

Abstract
Transcription by RNA polymerase III (pol III) is responsible for ~15% of total cellular transcription through the generation of small structured RNAs such as tRNA and 5S RNA. The coordinate synthesis of these molecules with ribosomal protein mRNAs and rRNA couples the production of ribosomes and their tRNA substrates and balances protein synthetic capacity with the growth requirements of the cell. Ribosome biogenesis in general and pol III transcription in particular is known to be regulated by nutrient availability, cell stress and cell cycle stage and is perturbed in pathological states. High throughput proteomic studies have catalogued modifications to pol III subunits, assembly, initiation and accessory factors but most of these modifications have yet to be linked to functional consequences. Here we review our current understanding of the major points of regulation in the pol III transcription apparatus, the targets of regulation and the signaling pathways known to regulate their function.
Keywords: Maf1, rapamycin-sensitive TOR signaling, Sch9, Kns1, Mck1, TFIIIB, protein kinase A, protein kinase CK2
1. Introduction
The major biosynthetic function of RNA polymerase III (pol III) is the production of small, untranslated structural RNAs for protein synthesis. The main gene products, 5S rRNA and tRNA, are highly abundant molecules comprising ~15% of total RNA by weight, and function respectively as part of the large ribosomal subunit and as amino acid-specific adapters for mRNA decoding. Several other pol III transcripts play functionally-related roles in pre-rRNA or pre-tRNA processing complexes (RNase MRP and RNase P RNAs), serve as guide RNAs for rRNA methylation (e.g. yeast snR52) or are required for co-translational insertion of nascent polypeptides across the membrane of the endoplasmic reticulum (7SL RNA). Numerous pol III transcripts have been ascribed functions in processes other than protein synthesis. Well known examples include U6 snRNA, involved in pre-mRNA splicing, the RNA component of telomerase (TERC) and the 7SK RNA regulatory complex of the pol II transcription elongation factor P-TEFb. These and other pol III transcripts of both cellular and viral origin have been described in many reports over the years [1]. Adding to this knowledge, recent efforts to understand the scope of the pol III transcriptome in mammalian cell lines and in mouse liver have identified a small number of new bona fide pol III-transcribed genes and a somewhat larger group of candidate pol III-transcribed genes for further study [2–4].
The pol III transcription apparatus is generally well conserved from yeast to man and in its simplest form comprises a DNA binding factor (TFIIIC), an initiation factor (TFIIIB) and the polymerase which at 17 subunits is the largest of the eukaryotic RNA polymerases in terms of molecular complexity and mass [5]. The six subunit TFIIIC complex binds to the two conserved internal promoter elements (A and B blocks) that characterize tRNA genes and directs the assembly of TFIIIB on the DNA upstream of the transcriptional start site. Concerted binding of the three subunits of TFIIIB (TBP, Brf1 and Bdp1) produces a very stable, kinetically-trapped DNA complex that recruits and orients the polymerase and contributes to the formation of the open promoter complex [6,7]. Other pol III templates have different promoter structures and factor requirements and these are described elsewhere in this issue. Once transcription has initiated, elongation by pol III does not require additional accessory factors (at least in vitro) and proceeds with high fidelity, displacing the TFIIIC in its path before terminating at a short run of T residues [8]. Subsequently, pol III can either dissociate from the DNA or it can reinitiate transcription on the same DNA-bound TFIIIB complex in a process of facilitated recycling [9]. This involves multiple cycles of transcription by a single polymerase molecule on the same gene. Relatively little is known about how this process occurs and to date there are no single molecule studies to support this concept. However, there is growing evidence that reinitiation by pol III may be an important point of regulation in this system.
Unlike the majority of transcripts generated by the other nuclear RNA polymerases, the primary transcripts synthesized by pol III are short, ranging from ~100 nucleotides for tRNA and 5S rRNA genes to 522 nucleotides for SCR1 in yeast [10] and robustly transcribed. The doubling of yeast cells every 90 minutes in rich medium requires an initiation rate of ~2–4 transcripts/gene/sec for the average tRNA gene (3–6 × 106 tRNAs per cell from 274 genes) versus ~0.5 transcripts/gene/sec for 35S rRNA (~200,000 ribosomes per cell and 75 active gene copies) [11,12]. The turnover rate of tRNA and ribosomes is low under physiological conditions and not considered here. The high rates of synthesis and the short size of pol III transcripts appear to favor initiation as the major point of regulation for pol III gene transcription. In general, pol III-transcribed genes are required for cell growth and proliferation and their expression is co-ordinately regulated in response to nutrients and cellular stress along with the other components of the ribosome. In this review we focus on recent insights, primarily from yeast, into the function of conserved regulators and signaling pathways that control pol III transcription.
2. Targets for Regulation of Pol III Transcription
2.1.1 Maf1 - Discovery of its Conserved Function
Three repressors of pol III transcription have been identified that bind and inhibit the transcription machinery in a manner that is regulated by upstream signaling pathways. Two of these proteins, RB and p53, are well known tumor suppressors and their effects on pol III (and pol I) transcription are reviewed in this volume and elsewhere [13]. The third protein, Maf1, is conserved throughout eukaryotes and was identified initially in yeast in a screen for mutants that decreased SUP11 tRNA nonsense suppressor activity and were unable to utilize glycerol as a carbon source at elevated temperatures [14]. In addition to these phenotypes, subsequent studies found that the original maf1-1 mutant as well as a MAF1 deletion strain had elevated levels of total tRNA and increased rates of tRNA synthesis [15]. A key genetic finding in these early studies was that the anti-suppressor phenotype (attributed to increased translational fidelity in the mutant strain) [16], and the inability to respire at elevated temperatures could be suppressed either by overexpression of Rpc160, the largest subunit of pol III (i.e. a dominant negative effect) or by mutations in Rpc160 that reduced pol III transcription [15,17]. These genetic interactions together with the physical association of Maf1 with the polymerase and the effect on tRNA synthesis provided strong support for the function of Maf1 as a negative regulator of pol III transcription [15]. Biological proof for this hypothesis came when a range of nutritional, environmental and cellular stress conditions that were demonstrated to repress pol III transcription, quantitatively failed to do so in strains deleted for Maf1 [18]. The block to repression was absolute and the scope of the involvement of Maf1 in the repression of pol III transcription was broad. All tested conditions of nutrient limitation, poor carbon sources and cellular stresses that directly or indirectly affect cell growth (e.g. secretory pathway and cell wall defects, oxidative and ER stress, DNA damage, etc.) require Maf1 to repress pol III transcription [14,18,19]. As discussed later in this review, the responses to these conditions involve many important and conserved signaling pathways in eukaryotic cells.
More recently, studies in several labs have shown that the negative role of Maf1 in pol III transcription is conserved in higher eukaryotes: Mammalian cells that have little or no Maf1 have elevated steady state levels of pol III transcripts [20–25]. A knock-down of Maf1 in developing Drosophila larvae also led to the accumulation of tRNA [25]. The requirement for Maf1 in signaling repression is also conserved in these systems: Efficient repression of pol III transcription in response to nutrient deprivation, inhibition of TORC1 and alkylating DNA damage was not seen in cell lines that either lack Maf1 or were treated with Maf1 siRNAs [20,23,24]. Additionally, knockdown of Maf1 in either Drosophila larvae or the fat body (an endocrine organ that regulates energy storage and utilization in insects) was sufficient to prevent the normal decrease in total tRNA caused by nutrient starvation or inhibition of TORC1 by rapamycin [25,26].
In contrast to the congruity of responses to Maf1 ablation/knockdown, elevated Maf1 expression produces different outcomes depending on the system and conditions. In yeast cells, greater than 10-fold overexpression of Maf1, achieved by elevated gene dosage, does not cause repression of pol III transcription on glucose-containing medium in the absence of exogenous stress [19]. However, Maf1 overexpression induced by the GAL1 promoter produces a significant (~two-fold) decrease in transcription [27] raising the possibility that the slower growth and/or metabolic changes that occur on a suboptimal carbon source (galactose) contribute to repression under these conditions. Ectopic expression of Maf1 in apparently unstressed mammalian cells has also been reported to decrease pol III transcription in some but not all cell lines [21,22,24,28]. Whether these observations reflect differences in the way that yeast and mammalian systems regulate Maf1-dependent repression is currently unknown. However, a unifying view might be that mammalian cells in culture have activated a cellular response that (like yeast on galactose) can affect repression when Maf1 levels are elevated.
2.1.2 Phosphoregulation of Maf1 Localization and Interactions
The yeast and mammalian Maf1 proteins are negatively regulated by phosphorylation at multiple sites, i.e. the protein is largely dephosphorylated under repressing conditions [20,22–24,29–34]. The results of several unbiased or Maf1-targeted phosphoproteomic studies [33,35–38] combined with in vitro phosphorylation assays and functional studies of phosphosite and kinase/signaling mutants [31–33,39] have found that the biologically important sites in yeast Maf1 are seven serine residues (amino acids 90, 101, 177–179, 209 and 210), six of which correspond to consensus PKA/Sch9 sites (RR/KXS/T)[40]. Phosphorylation of these sites changes the localization of Maf1 in the cell and alters the interaction of Maf1 with the polymerase [29–33,41,42]. Mutagenesis studies indicate that no single phosphosite is critical, their contributions are cumulative and that residues 209 and 210 account for most of the phosphorylation on Maf1 [32]. Indeed, recent experiments with an internally-deleted, functional MAF1 allele (MAF1-id) that is missing five of the seven phosphosites (noted above) suggest that phosphorylation of serines 209 and 210 is sufficient for the normal regulated localization of this protein [42]. Despite the fact that Maf1 is phosphorylated on many sites, the protein usually separates into just two species on high resolving one-dimensional SDS gels [29,30,32]. This complicates the interpretation of some experiments since the slow migrating species in this system is especially sensitive to phosphorylation at two sites (S177 and S178) that together contribute only ~35% of the total PKA/Sch9 consensus site phosphorylation [32]. Moreover, unphosphorylated Maf1 is not separable from fast-migrating multiply-phosphorylated forms of Maf1 at the other PKA/Sch9 consensus sites. Thus, Maf1 mobility in these gels does not correlate with the total level of phosphorylation of the protein. Immunoprecipitation and western blotting with a phospho-PKA substrate specific antibody [31,32,43] or the use of Phostag acrylamide gels are able to report the extent of Maf1 phosphorylation [42]. For human Maf1, a combination of mass spectrometry, Phostag acrylamide gel analysis and mutagenesis identified serines 60, 68 and 75 as the major regulated phosphosites [24]. The phospho-regulation of serine 75 has been independently examined in two other studies [23,34]. It is noteworthy that although the sequence in this region of human Maf1 is not conserved in the yeast protein, the sites are positionally similar to serines 209/210 in yeast Maf1 relative to the phylogenetically conserved domain B [42] and appear to be functionally equivalent [23,24,34].
Yeast Maf1 contains two nuclear localization sequences (NLSs). The N-terminal NLS (NtNLS) is located immediately adjacent to serines 209/210 and its function is regulated by phosphorylation at the consensus PKA/Sch9 sites [31,42]. A functionally weaker C-terminal NLS (CtNLS) is located within conserved domain C. Whether the CtNLS is regulated has not been examined. Both NLS sequences contribute to the localization and function of Maf1 in yeast [31]; either NLS is sufficient to localize Maf1 to the nucleus under appropriate conditions. Mutations that compromise the regulation and/or function of both NLSs fail to accumulate Maf1 in the nucleus under repressing conditions and are unable to repress transcription. In the same study, mutagenesis of six PKA/Sch9 consensus sites to alanine was found to promote the nuclear accumulation of Maf1 but did not induce repression under optimal growth conditions [31]. Thus, it was concluded that nuclear dephosphorylated Maf1 is not sufficient for repression and that an additional step or steps must therefore be necessary. As described below, recent work has identified one such step which involves phosphorylation of pol III.
Another way to accumulate Maf1 in the nucleus under normal growth conditions was revealed with the identification of MSN5 as the exportin for Maf1 [44]. In cells deleted for MSN5, Maf1 is predominantly nuclear and, like the phosphosite mutants described above, does not cause repression of pol III transcription in the absence of cellular stress. Moreover, despite its nuclear location in the msn5 strain, Maf1 was found to be phosphoregulated normally upon carbon source switching (glucose to glycerol and vice versa). These experiments and the phospho-regulated nuclear import of Maf1 [31] indicate that kinases and phosphatases in both the cytoplasm and the nuclear compartments control the localization of Maf1 (Figure 1). Together with variations in the cellular distribution of Maf1 in different strain backgrounds and growth conditions [27,29–31,33,44,45], the data suggest that, like other stress responsive regulators (e.g. Msn2), Maf1 may cycle between the nucleus and the cytoplasm during growth in unstressed conditions. Consistent with this idea, Maf1 can be ChiPed at a low level to pol III genes in the absence of any stress when it is primarily located in the cytoplasm [29,30]. Additional trafficking of Maf1 between the nucleoplasm and the nucleolus is suggested by studies on the association of rapamycin-resistant TORC1 mutants with 5S rDNA chromatin in the nucleolus and correlated changes in Maf1 phosphorylation, localization and pol III repression (Figure 1, see also section 3.2).
Figure 1. Multiple kinases and phosphatases determine the cellular localization of Maf1 in unstressed yeast cells.
