baseline comparator between the two papers.
- 12.Nair V, Rajesh C, Dhanya R, Rath NP. Tet. Lett. 2002;43:5349–5351. doi: 10.1021/ol025503j. [DOI] [PubMed] [Google Scholar]
- 13.It is worth noting that intermediates 13, 14, 16 and 17 (Schemes 2 and 3), which bear a close resemblance to the M1,3,5 PAM VU0119498, were found to be inactive at hM1.
- 14.For information on the MLPCN and information on how to request any of the ML# probe compounds, such as ML137, see: http://mli.nih.gov/mli/mlpcn/