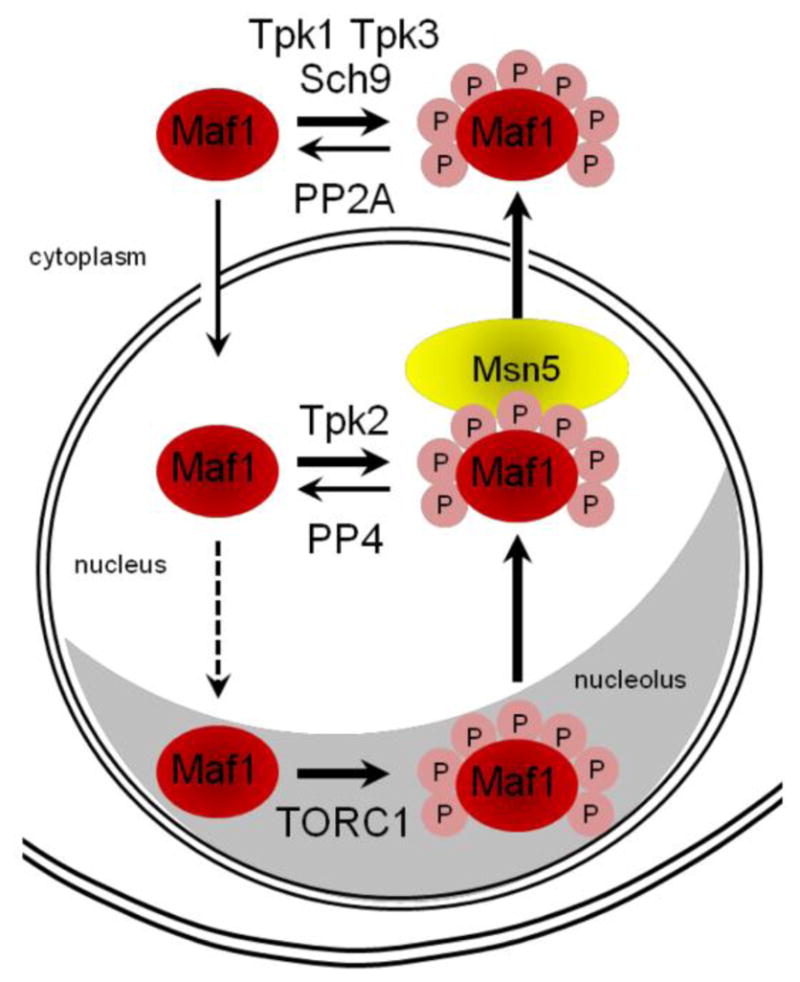
Sch9, PKA and TORC1 kinases modify Maf1 during exponential growth in rich media leading to its localization primarily in the cytoplasm in some but not all strains. Phosphorylation of Maf1 at seven PKA/Sch9 consensus sites is depicted by pink circles (annotated P). The PKA/Sch9 sites are immediately adjacent to or proximal to the NtNLS. Phosphorylation at these sites negatively regulates the nuclear accumulation of Maf1 in unstressed cells. The kinases and phosphatases known to modify Maf1 are annotated. The karyopherin Msn5 is required for export of Maf1 from the nucleus. See sections 2.1.2 and 3.2 for details.
Metazoan Maf1 retains the NLS located in conserved domain C (CtNLS) [15] and the protein is localized to the nucleus in many but not all cell lines [23,28,34]. Regardless of the extent of differential localization, repression of pol III transcription in all systems involves the nuclear accumulation of dephosphorylated Maf1 and an increase in the association of Maf1 with pol III genes [20,22,23,29–31,34,44,45]. Since Maf1 cannot bind specifically or non-specifically to nucleic acids [19], its interaction with pol III-transcribed genes is indirect and is mediated largely via the polymerase [15,19,20,29,30,33]. This is supported by biochemical experiments demonstrating a stable association of Maf1 with pol III bound to model DNA templates [46].
In addition to regulating its localization in the cell, phosphorylation of Maf1 changes its interaction with pol III. Reciprocal co-immunoprecipitations of Maf1 and pol III subunits from control versus nutrient-deprived or rapamycin-treated yeast cells show that only a small fraction of Maf1 corresponding to the fast-migrating hypo- or dephosphorylated protein is associated with pol III under optimal growth conditions. However, the amount of this complex increases significantly under robust repressing conditions where Maf1 is fully dephosphorylated [29,30]. Maf1 mutants with a diminished capacity to be dephosphorylated also show a lower level of association with pol III [29]. In addition, mutation of all seven sites targeted by PKA and Sch9 to alanine (Maf1 7SA) significantly enhanced the Maf1-pol III interaction under non-repressing conditions whereas a phosphomimetic mutant of the same sites (Maf1 7SE) significantly diminished the interaction under repressing conditions [33]. While these biochemical experiments clearly point to an affinity differential favoring the formation of a dephosphorylated Maf1-pol III complex under repressing conditions, the biological outcome is not so clear cut. A phosphomimetic mutant of Maf1 (6SE, which retains S179, a minor contributor to overall Maf1 phosphorylation) [33] is as active as wild-type Maf1 in repressing transcription in rapamycin-treated cells [31]. Moreover, ChIP assays in a human cell extract demonstrated that Maf1 can be cross-linked to tRNA and 5S rRNA genes with a phospho-specific antibody prepared against a phosphoserine 75 Maf1 peptide [34].
Given the essential role played by Maf1 in repressing pol III transcription, it is striking that none of the known regulatory steps that limit the access of Maf1 to its targets or affect the direct interaction of Maf1 with pol III are similarly essential for controlling its repressing function. These observations indicate the existence of failsafe mechanisms that prevent the repression of pol III transcription in the absence of cellular stress.
2.1.3 Inhibition of Pol III Transcription by Maf1 in vitro
Pol III transcription in crude in vitro systems is especially sensitive to changes in the amount or activity of TFIIIB subunits since the assembly of this initiation factor onto DNA is generally the limiting step for transcription in these systems [47]. Consistent with this, biochemical studies in extracts from control and repressed (chlorpromazine-treated) yeast cells initially identified TFIIIB as a target of Maf1-dependent repression [18,19]. Subsequently, a direct biochemical interaction was demonstrated between Maf1 and Brf1 and this interaction was shown to inhibit the recruitment of Brf1 to TFIIIC-DNA complexes [19]. The Maf1-Brf1 interaction is conserved in mammalian systems [12,20,22] and this supports the idea that its biological importance is the inhibition of de novo transcription complex assembly [48]. Further details about the interaction and its potential regulation remain to be elucidated.
Given the high stability of TFIIIB complexes on pol III-transcribed genes in vitro [6,49] and in vivo (even under repressing conditions [50,51], it was apparent that a different mechanism of repression was needed for acute responses. Focusing on the already known Maf1-pol III interaction, order-of-addition experiments in both yeast and human systems revealed that recombinant Maf1 inhibited transcription from preassembled TFIIIB-DNA complexes primarily by preventing pol III recruitment [12,19]. Conversely, addition of Maf1 after pol III binding to the TFIIIB-DNA complex or after the synthesis of a short RNA transcript did not inhibit initiation or elongation [12]. Consistent with this, factor-independent transcription on an elongation scaffold was also not inhibited despite the stable binding of Maf1 to pol III [46]. In addition, immobilized template assays were used to demonstrate that human pol III could undergo a limited number of rounds of facilitated recycling, extending findings established for the enzyme in yeast [9] and that this process was refractory to inhibition by Maf1 [12]. Thus, our current concept is that Maf1 binding to pol III in solution (i.e. off the DNA) inhibits its recruitment to the initiation complex. Maf1 binding to pol III after it has engaged the initiation complex or during elongation only inhibits transcription upon dissociation of the polymerase from the template [12,46]. Based on the cryo-EM structures of free pol III, Maf1-pol III and pol III in an elongation complex along with homology modeling, the structural basis for the preceding effects is thought to involve the rearrangement of the C82/C34/C31 subcomplex of polymerase as a consequence of Maf1 binding to the polymerase clamp domain [46].
The inability of Maf1 to inhibit facilitated recycling of pol III in vitro has important implications for transcription during the response to cellular stress. Consequently, it has become important to know whether facilitated recycling occurs stochastically, how it proceeds in relation to termination and if the process is regulated. These are challenging questions since the phenomenon of facilitated recycling has only been demonstrated biochemically and the regulatory systems that may influence the process operate within the context of the cell.
2.2 RNA Polymerase III
The concept that repression of pol III transcription requires additional steps beyond the nuclear localization of dephosphorylated Maf1 [31], along with the resistance of facilitated recycling to inhibition by Maf1 [12] led to the consideration of pol III as a target of repression signaling. Support for this idea (in principle) has existed for some time based on the results of high throughput proteomic screens as well as targeted studies that have identified post-translational modifications in several pol III subunits. Sumoylation of specific pol III subunits has been documented in multiple studies, one of which included stress conditions (see ref [52] and references therein). However, the significance of this modification for pol III function has yet to be established. Numerous pol III subunits are also known phosphoproteins [35,53–55].
A recent examination of the effect of rapamycin on in vivo 32P labeling of pol III identified Rpc53 as a differentially-labeled subunit [56]. Subsequently, it was found that phosphorylation of Rpc53 correlates with the inhibition of pol III transcription in response to diverse stresses [56]. A screen of viable yeast gene-deletion strains representing kinases, phosphatases and their regulatory proteins identified two conserved kinases, Kns1 and Mck1, both of which were required for Rpc53 hyperphosphorylation. This joint requirement was rationalized by in vivo and in vitro experiments showing that Kns1 phosphorylation of a single site (T232) primed Rpc53 for further phosphorylation by Mck1 at two adjacent sites (S228 and S224). Importantly, mutation of these phosphorylation sites to alanine attenuated rapamycin-mediated repression of pol III transcription although this result has so far only been demonstrated in a “sensitized” strain containing a hypomorphic allele of another polymerase subunit, Rpc11 [56]. The findings suggest that Kns1 and Mck1 phosphorylation of Rpc53 under repressing conditions cooperates with Rpc11 to allow repression by Maf1.
A model for how this may be achieved has been proposed based on the known functions of Rpc53 as part of the heterodimeric TFIIF-like complex (Rpc53/Rpc37) and Rpc11 which is homologous to Rpb9 and TFIIS. The Rpc53/37 complex functions to slow the elongation rate of pol III which is important for normal termination of transcription [57]. It also participates in promoter opening and interacts with subunits of TFIIIB and TFIIIC [57–60]. Known functions of Rpc11 include termination, exoribonucleolytic proofreading and facilitated reinitiation [57,61]. There is also evidence for physical associations between the Rpc53/Rpc37 complex and Rpc11, including the finding that a C-terminal truncation of Rpc37 leads to the loss of all three subunits from the polymerase [57]. Together these data suggest that phosphorylation of Rpc53, through allosteric or other mechanisms, leads to changes in Rpc11 that disrupt facilitated recycling or favor pol III dissociation from the DNA template after termination [56]. If this occurs when pol III is already in a complex with Maf1, reinitiation is immediately inhibited. Alternatively, pol III may interact with Maf1 off the DNA to prevent rebinding to the initiation complex [5,56].
Hyperphosphorylation of Rpc53 by Kns1 and Mck1 (driven by overexpression of Kns1) was found to be independent of the Maf1 repressor but was not sufficient to cause repression in the absence of cellular stress [56]. Similarly, dephosphorylation of Maf1 under the same repressing conditions was not dependent on either kinase and, as already noted, nuclear dephosphorylated Maf1 produces robust repression only under stress conditions. Together, these observations reveal some of the properties of the proposed failsafe mechanism that prevents transcriptional repression in the absence of stress signaling. Maf1 and pol III are controlled by separate branches of the TORC1 signaling pathway but they must function together in order to rapidly and efficiently repress transcription. Thus, control of pol III transcription is exercised by a network structure that is similar to an AND-gated feed-forward loop (Figure 2)[62].
Figure 2. A hypothetical network model for regulation of pol III transcription.
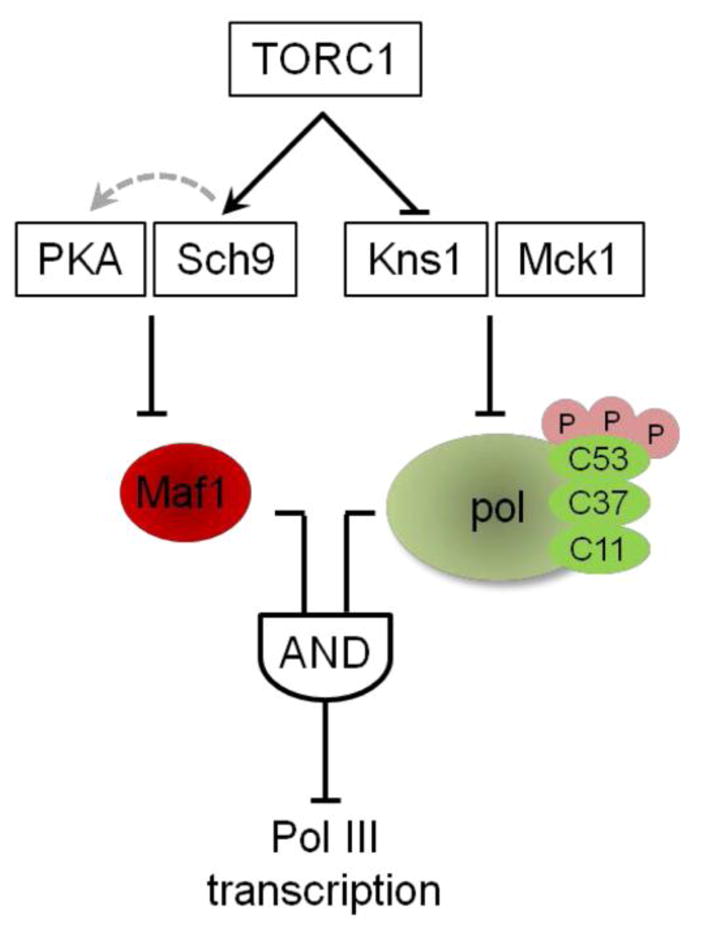
The feed forward loop has two inputs that independently regulate Maf1 and pol III function downstream of stress responsive signaling, in this case, TORC1. The branches control Maf1 function in repression and the sensitivity of the polymerase to Maf1 inhibition of function. Repression requires regulation from both inputs indicating an AND-gate feed forward loop. See section 2.2 for details. This figure was adapted from Willis and Lee [62].
The association of Rpc53/37 with the core polymerase can be destabilized by mutations in the C-termini of either protein and both the Rpc53/37 subcomplex and the TFIIE-like Rpc82/34/31 subcomplex can be separated from the core polymerase during purification [57,58,63]. These properties coupled with the homology of the Rpc53/37 and Rpc82/34/31 subcomplexes to the general pol II transcription factors TFIIF and TFIIE has led to the suggestion that they may bind dynamically during the transcription cycle [63]. However, there is no evidence to date that the stability of pol III is regulated in vivo.
Two forms of human pol III have been identified that differ in the composition of the TFIIE-like RPC362/39/32 subcomplex [64]. The pol III isoforms, PolIII alpha and PolIIIbeta, contain RPC32 subunits which share only 51% identity. Although both pol III isoforms support transcription in vitro, they differ in their expression and function in vivo: Pol IIIbeta is ubiquitously expressed and essential while Pol IIIalpha is dispensable for cell survival and is associated with cell proliferation and transformation and elevated transcription of the 7SL gene. These observations suggest that pol III composition, at least in higher eukaryotes, can alter pol III activity at specific loci.
2.3 TFIIIB
The three subunit initiation factor TFIIIB comprises the TATA-box binding protein TBP, Brf1 and Bdp1 in yeast and related subunits in metazoans. TFIIIB functions to recruit and position pol III appropriately for transcription and participates in promoter opening [7]. Studies in yeast initially found that overexpression of Brf1, or mutations in Tfc4 that facilitate Brf1 recruitment to TFIIIC-DNA complexes, increase TFIIIB assembly and pol III transcription from genes with defective or weak promoters [47]. This indicated that the assembly of TFIIIB on DNA was a key limiting step in transcription and therefore a likely target for regulation. Subsequent studies in higher eukaryotes have found that changes in the abundance, phosphorylation state and activity of TFIIIB affects pol III transcription. TFIIIB recruitment to pol III-transcribed genes is limited by the expression level of its subunits which are coordinately regulated in response to proliferative signals, e.g. in response to JNK signaling [65]. Oncogenic proteins, tumor suppressors, viral infection and the activation of specific signaling pathways can also alter the expression of specific TFIIIB components [66–68].
TFIIIB activity and pol III transcription is positively regulated by protein kinase CK2, in both yeast and mammalian cells. CK2 is physically associated with TFIIIB on pol III-transcribed genes in both systems and CK2 activity is required for efficient TFIIIB assembly and high levels of transcription [38,69–71]. While all of the subunits of TFIIIB can be phosphorylated by CK2 in vitro [69–71], it is not known which of these factors are modified by CK2 in vivo to promote transcription. Exposure of yeast to ultraviolet light or methane methylsulfonate (MMS) leads to inhibition of TFIIIB function and repression of pol III transcription. These changes correlate with the dissociation of the CK2 catalytic subunits from the regulatory beta subunit which remains bound to TBP [69]. The latter interaction is required for high TBP-associated CK2 activity and pol III transcription in unstressed yeast cells. Several high throughput studies have identified yeast TFIIIB subunits as phosphoproteins, but to date there is no evidence for their differential modification in vivo in response to stress.
Pol III transcription is repressed during mitosis in vertebrates. This response is linked to the phosphorylation of multiple components of the system [72] and to the inactivation of TFIIIB [73–76]. The Brf1 and Bdp1 subunits become hyperphosphorylated in mitosis and Bdp1 dissociates from the promoters of the U6, 5S and tRNA genes to limit TFIIIB function [75,76]. The phosphorylation of TFIIIB is not uniformly negative for pol III transcription as ERK phosphorylation of Brf1 increases TFIIIB interactions with both TFIIIC and pol III and stimulates transcription [77]. Also, inactivation of mTOR signaling by PTEN expression causes a decrease in Brf1 phosphorylation, disrupts TBP binding to Brf1, reduces TFIIIB occupancy on tRNA genes and inhibits pol III transcription [78]. Phosphorylation of Brf1 by the polo-like kinase Plk1 can either stimulate or repress transcription depending on the cell-cycle stage and the sites that are modified [79]. Plk1 physically associates with 5S and tRNA genes and promotes transcription by phosphorylating Brf1 at S450 during interphase. However, the abundance and specific activity of Plk1 increases during mitosis resulting in modification of Brf1 at T270 and inhibition of pol III recruitment [79]. The modification of Brf1 at different sites in response to various signaling pathways and cell cycle stage demonstrates the complexity of its phosphoregulation.
Bdp1 phosphorylation is so far only associated with negative effects on transcription and correlates with its dissociation from DNA during mitosis and following inactivation of mTOR signaling [75,76,78]. The mitotic effects on Bdp1 function are due to its hyperphosphorylation by CK2. This conclusion is supported by in vivo and in vitro studies using peptide and/or chemical inhibitors of CK2 [76]. Notably, the inhibitory effect of CK2 was demonstrated on a truncated Bdp1 protein in a reconstituted transcription system and could be blocked by mutation of five consensus CK2 sites; i.e. the mutant protein was no longer phosphorylated by CK2 and was as active in transcription as the unphosphorylated protein. Given the mitotic effect of Plk1 described above, it seems likely that Plk1 and CK2 function cooperatively to repress pol III transcription by targeting Brf1 and Bpd1, respectively [79].
2.4 TFIIIC
TFIIIC serves as a DNA binding factor for the assembly of TFIIIB and polymerase on pol III-transcribed genes [49]. The factor is also bound at genomic sites in yeast and mammals that do not encode pol III genes, the so-called ETC loci (extra TFIIIC sites, also called chromosome organizing clamps in S. pombe) [2,80,81]. The nucleosome-positioning and chromatin-boundary functions of pol III-transcribed genes and ETC loci, coupled with their cohesin and condensin binding properties, suggest that TFIIIC may directly contribute to chromatin organization [82]. TFIIIC is also required for efficient pol III reinitiation [83].
The activity and/or abundance of TFIIIC in higher eukaryotes is targeted for regulation in pathological states. Viral transformation models and human tumors have increased TFIIIC activity consistent with their increase in protein synthesis and proliferative capacity [84]. Adenovirus infection and E1A or SV40 transformation increase the abundance of the TFIIIC110 subunit (homologous to Tfc6 in yeast) and is correlated with elevated active, hyperphosphorylated TFIIIC and pol III transcription [85,86]. However, ecotopic expression of TFIIIC110 alone is not sufficient to cause increased transcription in non-transformed cells [87]. TFIIIC abundance is also controlled by proteolysis of specific subunits: The large subunit of human TFIIIC is targeted by the poliovirus-encoded 3C protease (3Cpro) both in vivo and in vitro and correlates with the accumulation of an inactive form of the factor [88]. Virulent Leishmania infection of macrophages results in the degradation of the TFIIIC110 subunit through thrombin receptor-mediated activation of the protease μ-calpain. This leads to a decrease in functional TFIIIC and the loss of pol III transcription of genes containing tRNA-type promoters [89].
The Tfc3, Tfc4 and Tfc1 subunits of yeast TFIIIC are phosphorylated in vivo but the functional significance of these modifications is unknown [90].
2.5 Other Potential Regulatory Targets
Sub1, a non-histone chromatin-associated protein in yeast with positive and negative roles in transcription by pol II, ChIPs to pol III-transcribed genes consistent with its role in transcription initiation and reinitiation by pol III [91–93]. Deletion of SUB1 reduces the level of the polymerase at pol III-transcribed genes suggesting a general role in pol III recruitment. Although the association of Sub1 with pol III genes decreases in stationary phase [92], Sub1 occupancy on osmoregulated pol II genes increases with gene induction under osmotic stress conditions suggesting a positive role in osmolarity tolerance and a potential for regulation by cellular stress signaling pathways [93]. In vitro studies show that Sub1 promotes TFIIIB and TFIIIC binding and increases initiation and reinitiation by pol III [92]. The DNA binding activity of the human homolog of Sub1, PC4, is affected by both phosphorylation and acetylation (reviewed in [94]), but regulation of Sub1 activity in yeast by differential modification has not been reported.
The abundant Nhp6 proteins bind and bend DNA and contribute to the maintenance of chromatin structure (reviewed in [95]). The Nhp6 proteins were originally implicated in pol III transcription through their positive effect on SNR6 gene transcription, which requires a positioned nucleosome for optimal expression from its atypically-spaced promoter elements [96–99]. The Nhp6 proteins also affect pol III transcription from tRNA genes through their effect on TFIIIB positioning, specifically TBP binding, and the fidelity of transcription initiation [98,100–102]. The chromatin remodeling complex RSC interacts with Nhp6 and is enriched at pol III-transcribed genes where it is thought to function in excluding nucleosomes from these loci [103,104]. The positive role of Nhp6 in promoting pol III transcription raises the possibility that changes in chromatin structure may be linked to transcriptional responses at these genes in vivo.
Genetic, biochemical and genome-wide studies in yeast and metazoans have expanded the number of proteins physically associated with pol III-transcribed genes and the basal transcription factors [3,105,106]. In yeast, known regulators of pol II transcription such as Reb1, Fkh1, Yap6 and Hda1, have been identified by genome-wide ChIP-chip and map immediately upstream of tRNA genes [106]. Other proteins that are physically associated with tRNA and 5S RNA genes include a subset of ribosomal proteins that copurify with the positively-acting, TFIIIE fraction [107]. The RPL6A gene in particular was identified as a strong multicopy suppressor of a DNA-binding mutation in the basal transcription factor TFIIIC. In addition, the general pol II transcription factor IIS which functions to promote elongation by stimulating the intrinsic endonucleolytic activity of pol II at promoter-proximal stall sites and at sites of DNA sequence-dependent polymerase pausing or arrest is also found at many pol III-transcribed genes in yeast and mouse ES cells [3,108]. Together with functional studies in yeast, the data support a role for TFIIS in pol III transcription. Collectively, these proteins provide additional layers of potential regulatory complexity in the control of pol III transcription.
3 Regulating Activities and pathways
3.1 Protein Kinase A
Cyclic AMP-dependent protein kinase (PKA) is a key glucose-sensitive regulator of cell growth, proliferation, metabolism and stress resistance in budding yeast (reviewed in [109]) and functions as part of a G-protein coupled receptor signaling pathway that is conserved throughout eukaryotes [110]. In the absence of cAMP, PKA is an inactive heterotetramer composed of two catalytic subunits, encoded by TPK1, TPK2 or TPK3 in S. cerevisiae, and two regulatory subunits, encoded by BCY1. cAMP-bound Bcy1 dissociates from the complex to release monomeric, active catalytic subunits (reviewed in [111]). Glucose promotes PKA activity through two G-protein based mechanisms: Ras GTP-binding proteins activate adenylate cyclase (Cyr1) to synthesize cAMP [112] and Gpa2, the GTP-binding ligand for the plasma membrane sensors of extracellular glucose, activates Cyr1 and blocks the inhibitory action of Gpb1/2 on PKA activity [113]. PKA also negatively regulates cAMP production by feedback inhibition [114] and in glucose-replete media responds to essential nutrients, such as nitrogen, phosphate and amino acids, in a cAMP-independent manner (reviewed in [115]).
Post-translational modification and sub-cellular localization of both catalytic and regulatory subunits also control PKA function. Phosphorylation of the Tpk isozymes affects substrate specificity and phosphorylation of Bcy1 affects the extent of PKA activity in response to cell stresses from carbon source, heat shock, DNA damage and loss of TORC1 activity [43, 116–118, 189]. In exponentially growing cells Tpk2 and Bcy1 are nuclear while Tpks1 and 3 are distributed throughout the cell. All PKA components are redistributed to the cytoplasm on inhibition of PKA activity [119–121]. The catalytic subunits have overlapping substrate specificities and are functionally redundant for cell viability [122]. Deletion of all three Tpk isoforms is lethal unless specific substrates that are negatively regulated by PKA are also deleted (e.g. the downstream kinases Rim15 or Yak1, or the stress-responsive transcription factors Msn2 and Msn4) (reviewed in [123]).
Maf1 was identified as a phosphoprotein and a substrate of PKA through an evolutionary proteomics approach: Maf1 proteins from different budding yeast species were found have conserved PKA consensus phosphorylation sites and S. cerevisiae Maf1 was robustly phosphorylated by PKA in vitro [124]. The protein was subsequently shown to be differentially modified at PKA consensus sites in vivo [31]. Consistent with these observations, PKA activity is required for normal regulation of pol III transcription in glucose-replete media: Deletion of BCY1, which constitutively activates PKA, blocks efficient repression of pol III transcription by various treatments and this response correlates with an increase in Maf1 phosphorylation (at least on serines 177/178, if not other sites as well) [31]. Constitutive production of cAMP, caused by a dominant allele of RAS2 (RAS2Val19) also maintains high PKA activity and this too blocks repression of pol III transcription [31]. Conversely, a tpk1wi strain, which contains a debilitated but constitutively active kinase as the source of PKA activity [125], allowed efficient repression of transcription whereas eliminating all three catalytic subunits of PKA (in tpk1 tpk2 tpk3 yak3 or tpk1 tpk2 tpk3 msn2 msn4 strains) allowed more potent repression of transcription than in control strains [31]. These observations led to the conclusion that PKA phosphorylation of Maf1 in vivo counteracts its function as a repressor of transcription and that the loss of PKA activity (like the Maf1 6SA mutation) was not sufficient to cause repression under otherwise optimal growth conditions.
The switch from glucose to glycerol as a carbon source lowers PKA activity, changes the localization of the catalytic and regulatory subunits, shifts the cell from fermentation to respiratory metabolism and alters global transcription to induce general stress, oxidative and osmotic stress responses [121,126]. Growth in glycerol also results in dephosphorylation and nuclear accumulation of Maf1 and lower pol III transcription [127]. The ability of these Maf1-related effects of glycerol to be suppressed by elevated PKA activity has been tested using the RAS2Val19 mutation. Despite the severe growth defect of the RAS2Val19 strain under these conditions [128], the mutation blocked repression of pol III transcription upon transfer of cells from glucose to glycerol as predicted [127]. However, the nuclear accumulation of Maf1 was not prevented and the protein appeared to be dephosphorylated based on its mobility in one dimensional SDS gels. At the time, these data suggested that elevated PKA activity did not affect Maf1 localization or phosphorylation [127]. However, current understanding suggests that more complicated explanations may apply. The previously noted difficulty in assessing Maf1 phosphorylation by SDS-PAGE coupled with the knowledge that Maf1 can be phosphoregulated normally when restricted to the nucleus [44] indicates that the extent of Maf1 dephosphorylation might not be as great in the RAS2Val19 mutant (compared to a wild-type strain) on glycerol.
Additional experiments supporting a role for PKA in the phosphorylation of Maf1 in vivo have employed a cell permeable phosphodiesterase-resistant analogue of cAMP, 8-bromo-cAMP [43]. Treatment of yeast cells with 8-bromo-cAMP leads to elevated PKA activity and was shown to reverse the loss of PKA-site phosphorylation on Maf1 caused by both glucose starvation and rapamycin treatment. While recognizing the data supporting the role of PKA in regulating pol III transcription and Maf1 function, it is important to keep in mind that Sch9, a nutrient and stress-responsive kinase regulated by TORC1, also has a well documented role in phosphorylating Maf1 at PKA consensus sites [32,33,41]. PKA and TORC1 regulated pathways function in parallel to promote cell growth and the activity of each pathway has been reported to antagonize or restrain the activity of the other [43,129–131]. Thus, deletion or inactivation of one pathway can result in hyperactivation and compensation by the other pathway. The following sections consider the contribution of TORC1 and Sch9 in regulating pol III transcription.
3.2 Target of Rapamycin Complex 1
The TOR signaling pathway contributes to cell growth and expansion. The two functionally distinct TOR complexes differ in their sensitivity to rapamycin: TORC1 is inhibited by rapamycin and contains either of the homologous kinases, Tor1 or Tor2 in yeast, together with the Kog1, Lst8 and Tco89 proteins. TORC1 functions to regulate protein synthesis, nutrient transport, cell cycle progression and autophagy in response to nutrient status and cell stress. TORC2 is insensitive to acute rapamycin treatment and contains the Tor2 kinase complexed with Avo1, Avo2, Tsc11, Lst8, Bit61 and Bit2. TORC2 functions to regulate polarized cell growth and the actin cytoskeleton among other processes (reviewed in [132]). In mammalian cells a single kinase is partitioned between the two TOR complexes. A phylogenetic analysis of TOR components indicates that while the subunits of the TORC1 and TORC2 complexes have been conserved through evolution, regulatory inputs differ between organisms: for example, the TOR pathway in animals responds to insulin and growth factor receptor signaling pathways that are not present in yeast [133]. TORC1 is predominantly localized to the vacuole in unstressed yeast cells but is also detected at mitochondria, endoplasmic reticular membranes and in the nucleus [134–137]. Ongoing TORC1 signaling is required for Brf1 phosphorylation, TFIIIB function and pol III transcription in mammalian cells (see section 2.3 [78]). Disruption of TORC1 signaling through chemical inhibition or nutrient-deprivation inhibits pol III transcription in both yeast and higher eukaryotic cells in a Maf1-dependent manner (see section 2.1.1).
TORC1 physically associates with pol III-transcribed genes in both yeast and mammalian cells. mTOR and the mTORC1-specific subunit Raptor (Kog1 in yeast) have been ChiPed to tRNA, U6 and 5S RNA genes in proliferating human cells [23,34,138] and in two of these reports, the association was dependent on TORC1 activity. The recruitment of TORC1 to tRNA and 5S genes is directed by its interaction with TFIIIC, as demonstrated by reciprocal coimmunoprecipitation of the endogenous proteins. A role for the 63 kDa subunit of TFIIIC (analogous to Tfc1 in yeast) in this process was suggested by mutagenesis of a predicted TOR signaling motif (Phe109 to Ile in TFIIIC63) which compromised the interaction with mTOR in transfected cells. These interactions are thought to occur on the DNA and thus provide direct access of the kinase to its regulatory targets [34]. TORC1 is also detected at pol III-transcribed genes in yeast although the association is limited to 5S RNA genes which are localized to the nucleolus as part of the rDNA repeat. Interestingly, the interaction in this case is also sensitive to TORC1 activity [45].
One critical target of mTORC1 at pol III genes is the Maf1 repressor. Consistent with the localization of mTORC1 to pol III genes, mTOR and Raptor immunoprecipitates obtained using epitope-tagged constructs and transiently transfected cells were shown to phosphorylate recombinant human Maf1 in vitro at the same sites that are known to be regulated by mTORC1 activity in vivo. This in vitro activity of mTORC1 was specific; it was inhibited by rapamycin, abolished by a kinase-dead mTOR mutation and was not observed with immunoprecipitates of mTORC2 [24]. In an independent study, a recombinant fragment of mTOR was found to phosphorylate recombinant human Maf1 on S75 – one of the major mTORC1-sensitive sites [34]. Similar results have been obtained in yeast with TORC1 immunoprecipitates and recombinant yeast Maf1. Although the in vitro activity of yeast TORC1 was relatively weak in these assays, the signal was reduced to background levels by pretreatment of the cells with rapamycin or in a kinase dead mutant and was unaffected in a rapamycin-resistant TOR1 mutant [45].
The nucleolar localization of TORC1 correlates with ongoing pol I transcription and the physical association of Tor1 at the 35S rDNA promoter in yeast [136]. The association of Tor1 at the rDNA promoter and at 5S RNA genes in the rDNA repeat is driven by the dynamics of nuclear import and export through the NLS and NES sequences within the Tor1 protein [45,136]. An analysis of Maf1 distribution, phosphorylation and binding to 5S RNA genes using a rapamycin-resistant allele of TOR1 showed that nuclear Tor1 (containing a functional DNA binding domain) was required for Maf1 phosphorylation and subsequent export of Maf1 to the cytoplasm [45]. Loss of TORC1 activity caused Maf1 to become associated with the nucleolus. These studies led to a model in which TORC1 regulates Maf1 phosphorylation both directly and indirectly. In unstressed cells, TORC1 activation of the Sch9 kinase (which is primarily in the cytoplasm, http://yplp.uni-graz.at/index.php) promotes Maf1 phosphorylation and its retention in this compartment (Figure 1). TORC1 at the rDNA locus contributes to this partitioning by directly phosphorylating any Maf1 that enters the nucleolus (Figure 1)[139]. Loss of TORC1 activity allows Maf1 to both accumulate in the nucleus and at the nucleolus to repress pol III transcription at tRNA and 5S RNA genes [45,139].
3.3 Sch9
The Sch9 kinase is a well-characterized downstream effector of TORC1 (analogous to the S6K kinases in mammalian cells) that is phosphorylated and activated directly by TORC1 [137]. Sch9 was originally identified as a multi-copy suppressor of PKA loss of function mutations [140]. Consistent with this phenotype, microarray analysis indicates that PKA activity is responsible for the bulk of the transcriptional response to glucose addition and that Sch9, when overexpressed in a tpk analog-sensitive mutant, induces a comparable transcriptional output [130]. However, Sch9 activity contributes relatively little to this transcriptional program in otherwise wild-type cells. Thus, Sch9 primarily promotes cell growth in response to nutritional inputs other than glucose. In addition to its phosphorylation by TORC1 (which involves multiple residues in the C-terminus), Sch9 activity is dependent on activation loop phosphorylation by the redundant 3-phosphoinositide-dependent kinase 1 (PDK1) orthologs, Pkh1 and Pkh2 [137]. Thus, nitrogen and stress-sensitive regulation of TORC1 activity and sphingolipid regulation of the Pkh1/2 kinases, link Sch9 activation to multiple signaling pathways including the UPR [141](see Section 3.8).
The role of TORC1 signaling in nutrient and stress responses, coupled with the complementation of PKA defects by Sch9, suggested that Sch9 might also contribute to the regulation of pol III transcription in yeast. Indeed, deletion of SCH9, or its inactivation in a sch9-as strain with the ATP analog 1NM-PP1, severely compromises pol III transcription indicating that TORC1 activity, and Sch9 activity in particular, is required for robust pol III transcription [32,33,41]. Deletion of MAF1 completely restored transcription in an sch9Δ strain, suggesting that Sch9 functions to promote pol III transcription by inactivation of Maf1 [32]. Transcription in an sch9Δ strain could be further decreased by multiple, diverse repressing conditions such as entry into stationary phase and treatments with rapamycin, tunicamycin, CPZ and MMS indicating that additional mechanisms exist to further repress pol III transcription in the absence of Sch9 [32]. Conversely, expression of a constitutively active allele of SCH9 (achieved using phosphomimetic activating mutations) which separates nutrient-signaling to TORC1 from activation of Sch9, blocked repression of pol III transcription by rapamycin [33,41].
The Sch9 kinase phosphorylates Maf1 and other substrates at an RxxS/T motif (identified by mass spectrometry and phospho-specific antibodies) and overlaps the consensus site for PKA phosphorylation [32,33,41,43]. Sch9 coimmunoprecipitated with Maf1 in a rapamycin-sensitive manner and yeast-purified Sch9 phosphorylated Maf1 in vitro. These modifications were at PKA consensus sites as detected by phospho-specific antibodies. However, mutation of the PKA consensus sites to glutamic acid residues (either MAF1-6SE or -7SE alleles) did not prevent a shut-down of pol III transcription under stress [31,33]. Moreover, the MAF1-7SE allele did not prevent the repression of transcription in an sch9-as analog-treated strain [33]. Together, these data indicate that Sch9 has additional substrates that promote pol III transcription.
Coimmunoprecipitation assays demonstrate that phosphorylation by Sch9 affects the extent to which Maf1 binds pol III [33]. Rapamycin treatment increased the association between pol III and Maf1, while the constitutively active Sch9DE mutant blocked this interaction. Maf1 phosphorylation altered the extent of Maf1 binding to pol III: The phosphomimetic substitutions in Maf1 (MAF1-7SE) showed no physical association with pol III while the non-phosphorylatable allele, MAF1-7SA, bound even in unstressed conditions. The enhanced binding of MAF1-7SA to pol III was strain specific and could be further increased by inactivation of PKA [33] consistent with multiple effectors contributing to inhibit pol III function. Sch9 activity modified the intracellular localization of Maf1, as shown by the nuclear accumulation of Maf1 in sch9Δ and sch9-as inactivated strains [33,41]. The distribution of Maf1 between the nucleoplasm and the nucleolus was Sch9-independent as Maf1 accumulated at the nucleolus on rapamycin treatment [41].
The relative contribution of Sch9 and PKA to Maf1 phosphorylation was assessed using PKA consenus site phospho-specific antibodies and analog-sensitive mutants of the PKA and Sch9 kinases [32]. Inactivation of PKA in a triple tpk analog-sensitive strain or inactivation of Sch9 in an sch9-as strain caused little effect on the level of Maf1 phosphorylation at PKA/Sch9 sites. Inactivation of both PKA and Sch9 enzymes caused almost total loss of Maf1 phosphorylation at the consensus sites, consistent with the independent and compensatory phosphorylation by PKA and Sch9 [32]. A comparable analysis that assessed Maf1 phosphorylation by SDS-PAGE did not detect such compensatory effects [33].
The genetic and biochemical data demonstrating that the Sch9 kinase directly phosphorylates Maf1 in yeast, did not extrapolate to mammalian Maf1 phosphorylation by the metazoan homolog, S6K1 [24]. Immuno-purified S6K1 preparations did not phosphorylate recombinant human Maf1 in vitro. Moreover, the extent of Maf1 phosphorylation and sensitivity to rapamycin was comparable between wild-type and S6K1/2 double knockout cell extracts. These observations suggest that substrate preferences of TORC1 and Sch9 differ between yeast and mammalian cells: yeast Maf1 is a poor substrate for TORC1 and is robustly phosphorylated by Sch9 in vitro while human Maf1 is a good substrate for mTOR and is not detectably phosphorylated by S6K1 in vitro.
3.4 Other TORC1 effectors
TORC1 regulates multiple phosphatases (PP2A, PP4, Ppg1 and Sit4) in part through the Tap42 protein. Conditions that activate TOR promote Tap42 phosphorylation and its association with the catalytic subunits of PP2A (Pph21 and Pph22) and Sit4 [142]. Tap42 can function both positively and negatively on readouts for phosphatase activity [143]. A recent model posits that inactive Tap42-phosphatase complexes associate with membrane-bound active TORC1. On rapamycin treatment or nutrient starvation, the Tap42-phosphatase complex is dissociated from TORC1 and activated, remaining active to dephosphorylate substrates until Tap42 is dephosphorylated by PP2A [144]. The Tap42 pathway downstream of TORC1 does not affect the transcription of ribosomal protein genes or their repression by rapamycin [143]. Similar experiments with conditional alleles of SIT4 (sit4-102) and TAP42 (tap42-109) showed comparable results for the regulation of pol III transcription: loss of either Tap42 or Sit4 function did not affect transcription or its repression by rapamycin [145]. Thus rapamycin-induced repression of pol III transcription occurs independently of Tap42 and Sit4.
Phenotypes associated with mutant alleles of Tap42 are sensitive to strain-specific polymorphisms in SSD1 [146]. Ssd1 associates with specific mRNAs and affects the transcription and translation of daughter-specific mRNA and polarized cell growth [147]. Ssd1 also cooperates with TORC1 to maintain cell integrity through the TORC1-associated protein Tco89 [146]. Wei et al [45] reported that the appearance of cytoplasmic Maf1 in unstressed cells and its nucleolar localization under repressing conditions required a wild-type allele of SSD1. An analysis of rapamycin-dependent phosphopeptides in an SSD1-wildtype background showed that Maf1 dephosphorylation was Tap42-independent [33]. Although a tap42-11 allele in this strain background caused a decrease in pol III transcription (unstressed), repression of transcription by rapamycin was not compromised, consistent with regulation of pol III transcription being Tap42-independent [33].
The sensitivity of pol III transcription in an sch9Δ strain to further repression by rapamycin (and other stresses) led to the conclusion that regulation of pol III transcription by TORC1 also can also occur via an sch9Δ-independent mechanism [32,33,41]. A recent study showed that rapamycin and other stresses activate Rho1 directly, to promote Rho1 binding to Kog1 causing disruption of the membrane association of the Kog1/TORC1 complex and an effect on TORC1 downstream signaling [148]. Activation of Rho1 GTPase activates Pkc1 and the CWI pathway which in turn affects the regulation of ribosome and tRNA synthesis in a manner that is not understood (see section 3.5).
3.5 PKC and cell wall integrity
The cell wall integrity (CWI) signaling pathway maintains yeast cell wall structure and shape during cell division and polarized cell growth and protects the cell against osmotic and mechanical stress [149]. Cell wall integrity sensors bind guanine nucleotide exchange factors (Rom1/2 and Tus1) to activate the Rho1 GTPase which in turn affects cell wall synthesis, actin polarization and exocytosis. An important part of this response involves the activation of protein kinase C (Pkc1) by GTP-bound Rho1 to trigger a MAP kinase signaling cascade (Bck1 → Mkk1/2 → Slt2). Subsequently, activated Slt2 induces the expression of cell wall stress-regulated genes through catalytic and non-catalytic mechanisms involving the transcription factors Rlm1 and SBF (Swi4 and Swi6), respectively [149].
Disruption of the secretory pathway at any step from peptide insertion into the endoplasmic reticulum to vesicle fusion with the plasma membrane results in transcriptional repression of ribosome and tRNA synthesis [150,151]. The arrest of secretion effect on transcription, caused by inactivation of sec mutants or tunicamycin treatment, was shown to be independent of the unfolded protein response (UPR), high-osmolarity glycerol (HOG) and stringent response pathways (reviewed in [145]). However, components of the CWI pathway including the cell wall stress sensors and Pkc1, but not the MAPK signaling cascade downstream of Pkc1, contribute to repression of transcription under these conditions [151,152]. Activation of the CWI pathway (and Pkc1) is thought to downregulate ribosome biogenesis to limit protein synthesis when vesicular transport or expansion of the plasma membrane is blocked (reviewed in [153]).
The substrates of Pkc1 and the role of the kinase outside the CWI pathway are not well defined and it is not known how Pkc1 and/or CWI signaling are connected to transcriptional regulation of ribosome and tRNA synthesis. Pkc1 is predominantly localized to the bud neck and sites of polarized growth. However, functional nuclear exit and localization motifs have been identified in the protein [154]. The physiological significance of nuclear Pkc1 is poorly understood. However, a recent report links ongoing membrane traffic and Pkc1 to control of mitosis: activated Pkc1 binds to the phosphatase PP2ACdc55-Zds1/2 complex to dephosphorylate Mih1 and relieve the cell size checkpoint control that blocks mitotic entry [155]. As with all down-regulation of pol III transcription, a block in the secretion pathway and the transcriptional repression that follows requires the Maf1 protein, its dephosphorylation and accumulation in the nucleus [18]. Whether Pkc1 contributes to the targeting of PP2A to Maf1, or other regulators of pol III transcription, specifically in secretion-blocked cells is not known.
Rapamycin treatment and nutrient starvation activates the CWI pathway [156]. Consistent with this, components of the pathway from the cell surface sensors to Rho1 are required for Sch9 dephosphorylation (and loss of function) following nitrogen starvation, rapamycin treatment and caffeine inhibition of TORC1 [148]. The CWI pathway also connects TORC1 and PKA through the TORC1 and CWI downstream effector kinases, Sch9 and Slt2: activated (phosphorylated) Slt2 MAP kinase phosphorylates Bcy1 at residue T129 (amongst other sites) and limits PKA activity. TORC1 signaling and Sch9 activity prevents Slt2 phosphorylation thereby promoting PKA activity (see section 3.8 on Crosstalk, below) [43].
3.6 Other Kinases
3.6.1 Protein Kinase CK2
CK2 is a highly conserved, Ser/Thr protein kinase with roles in multiple cellular processes including cell growth and proliferation, apoptosis and cell survival (reviewed in [157]). The enzyme forms a tetramer of two constitutively active catalytic (α) subunits and two regulatory (β) subunits. The physiological targets of CK2, which contain a canonical CK2 motif (pS/pT-x1-x2-D/E/pS), have been estimated to represent a significant portion of the phosphoproteome [158]. CK2 activity is directed by substrate-dependent localization of the enzyme and the dynamics of holoenzyme formation [159]. In yeast, the catalytic activity of CK2 is essential and is influenced by growth rate and nutrients in a substrate-specific manner [160]. The β-subunits are non-essential in yeast but contribute to full activity of the holoenzyme, the formation of self-aggregated inactive CK2 structures and regulation of CK2 activity in response to nutrients [161].
CK2 has been linked to the phosphorylation of many transcription factors and all three nuclear RNA polymerases [162]. High levels of pol III transcription require CK2 activity suggesting a stimulatory function for CK2 in both yeast and mammalian cells [38,70,71,163]. The physical localization of CK2 at pol III-transcribed genes by chromatin immunoprecipitation links CK2 activity to regulation of ongoing transcription [38,71,76]. In yeast, CK2 positively affects the activity of the initiation factor TFIIIB and is required for restoration of robust transcription after carbon source stress [38,69]. Although CK2 phosphorylation of TBP has been shown to inhibit TBP recruitment to DNA and to limit pol II transcription in vitro [164], the regulatory subunits of CK2 bind TBP and stimulate the assembly of TFIIIB on pol III-transcribed genes [69]. DNA damage caused by MMS treatment correlates with loss of CK2 activity and the dissociation of the catalytic subunits from the regulatory subunits. The dynamic association of CK2 with TFIIIB suggests that the catalytic activity of CK2 maintains TFIIIB function and transcription and opposes repression of transcription. The role of CK2 in higher eukaryotes is more complex: CK2 stimulates the assembly of TFIIIB in vitro, but during mitosis CK2 hyperphosphorylation of human Bdp1 leads to the dissociation of Bdp1 from chromatin [70,76] (see section 2.3).
Recently, the role of CK2 was evaluated in the restoration of pol III transcription following carbon source repression [38,42]. In glycerol medium, transcription is repressed coincident with the elevated occupancy of Maf1 at tRNA genes and Maf1 binding to pol III [38,127]. Genetic and/or chemical inhibition of CK2 prevented the recovery of transcription and impaired the dissociation of Maf1 from pol III upon switching from glycerol to glucose-containing medium [38]. Maf1 was shown to be phosphorylated by CK2 in vitro and on CK2 sites in vivo suggesting that CK2 opposed Maf1 function directly on tRNA genes to reverse repression [38]. However, a Maf1 mutant lacking all five CK2 sites implicated in this response remained fully functional in repressing transcription and exhibited normal restoration of transcription upon carbon source switching [42]. Subsequent analysis showed that CK2 activity indirectly affects the extent to which consensus PKA/Sch9 sites on Maf1 are phosphorylated [42] suggesting that CK2 activity is required for optimal signaling to Maf1 via these pathways.
3.6.2 Lammer protein kinases and Kns1
The LAMMER Cdc-like (Clk) family of kinases are dual specificity protein kinases that act on serine/threonine and tyrosine residues [165]. The defining signature sequence of the family (EHLAMMERILG) forms a solvent-inaccessible structural motif within the kinase core rather than a substrate interaction domain as first thought [166]. The mammalian and Drosophila LAMMER kinases have been studied initially in the context of regulated pre-mRNA splicing through their phosphorylation of SR (serine-arginine rich) proteins [167,168]. However, a growing number of LAMMER/Clk substrates have been identified with functions related to signal transduction and transcriptional regulation. Examples include PTP-1B phosphatase and the WD-40 repeat-containing transcriptional repressors Tup11 and Tup12 which are positively regulated by LAMMER/Clk kinase phosphorylation [169,170]. Additionally, Clk2, one of four mammalian LAMMER kinases, is known to be activated by Akt phosphorylation and mediates insulin signaling by phosphorylating and repressing PGC1-α, a critical transcriptional coactivator of energy metabolism [171]. Interestingly, Clk2 also phosphorylates the PP2A regulatory subunit, B56β, which promotes the association of PP2A phosphatase with Akt leading to its dephosphorylation and attenuation of insulin signaling [172].
Kns1 is the sole member of the LAMMER/Clk kinase family in S. cerevisiae and was identified recently in a screen for kinases that are required for rapamycin-induced hyperphosphorylation of the pol III-specific subunit, Rpc53 [56]. As noted earlier, Kns1 phosphorylates a single threonine residue in Rpc53 as part of the signaling response to nutritional and cellular stress that represses pol III gene transcription. This role of Kns1 is not limited to the pol III system since deletion of KNS1 attenuates transcriptional repression by all three nuclear RNA polymerases with respect to their functions in ribosome and tRNA synthesis. The substrates mediating this effect in the pol I and pol II systems have not yet been identified and substrates in addition to Rpc53 may also exist in the pol III system [56]. Further study is also needed to understand how Kns1 function is regulated by upstream signaling. One indication is provided by the finding that alkylating DNA damage and rapamycin inhibition of TORC1 increases the total amount of Kns1 protein. This in turn promotes the largely autocatalytic phosphorylation of Kns1 at multiple sites and its accumulation in the nucleus [56]. Notably, regulation of kinase abundance in response to insulin signaling is also a feature of mammalian Clk2 [171].
3.6.3 Glycogen Synthase Kinase-3 (GSK-3) and Mck1
GSK-3 was first described as a regulator of glycogen metabolism in 1980. More than 30 years later, the enzyme is known for its broad regulatory influence in cellular signaling and for the myriad of processes that it impacts. GSK-3 kinase activity in higher eukaryotes is regulated by phosphorylation, by its sequestration into macromolecular complexes and by its subcellular localization [173]. Like the LAMMER/Clk kinases described above, GSK-3 family members are dual specificity kinases that are positively regulated by phosphorylation (either autocatalytic or by another kinase) on a T-loop tyrosine residue and negatively regulated by phosphorylation at sites in the N- and C-termini. Regulation of GSK-3 activity is also achieved through its preference for a “primed” or pre-phosphorylated residue within the consensus motif S/TXXXpS/T, where the pS/T primed site orients the substrate in the enzyme active site [174]. This substrate preference also accounts for the inhibitory effect of N-terminal phosphorylation of GSK-3 through the creation of an intramolecular pseudo-substrate which competes with the priming phosphate for substrate binding. GSK-3 kinases function downstream of multiple signaling cascades that include Akt, TOR, PKC, PKA and stress response pathways [173]. The phosphorylation of some GSK-3 substrates, such as β-catenin, cyclins, transcription factors and oncogenes promotes their ubiquitination and subsequent degradation [175]. Collectively, the action of the GSK-3 kinases negatively regulates the function or stability of their protein substrates.
S. cerevisiae contains four GSK-3 homologs, Mck1, Rim11, Mrk1 and Ygk3 with functions ranging from transcriptional control of stress responsive genes, protein degradation, entry into meiosis and other processes [176–178]. More recently, Mck1 was identified along with Kns1 as kinases that differentially modify Rpc53 in response to a various types of cellular stress [56]. Mck1-directed phosphorylation of Rpc53 in vivo and in vitro at serines 224 and 228 required the priming activity of Kns1 [56] and was highly specific: Other family members could not complement this function of Mck1 in cells treated with rapamycin, MMS, tunicamycin or CPZ treatment [56]. The mechanism(s) that limit the phosphorylation of Kns1-primed Rpc53 to Mck1 and not the other family members are currently unknown. Similarly it is not clear if or how Mck1 activity is regulated by upstream signaling. Deletion of MCK1, like deletion of KNS1, compromised transcriptional repression of all three RNA polymerases revealing a common role for both kinases in the down-regulation of ribosome and tRNA synthesis. Moreover, the level of transcription attenuation in all three polymerase systems was greater in the double kinase deletion strain than in either single mutant. These data suggest that one or both kinases function, to some extent, independently of one another to affect repression. This raises the possibility that other priming kinases may also direct the activity of Mck1 in the transcriptional response to nutrient and stress signaling.
3.7 Phosphatases
Phosphoregulation of Maf1, Rpc53 and various subunits of TFIIIB has been demonstrated in yeast and/or mammalian cells and much has been learned about the kinases responsible for these modifications. By comparison, less is known about the protein phosphatases associated with transcriptional control in the pol III system. Our current knowledge is this area is based on studies in yeast and centers on the dephosphorylation of Maf1 by PP2A and PP4. While considering this work, it is important to keep in mind that the patterns of phosphorylation of Maf1 and Rpc53 are oppositely correlated with respect to transcription [56]. Hence, the action of phosphatases on Rpc53 would be expected to positively affect transcription and their simultaneous action on Maf1 would promote its repressing function. To avoid these opposing effects, mechanisms must exist to control phosphatase substrate specificity, localization or activity in order to achieve the appropriate modification state of these proteins under a specific set of conditions.
Phosphatases of the PP2A family are heterotrimeric enzymes comprising a scaffolding A subunit, a catalytic C subunit and a regulatory B subunit which determines substrate specificity and subcellular targeting [179]. PP2A in yeast is assembled from a single scaffold protein, Tpd3, one of two catalytic subunits, Pph21 or Pph22, and either Cdc55 or Rts1 as the regulatory subunit. The assembly and activation of the heterotrimeric holoenzyme is controlled by post-translational modifications. This includes reversible carboxymethylation at the C-terminus of the catalytic subunit by the methyltransferase Ppm1 and the esterase Ppe1 and activation of the catalytic subunit by the partially redundant peptidyl-prolyl cis/trans-isomerases Rrd1 and Rrd2 [179].
Tpd3 and both catalytic subunits are distributed throughout the cytoplasm and the nucleus [180] and function in a variety of processes including cell-cycle progression, metabolic regulation, transcription and translation. Strains deleted for TPD3 grow poorly, have defects in cytokinesis and exhibit low levels of pol III transcription [181]. As the Tpd3 scaffold is required for appropriate positioning of the catalytic subunits for their Rrd activators, the TPD3 deletion strain has unregulated low phosphatase activity rather than high activity as first thought [182]. Previous work demonstrated that defective pol III transcription in extracts prepared from a tpd3Δ strain could be restored to normal levels by the addition of purified pol III or TFIIIB [181]. These observations can now be interpreted as a requirement for PP2A activity in the synthesis, stability or function of these components.
Deletion of both PP2A catalytic subunits causes severe morphological and growth defects with strain viability maintained due to partial complementation by PPH3, which encodes the catalytic subunit of PP4 [183]. In this strain and in the slow growing tpd3Δ strain, Maf1 is at least partially dephosphorylated under growth conditions that would otherwise support full phosphorylation of the protein [27,29]. Whether the growth defects of these strains cause cell stress leading to Maf1 dephosphorylation and downregulation of pol III transcription or whether PP2A directly phosphoregulates Maf1 was not resolved by these studies. Maf1 dephosphorylation, nuclear localization and transcriptional repression has also been evaluated in a double mutant strain lacking the PP2A catalytic subunit PPH21 and the PP4 catalytic subunit, PPH3, i.e. PP2A activity is provided by the remaining a wild-type copy of PPH22 [30]. Some Maf1 dephosphorylation and transcriptional repression was observed in this strain upon rapamycin treatment, albeit with very poor kinetics and low efficiency compared with a wild-type strain. The role of PP2A was not addressed in a PP4 wild-type background in this study. Nonetheless, the work showed that the Pph22 catalytic subunit of PP2A can contribute to the regulation of Maf1 and to repression of pol III transcription in the absence of PP4 [30].
The PP4 phosphatase also contributes to the phosphoregulation of Maf1 [27]. PP4 is composed of three subunits; the catalytic subunit Pph3, the coactivator Psy2, and an additional substrate specificity factor Psy4 [184]. Tip41, a known binding partner of Tap42 and a component of the TOR signaling pathway is also associated with PP4 [184]. A fusion reporter assay has been used to evaluate the contributions of various phosphatases and their binding partners to Maf1 function [27]. In this reporter assay, the entire Maf1 protein was fused to the amino-terminus of Rpc160, the largest pol III subunit, under the control of a galactose-inducible promoter. Hybrid protein production inhibited cell growth, a phenotype termed the fusion growth defect, and caused a loss of pol III transcription. GAL1-driven expression of the Maf1 protein alone was sufficient to cause a significant decrease in transcription suggesting that both Maf1 and the Maf1-Rpc160 fusion impaired transcription in apparently unstressed cells. The fusion growth defect and repression by Maf1 was lost in strains deleted for PPH3, PSY2 and (to a lesser extent) PSY4 and in a Maf1 mutant that is compromised in its ability to be dephosphorylated [27]. Conversely, deletions of the PP2A components did not relieve the fusion growth defect. Rrd1 and Tip41, which are associated with Tap42 activation of PP2A and PP2A-like phosphatases but not the PP2A-associated Rrd2 protein were also linked to the Maf1 fusion phenotype [27]. The physical association of Pph3 with Maf1 under non-stress conditions and the effects of deleting PPH3 on Maf1 dephosphorylation, nuclear localization and transcriptional repression upon nutrient deprivation and/or other stress conditions are all consistent with a significant role for PP4 in Maf1 phosphoregulation.
Curiously, strains containing the 7SA mutant of Maf1 (which cannot be phosphorylated by PKA or Sch9) also showed a dependence on Pph3 for decreased transcription [27]. The Maf1 7SA protein has little to no residual phosphorylation as assayed by Phos-tag gel separation, and functions to repress transcription as effectively as the wild-type protein in otherwise normal cells [33,42]. The requirement for Pph3 in the Maf1 7SA strain suggests that additional components of the pol III transcription machinery are targeted by PP4 and contribute to the decrease in transcription.
A survey of Maf1 phosphoregulation in strains deleted for the SIT4, YVH1, PPZ1, HIS2 and MSG5 phosphatases or containing a conditional CDC14 allele, showed that none of these phosphatases altered Maf1 dephosphorylation caused by nutrient depletion [29]. Thus, collectively, the data point to contributions from both PP2A and PP4 as phosphatases that regulate pol III transcription.
3.8 Crosstalk
Multiple studies demonstrate that the TORC1 and PKA pathways, despite responding to different nutritional signals, positively regulate growth and attenuate stress responses in exponentially growing cells (Figure 3). Functional overlap between these pathways is well-known and is reflected in many ways including their common targets and the overlapping substrate specificity exhibited by the TORC1 downstream effector kinase Sch9 and PKA [129,137,185,186]. In general, PKA and TORC1 appear to function in parallel although in specific cases, limiting the activity of one pathway results in increased signaling through the other [131]. In the case of Maf1, both pathways function in the same direction with Sch9 and PKA targeting overlapping sites to negatively regulate its function. While loss of signaling through either pathway has quantitatively different effects on the extent of Maf1 phosphorylation in a strain background, growth condition and assay-specific manner [32,33], the net effect is that pol III transcription is reduced when one or the other pathway is compromised [31,32].
Figure 3. A schematic representation of signaling pathways known to regulate pol III transcription in S.cerevisiae.
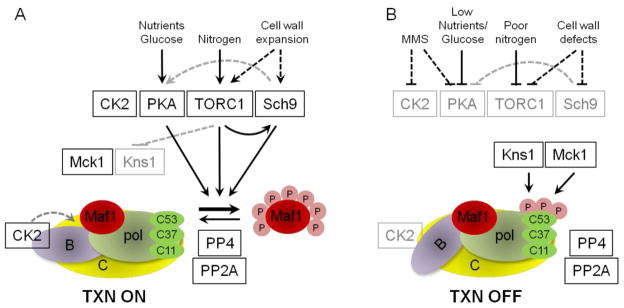
A. Transcription by pol III is sensitive to inputs from multiple signaling pathways that sense nutrients, glucose and cell wall expansion. In logarithmically growing cells in nutrient replete media the activities of PKA, TORC1 and CK2 promote transcription. CK2 activity is required for pol III transcription through a mechanism that affects TFIIIB activity. The Tpk, Sch9 and TORC1 kinases negatively regulate the repressor of pol III, Maf1, by phosphorylation at its seven PKA/Sch9-consensus sites. It is not known if there is preferential modification of specific sites in vivo by either enzyme. CK2 also promotes Maf1 phosphorylation at PKA/Sch9 sites through an unknown mechanism. Maf1 is dephosphorylated by the phosphatases PP4 and PP2A. Dotted lines indicate that the mechanism of inhibition is either not completely defined or is not direct. Black boxes indicate active enzymes while gray boxes indicate in inactive enzymes. Phosphorylation is indicated by the pink circles (annotated P); and the transcription components TFIIIC, yellow; pol III, light green; Rpc53/37/11 pol subunits, dark green; TFIIIB, purple and Maf1, red. B. Pol III transcription is shut-down in response to cellular stress. Maf1 is dephosphorylated and binds pol III in a manner that interferes with the C82/34/31 subcomplex, function in transcription initiation and binding to TFIIIB. Actively transcribing and reinitiating pol III is unaffected by Maf1. Cell stress and loss of transcription correlate with phosphorylation of Rpc53 and activation of the Kns1 kinase. Kns1 primes the pol III subunit Rpc53 for further phosphorylation by Mck1 in response to stress signaling pathways. Phosphorylation of Rpc53 occurs independently of Maf1 and Maf1 is not phosphorylated by Kns1 or Mck1. CK2 activity is impaired by DNA damage by a mechanism that promotes the dissociation of the catalytic subunits from TFIIIB. See section 3.6.1 for details.
Although PKA activity is regulated largely by the cAMP-dependent dissociation of its regulatory Bcy1 subunit in glucose replete media, other inputs include the stimulation of regulatory and catalytic subunit association by the Gα proteins Gpb1 and Gpb2 [187], phosphoregulation of Bcy1 [43] and nutrient availability [115]. The mechanism by which PKA activity is altered in response to phosphate, amino acid or ammonium levels is undefined. Multiple signaling pathways affect the phosphorylation of Bcy1 to alter PKA localization and the specificity of PKA substrate recognition. TORC1, for example, can control PKA activity toward some substrates [43]: Rapamycin inhibition of TORC1 results in the phosphorylation of Bcy1 at T129 and other sites, promoting the negative regulatory function of Bcy1 that inhibits PKA activity. Bcy1 T129 phosphorylation is affected by the CWI pathway MAP kinase Slt2 which is negatively regulated indirectly by Sch9. Disruption of TORC1 function also activates the CWI pathway and Slt2 through the TOR effectors Tap42 and Sit4 [156]. TORC1 therefore promotes PKA activity toward some substrates by preventing Slt2-mediated modification of Bcy1. This crosstalk between the TOR pathway, Sch9 and the CWI pathway via Slt2 alters the extent of Maf1 phosphorylation: a Slt2 deletion strain caused elevated phospho-Maf1 but did not affect pol III transcription [43,151]. In contrast, the phosphorylation of Bcy1 at other sites appears to have different effects on PKA activity towards other substrates [117,118,188,189]. For example, in response to DNA damage, Mec1-dependent phosphorylation of Bcy1 by Mck1 enables PKA regulation of the DNA damage checkpoint and inhibition of mitotic exit. This is achieved by PKA phosphorylation of Cdc20, an activator of the anaphase promoting complex, and the stabilization of securin and the mitotic cyclin Clb2 [188,189]. The phosphorylation of Bcy1 at T129 and other sites by multiple kinases (as detected by phosphoproteomic analysis, [43,55]) potentially links the regulation of PKA activity to multiple signaling pathways and processes. Significantly, deletion of BCY1 disconnects pol III transcription from repression signaling caused by multiple stresses [31].
Cell wall stress activation of the Rho1 GTPase at the cell surface regulates the CWI pathway and Pkc1 [149] and also affects the activity of TORC1. Activated Rho1 binds directly to the Kog1 component of TORC1, decreasing TORC1 activity and disrupting its membrane association. Since the Rho1 binding site on Kog1 overlaps with the binding site of Tap42, the Rho1-Kog1 interaction also releases and thereby activates the Tap42-PP2A phosphatase [148]. Loss of TORC1 activity also activates the CWI pathway upstream of Rho1, although the molecular details through which this is achieved are less clear [43,156]. Thus, TORC1 functions both upstream and downstream of Rho1 linking the signaling of nutrients and cell wall stress [148].
The Sch9 and PKA kinases that regulate Maf1 phosphorylation and Pkc1 are all phosphorylated by members of the 3-phosphoinositide-dependent protein kinase-1 (PDK1) family in yeast [190]. Pkh1, one of three PDK1 orthologs in yeast, phosphorylates Sch9, Tpk1 and Pkc1 at the PDK1 motif in their activation loop [137,190]. Phosphorylation of Sch9 by Pkh1 is independent of TORC1 phosphorylation, is nitrogen-sensitive and in all cases is required for full kinase activity [190]. Since the Pkh1/2 kinases are themselves regulated by sphingolipids, membrane behavior and function is thereby linked to the CWI, TORC1 and PKA pathways.
4. Conclusion
Repression of pol III transcription couples the dephosphorylation and nuclear accumulation of Maf1 with its binding to pol III. However, there are now many lines of evidence indicating that a shutdown of pol III transcription is subject to additional regulatory steps. In addition to the function of the phosphomimetic MAF1-7SE mutant under cellular stress and the dependence of the MAF1-7SA mutant on cellular stress for repression, the PKA and Sch9 kinases show compensatory phosphorylation of Maf1, yet pol III transcription is sensitive to a loss of signaling through either pathway. Collectively these observations point to additional modifications to Maf1 and/or additional targets of the PKA, TORC1 and stress signaling pathways to effect repression of pol III transcription. Consistent with this, it has been known for some time that the initiation factor TFIIIB requires ongoing CK2 activity for function. More recently, the polymerase itself was shown to be modified in response to the loss of TORC1 signaling in a new branch of the pathway that requires the concerted action of LAMMER/Clk and GSK-3 family kinases. However, a detailed understanding of how cells repress, maintain and re-activate pol III transcription in response to environmental cues is still some way off. Additional research is needed to (i) identify all of the regulated targets in the polymerase, the basal factors and associated proteins, (ii) understand how both positive and negative nutrient and stress signals modify each component and (iii) provide mechanistic explanations for how these events lead to or prevent Maf1-dependent repression of transcription. Together these approaches will provide knowledge of the structure and function of the regulatory networks that control pol III transcription and the specific nature of the failsafe mechanisms that prevent its repression in the absence of cell stress signaling. Understanding the control and deregulation of pol III transcription ultimately will drive innovative approaches to combat aberrant cell growth and proliferation, cell transformation and cancer.
Highlights.
Pol III transcription is regulated by multiple conserved signaling pathways
The Maf1 repressor is phosphoregulated in response to nutrient and stress signals
TFIIIB and pol III are also differentially modified under stress conditions
Changes to TFIIIB and pol III function allow Maf1-dependent repression
Failsafe mechanisms prevent pol III repression in the absence of cell stress
Acknowledgments
We thank past and present members of our lab for their contributions to the research described here. This work was support by NIH grant GM085177.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Dieci G, Fiorino G, Castelnuovo M, Teichmann M, Pagano A. The expanding RNA polymerase III transcriptome. Trends Genet. 2007;23:614–622. doi: 10.1016/j.tig.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 2.White RJ. Transcription by RNA polymerase III: more complex than we thought. Nat Rev Genet. 2011;12:459–463. doi: 10.1038/nrg3001. [DOI] [PubMed] [Google Scholar]
- 3.Carriere L, Graziani S, Alibert O, Ghavi-Helm Y, Boussouar F, Humbertclaude H, Jounier S, Aude JC, Keime C, Murvai J, Foglio M, Gut M, Gut I, Lathrop M, Soutourina J, Gerard M, Werner M. Genomic binding of Pol III transcription machinery and relationship with TFIIS transcription factor distribution in mouse embryonic stem cells. Nucleic Acids Res. 2012;40:270–283. doi: 10.1093/nar/gkr737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Canella D, Bernasconi D, Gilardi F, LeMartelot G, Migliavacca E, Praz V, Cousin P, Delorenzi M, Hernandez N. A multiplicity of factors contributes to selective RNA polymerase III occupancy of a subset of RNA polymerase III genes in mouse liver. Genome Res. 2012;22:666–680. doi: 10.1101/gr.130286.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vannini A, Cramer P. Conservation between the RNA polymerase I, II, and III transcription initiation machineries. Mol Cell. 2012;45:439–446. doi: 10.1016/j.molcel.2012.01.023. [DOI] [PubMed] [Google Scholar]
- 6.Cloutier TE, Librizzi MD, Mollah AK, Brenowitz M, Willis IM. Kinetic trapping of DNA by transcription factor IIIB. Proc Natl Acad Sci U S A. 2001;98:9581–9586. doi: 10.1073/pnas.161292298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kassavetis GA, Geiduschek EP. Transcription factor TFIIIB and transcription by RNA polymerase III. Biochem Soc Trans. 2006;34:1082–1087. doi: 10.1042/BST0341082. [DOI] [PubMed] [Google Scholar]
- 8.Schramm L, Hernandez N. Recruitment of RNA polymerase III to its target promoters. Genes Dev. 2002;16:2593–2620. doi: 10.1101/gad.1018902. [DOI] [PubMed] [Google Scholar]
- 9.Dieci G, Sentenac A. Facilitated recycling pathway for RNA polymerase III. Cell. 1996;84:245–252. doi: 10.1016/s0092-8674(00)80979-4. [DOI] [PubMed] [Google Scholar]
- 10.Dieci G, Giuliodori S, Catellani M, Percudani R, Ottonello S. Intragenic promoter adaptation and facilitated RNA polymerase III recycling in the transcription of SCR1, the 7SL RNA gene of Saccharomyces cerevisiae. J Biol Chem. 2002;277:6903–6914. doi: 10.1074/jbc.M105036200. [DOI] [PubMed] [Google Scholar]
- 11.Warner J. The economics of ribosome biosynthesis in yeast. Trends Biochem Sci. 1999;24:437–440. doi: 10.1016/s0968-0004(99)01460-7. [DOI] [PubMed] [Google Scholar]
- 12.Cabart P, Lee J, Willis IM. Facilitated Recycling Protects Human RNA Polymerase III from Repression by Maf1 in Vitro. J Biol Chem. 2008;283:36108–36117. doi: 10.1074/jbc.M807538200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.White RJ. RNA polymerases I, III growth control and cancer. Nat Rev Mol Cell Biol. 2005;6:69–78. doi: 10.1038/nrm1551. [DOI] [PubMed] [Google Scholar]
- 14.Ciesla M, Boguta M. Regulation of RNA polymerase III transcription by Maf1 protein. Acta Biochim Pol. 2008;55:215–225. [PubMed] [Google Scholar]
- 15.Pluta K, Lefebvre O, Martin NC, Smagowicz WJ, Stanford DR, Ellis SR, Hopper AK, Sentenac A, Boguta M. Maf1p, a negative effector of RNA polymerase III in Saccharomyces cerevisiae. Mol Cell Biol. 2001;21:5031–5040. doi: 10.1128/MCB.21.15.5031-5040.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kwapisz M, Smagowicz WJ, Oficjalska D, Hatin I, Rousset JP, Zoladek T, Boguta M. Up-regulation of tRNA biosynthesis affects translational readthrough in maf1-delta mutant of Saccharomyces cerevisiae. Curr Genet. 2002;42:147–152. doi: 10.1007/s00294-002-0342-7. [DOI] [PubMed] [Google Scholar]
- 17.Boguta M, Czerska K, Zoladek T. Mutation in a new gene MAF1 affects tRNA suppressor efficiency in Saccharomyces cerevisiae. Gene. 1997;185:291–296. doi: 10.1016/s0378-1119(96)00669-5. [DOI] [PubMed] [Google Scholar]
- 18.Upadhya R, Lee J, Willis IM. Maf1 Is an Essential Mediator of Diverse Signals that Repress RNA Polymerase III Transcription. Mol Cell. 2002;10:1489–1494. doi: 10.1016/s1097-2765(02)00787-6. [DOI] [PubMed] [Google Scholar]
- 19.Desai N, Lee J, Upadhya R, Chu Y, Moir RD, Willis IM. Two Steps in Maf1-dependent Repression of Transcription by RNA Polymerase III. J Biol Chem. 2005;280:6455–6462. doi: 10.1074/jbc.M412375200. [DOI] [PubMed] [Google Scholar]
- 20.Reina JH, Azzouz TN, Hernandez N. Maf1, a New Player in the Regulation of Human RNA Polymerase III Transcription. PLoS ONE. 2006;1 :e134. doi: 10.1371/journal.pone.0000134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johnson SS, Zhang C, Fromm J, Willis IM, Johnson DL. Mammalian Maf1 is a negative regulator of transcription by all three nuclear RNA polymerases. Mol Cell. 2007;26:367–379. doi: 10.1016/j.molcel.2007.03.021. [DOI] [PubMed] [Google Scholar]
- 22.Goodfellow SJ, Graham EL, Kantidakis T, Marshall L, Coppins BA, Oficjalska-Pham D, Gerard M, Lefebvre O, White RJ. Regulation of RNA polymerase III transcription by Maf1 in mammalian cells. J Mol Biol. 2008;378:481–491. doi: 10.1016/j.jmb.2008.02.060. [DOI] [PubMed] [Google Scholar]
- 23.Shor B, Wu J, Shakey Q, Toral-Barza L, Shi C, Follettie M, Yu K. Requirement of the mTOR Kinase for the Regulation of Maf1 Phosphorylation and Control of RNA Polymerase III-dependent Transcription in Cancer Cells. J Biol Chem. 2010;285:15380–15392. doi: 10.1074/jbc.M109.071639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Michels AA, Robitaille AM, Buczynski-Ruchonnet D, Hodroj W, Reina JH, Hall MN, Hernandez N. mTORC1 directly phosphorylates and regulates human MAF1. Mol Cell Biol. 2010;30:3749–3757. doi: 10.1128/MCB.00319-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rideout EJ, Marshall L, Grewal SS. Drosophila RNA polymerase III repressor Maf1 controls body size and developmental timing by modulating tRNAiMet synthesis and systemic insulin signaling. Proc Natl Acad Sci U S A. 2012;109:1139–1144. doi: 10.1073/pnas.1113311109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marshall L, Rideout EJ, Grewal SS. Nutrient/TOR-dependent regulation of RNA polymerase III controls tissue and organismal growth in Drosophila. EMBO J. 2012;31:1916–1930. doi: 10.1038/emboj.2012.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oler AJ, Cairns BR. PP4 dephosphorylates Maf1 to couple multiple stress conditions to RNA polymerase III repression. EMBO J. 2012;31:1440–1452. doi: 10.1038/emboj.2011.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rollins J, Veras I, Cabarcas S, Willis I, Schramm L. Human Maf1 negatively regulates RNA polymerase III transcription via the TFIIB family members Brf1 and Brf2. Int J Biol Sci. 2007;3:292–302. doi: 10.7150/ijbs.3.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roberts DN, Wilson B, Huff JT, Stewart AJ, Cairns BR. Dephosphorylation and Genome-Wide Association of Maf1 with Pol III-Transcribed Genes during Repression. Mol Cell. 2006;22:633–644. doi: 10.1016/j.molcel.2006.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oficjalska-Pham D, Harismendy O, Smagowicz WJ, Gonzalez dP, Boguta M, Sentenac A, Lefebvre O. General Repression of RNA Polymerase III Transcription Is Triggered by Protein Phosphatase Type 2A-Mediated Dephosphorylation of Maf1. Mol Cell. 2006;22:623–632. doi: 10.1016/j.molcel.2006.04.008. [DOI] [PubMed] [Google Scholar]
- 31.Moir RD, Lee J, Haeusler RA, Desai N, Engelke DR, Willis IM. Protein kinase A regulates RNA polymerase III transcription through the nuclear localization of Maf1. Proc Natl Acad Sci U S A. 2006;103:15044–15049. doi: 10.1073/pnas.0607129103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee J, Moir RD, Willis IM. Regulation of RNA polymerase III transcription involves SCH9-dependent and SCH9-independent branches of the target of rapamycin (TOR) pathway. J Biol Chem. 2009;284:12604–12608. doi: 10.1074/jbc.C900020200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huber A, Bodenmiller B, Uotila A, Stahl M, Wanka S, Gerrits B, Aebersold R, Loewith R. Characterization of the rapamycin-sensitive phosphoproteome reveals that Sch9 is a central coordinator of protein synthesis. Genes Dev. 2009;23:1929–1943. doi: 10.1101/gad.532109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kantidakis T, Ramsbottom BA, Birch JL, Dowding SN, White RJ. mTOR associates with TFIIIC, is found at tRNA and 5S rRNA genes, and targets their repressor Maf1. Proc Nat Acad Sci USA. 2010;107:11823–11828. doi: 10.1073/pnas.1005188107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chi A, Huttenhower C, Geer LY, Coon JJ, Syka JE, Bai DL, Shabanowitz J, Burke DJ, Troyanskaya OG, Hunt DF. Analysis of phosphorylation sites on proteins from Saccharomyces cerevisiae by electron transfer dissociation (ETD) mass spectrometry. Proc Natl Acad Sci U S A. 2007;104:2193–2198. doi: 10.1073/pnas.0607084104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li X, Gerber SA, Rudner AD, Beausoleil SA, Haas W, Villen J, Elias JE, Gygi SP. Large-scale phosphorylation analysis of alpha-factor-arrested Saccharomyces cerevisiae. J Proteome Res. 2007;6:1190–1197. doi: 10.1021/pr060559j. [DOI] [PubMed] [Google Scholar]
- 37.Albuquerque CP, Smolka MB, Payne SH, Bafna V, Eng J, Zhou H. A multidimensional chromatography technology for in-depth phosphoproteome analysis. Mol Cell Proteomics. 2008;7:1389–1396. doi: 10.1074/mcp.M700468-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Graczyk D, Debski J, Muszynska G, Bretner M, Lefebvre O, Boguta M. Casein kinase II-mediated phosphorylation of general repressor Maf1 triggers RNA polymerase III activation. Proc Natl Acad Sci U S A. 2011;108:4926–4931. doi: 10.1073/pnas.1010010108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nika H, Lee J, Willis IM, Angeletti RH, Hawke DH. Phosphopeptide Characterization by Mass Spectrometry using Reversed-Phase Supports for Solid-Phase Beta-Elimination/Michael Addition. J Biomol Tech. 2012;23 doi: 10.7171/jbt.2012-2302-002. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mok J, Kim PM, Lam HYK, Piccirillo S, Zhou X, Jeschke GR, Sheridan DL, Parker SA, Desai V, Jwa M, Cameroni E, Niu H, Good M, Remenyi A, Ma JLN, Sheu YJ, Sassi HE, Sopko R, Chan CSM, De Virgilio C, Hollingsworth NM, Lim WA, Stern DF, Stillman B, Andrews BJ, Gerstein MB, Snyder M, Turk BE. Deciphering Protein Kinase Specificity Through Large-Scale Analysis of Yeast Phosphorylation Site Motifs. Sci Signal. 2010;3:ra12. doi: 10.1126/scisignal.2000482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wei Y, Zheng XF. Sch9 partially mediates TORC1 signaling to control ribosomal RNA synthesis. Cell Cycle. 2009;8:4085–4090. doi: 10.4161/cc.8.24.10170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moir RD, Lee J, Willis IM. Recovery of RNA Polymerase III Transcription from the Glycerol-repressed State: Revisiting the Role of Protein Kinase CK2 in Maf1 Phosphoregulation. J Biol Chem. 2012;287:30833–30841. doi: 10.1074/jbc.M112.378828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Soulard A, Cremonesi A, Moes S, Schutz F, Jeno P, Hall MN. The rapamycin-sensitive phosphoproteome reveals that TOR controls protein kinase A toward some but not all substrates. Mol Biol Cell. 2010;21:3475–3486. doi: 10.1091/mbc.E10-03-0182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Towpik J, Graczyk D, Gajda A, Lefebvre O, Boguta M. Derepression of RNA Polymerase III Transcription by Phosphorylation and Nuclear Export of Its Negative Regulator, Maf1. J Biol Chem. 2008;283:17168–17174. doi: 10.1074/jbc.M709157200. [DOI] [PubMed] [Google Scholar]
- 45.Wei Y, Tsang CK, Zheng XF. Mechanisms of regulation of RNA polymerase III-dependent transcription by TORC1. EMBO J. 2009;28:2220–2230. doi: 10.1038/emboj.2009.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vannini A, Ringel R, Kusser AG, Berninghausen O, Kassavetis GA, Cramer P. Molecular basis of RNA polymerase III transcription repression by Maf1. Cell. 2010;143:59–70. doi: 10.1016/j.cell.2010.09.002. [DOI] [PubMed] [Google Scholar]
- 47.Moir RD, Willis IM. Tetratricopeptide repeats of Tfc4 and a limiting step in the assembly of the initiation factor TFIIIB. Adv Protein Chem. 2004;67:93–121. doi: 10.1016/S0065-3233(04)67004-5. [DOI] [PubMed] [Google Scholar]
- 48.Willis IM, Moir RD. Integration of nutritional and stress signaling pathways by Maf1. Trends Biochem Sci. 2007;32:51–53. doi: 10.1016/j.tibs.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 49.Geiduschek EP, Kassavetis GA. The RNA polymerase III transcription apparatus. J Mol Biol. 2001;310:1–26. doi: 10.1006/jmbi.2001.4732. [DOI] [PubMed] [Google Scholar]
- 50.Huibregtse JM, Engelke DR. Genomic footprinting of a yeast tRNA gene reveals stable complexes over the 5′-flanking region. Mol Cell Biol. 1989;9:3244–3252. doi: 10.1128/mcb.9.8.3244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Roberts DN, Stewart AJ, Huff JT, Cairns BR. The RNA polymerase III transcriptome revealed by genome-wide localization and activity-occupancy relationships. Proc Natl Acad Sci U S A. 2003;100:14695–14700. doi: 10.1073/pnas.2435566100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Makhnevych T, Sydorskyy Y, Xin X, Srikumar T, Vizeacoumar FJ, Jeram SM, Li Z, Bahr S, Andrews BJ, Boone C, Raught B. Global map of SUMO function revealed by protein-protein interaction and genetic networks. Mol Cell. 2009;33:124–135. doi: 10.1016/j.molcel.2008.12.025. [DOI] [PubMed] [Google Scholar]
- 53.Chedin S, Ferri ML, Peyroche G, Andrau JC, Jourdain S, Lefebvre O, Werner M, Carles C, Sentenac A. The yeast RNA polymerase III transcription machinery: a paradigm for eukaryotic gene activation. Cold Spring Harb Symp Quant Biol. 1998;63:381–389. doi: 10.1101/sqb.1998.63.381. [DOI] [PubMed] [Google Scholar]
- 54.Mohammed S, Lorenzen K, Kerkhoven R, Breukelen Bv, Vannini A, Cramer P, Heck AJR. Multiplexed Proteomics Mapping of Yeast RNA Polymerase II and III Allows Near-Complete Sequence Coverage and Reveals Several Novel Phosphorylation Sites. Anal Chem. 2008;80:3584–3592. doi: 10.1021/ac7024283. [DOI] [PubMed] [Google Scholar]
- 55.Bodenmiller B, Wanka S, Kraft C, Urban J, Campbell D, Pedrioli PG, Gerrits B, Picotti P, Lam H, Vitek O, Brusniak MY, Roschitzki B, Zhang C, Shokat KM, Schlapbach R, Colman-Lerner A, Nolan GP, Nesvizhskii AI, Peter M, Loewith R, von Mering C, Aebersold R. Phosphoproteomic Analysis Reveals Interconnected System-Wide Responses to Perturbations of Kinases and Phosphatases in Yeast. Sci Signal. 2010;3:rs4. doi: 10.1126/scisignal.2001182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee J, Moir RD, McIntosh KB, Willis IM. TOR Signaling Regulates Ribosome and tRNA Synthesis via LAMMER/Clk and GSK-3 Family Kinases. Mol Cell. 2012;45:836–843. doi: 10.1016/j.molcel.2012.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Landrieux E, Alic N, Ducrot C, Acker J, Riva M, Carles C. A subcomplex of RNA polymerase III subunits involved in transcription termination and reinitiation. EMBO J. 2006;25:118–128. doi: 10.1038/sj.emboj.7600915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lorenzen K, Vannini A, Cramer P, Heck AJ. Structural biology of RNA polymerase III: mass spectrometry elucidates subcomplex architecture. Structure. 2007;15:1237–1245. doi: 10.1016/j.str.2007.07.016. [DOI] [PubMed] [Google Scholar]
- 59.Kassavetis GA, Prakash P, Shim E. The C53/C37 subcomplex of RNA polymerase III lies near the active site and participates in promoter opening. J Biol Chem. 2010;285:2695–2706. doi: 10.1074/jbc.M109.074013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wu CC, Lin YC, Chen HT. The TFIIF-like Rpc37/53 dimer lies at the center of a protein network to connect TFIIIC, Bdp1, and the RNA polymerase III active center. Mol Cell Biol. 2011;31:2715–2728. doi: 10.1128/MCB.05151-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Iben JR, Mazeika JK, Hasson S, Rijal K, Arimbasseri AG, Russo AN, Maraia RJ. Point mutations in the Rpb9-homologous domain of Rpc11 that impair transcription termination by RNA polymerase III. Nucleic Acids Res. 2011;39:6100–6113. doi: 10.1093/nar/gkr182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Willis IM, Lee J. Two new kinases in the TOR signaling network regulate ribosome and tRNA synthesis. Cell Cycle. 2012;11:2769–2770. doi: 10.4161/cc.21257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lane LA, Fernandez-Tornero C, Zhou M, Morgner N, Ptchelkine D, Steuerwald U, Politis A, Lindner D, Gvozdenovic J, Gavin AC, Muller CW, Robinson CV. Mass spectrometry reveals stable modules in holo and apo RNA polymerases I and III. Structure. 2011;19:90–100. doi: 10.1016/j.str.2010.11.009. [DOI] [PubMed] [Google Scholar]
- 64.Haurie V, Durrieu-Gaillard S, Dumay-Odelot H, Da Silva D, Rey C, Prochazkova M, Roeder RG, Besser D, Teichmann M. Two isoforms of human RNA polymerase III with specific functions in cell growth and transformation. Proc Natl Acad Sci U S A. 2010;107:4176–4181. doi: 10.1073/pnas.0914980107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhong S, Johnson DL. The JNKs differentially regulate RNA polymerase III transcription by coordinately modulating the expression of all TFIIIB subunits. Proc Natl Acad Sci U S A. 2009;106:12682–12687. doi: 10.1073/pnas.0904843106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang HD, Trivedi A, Johnson DL. Hepatitis B virus X protein induces RNA polymerase III-dependent gene transcription and increases cellular TATA-binding protein by activating the Ras signaling pathway. Mol Cell Biol. 1997;17:6838–6846. doi: 10.1128/mcb.17.12.6838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Felton-Edkins ZA, White RJ. Multiple mechanisms contribute to the activation of RNA polymerase III transcription in cells transformed by papovaviruses. J Biol Chem. 2002;277:48182–48191. doi: 10.1074/jbc.M201333200. [DOI] [PubMed] [Google Scholar]
- 68.Daly NL, Arvanitis DA, Fairley JA, Gomez-Roman N, Morton JP, Graham SV, Spandidos DA, White RJ. Deregulation of RNA polymerase III transcription in cervical epithelium in response to high-risk human papillomavirus. Oncogene. 2005;24:880–888. doi: 10.1038/sj.onc.1208031. [DOI] [PubMed] [Google Scholar]
- 69.Ghavidel A, Schultz MC. TATA binding protein-associated CK2 transduces DNA damage signals to the RNA polymerase III transcriptional machinery. Cell. 2001;106:575–584. doi: 10.1016/s0092-8674(01)00473-1. [DOI] [PubMed] [Google Scholar]
- 70.Johnston IM, Allison SJ, Morton JP, Schramm L, Scott PH, White RJ. CK2 forms a stable complex with TFIIIB and activates RNA polymerase III transcription in human cells. Mol Cell Biol. 2002;22:3757–3768. doi: 10.1128/MCB.22.11.3757-3768.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hu P, Wu S, Hernandez N. A minimal RNA polymerase III transcription system from human cells reveals positive and negative regulatory roles for CK2. Mol Cell. 2003;12:699–709. doi: 10.1016/j.molcel.2003.08.011. [DOI] [PubMed] [Google Scholar]
- 72.Dephoure N, Zhou C, Villen J, Beausoleil SA, Bakalarski CE, Elledge SJ, Gygi SP. A quantitative atlas of mitotic phosphorylation. Proc Natl Acad Sci U S A. 2008;105:10762–10767. doi: 10.1073/pnas.0805139105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gottesfeld JM, Wolf VJ, Dang T, Forbes DJ, Hartl P. Mitotic repression of RNA polymerase III transcription in vitro mediated by phosphorylation of a TFIIIB component. Science. 1994;263:81–84. doi: 10.1126/science.8272869. [DOI] [PubMed] [Google Scholar]
- 74.White RJ, Gottlieb TM, Downes CS, Jackson SP. Mitotic regulation of a TATA-binding-protein-containing complex. Mol Cell Biol. 1995;15:1983–1992. doi: 10.1128/mcb.15.4.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fairley JA, Scott PH, White RJ. TFIIIB is phosphorylated, disrupted and selectively released from tRNA promoters during mitosis in vivo. EMBO J. 2003;22:5841–5850. doi: 10.1093/emboj/cdg544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hu P, Samudre K, Wu S, Sun Y, Hernandez N. CK2 phosphorylation of Bdp1 executes cell cycle-specific RNA polymerase III transcription repression. Mol Cell. 2004;16:81–92. doi: 10.1016/j.molcel.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 77.Felton-Edkins ZA, Fairley JA, Graham EL, Johnston IM, White RJ, Scott PH. The mitogen-activated protein (MAP) kinase ERK induces tRNA synthesis by phosphorylating TFIIIB. EMBO J. 2003;22:2422–2432. doi: 10.1093/emboj/cdg240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Woiwode A, Johnson SA, Zhong S, Zhang C, Roeder RG, Teichmann M, Johnson DL. PTEN represses RNA polymerase III-dependent transcription by targeting the TFIIIB complex. Mol Cell Biol. 2008;28:4204–4214. doi: 10.1128/MCB.01912-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fairley JA, Mitchell LE, Berg T, Kenneth NS, von Schubert C, Sillje HH, Medema RH, Nigg EA, White RJ. Direct regulation of tRNA and 5S rRNA gene transcription by Polo-like kinase 1. Mol Cell. 2012;45:541–552. doi: 10.1016/j.molcel.2011.11.030. [DOI] [PubMed] [Google Scholar]
- 80.Moqtaderi Z, Struhl K. Genome-wide occupancy profile of the RNA polymerase III machinery in Saccharomyces cerevisiae reveals loci with incomplete transcription complexes. Mol Cell Biol. 2004;24:4118–4127. doi: 10.1128/MCB.24.10.4118-4127.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Noma K, Cam HP, Maraia RJ, Grewal SI. A role for TFIIIC transcription factor complex in genome organization. Cell. 2006;125:859–872. doi: 10.1016/j.cell.2006.04.028. [DOI] [PubMed] [Google Scholar]
- 82.Donze D. Extra-transcriptional functions of RNA Polymerase III complexes: TFIIIC as a potential global chromatin bookmark. Gene. 2012;493:169–175. doi: 10.1016/j.gene.2011.09.018. [DOI] [PubMed] [Google Scholar]
- 83.Ferrari R, Rivetti C, Acker J, Dieci G. Distinct roles of transcription factors TFIIIB and TFIIIC in RNA polymerase III transcription reinitiation. Proc Natl Acad Sci U S A. 2004;101:13442–13447. doi: 10.1073/pnas.0403851101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Marshall L, White RJ. Non-coding RNA production by RNA polymerase III is implicated in cancer. Nat Rev Cancer. 2008;8:911–914. doi: 10.1038/nrc2539. [DOI] [PubMed] [Google Scholar]
- 85.Hoeffler WK, Kovelman R, Roeder RG. Activation of transcription factor IIIC by the adenovirus E1A protein. Cell. 1988;53:907–920. doi: 10.1016/s0092-8674(88)90409-6. [DOI] [PubMed] [Google Scholar]
- 86.White RJ, Stott D, Rigby PW. Regulation of RNA polymerase III transcription in response to F9 embryonal carcinoma stem cell differentiation. Cell. 1989;59:1081–1092. doi: 10.1016/0092-8674(89)90764-2. [DOI] [PubMed] [Google Scholar]
- 87.Innes F, Ramsbottom B, White RJ. A test of the model that RNA polymerase III transcription is regulated by selective induction of the 110 kDa subunit of TFIIIC. Nucleic Acids Res. 2006;34:3399–3407. doi: 10.1093/nar/gkl432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Clark ME, Hammerle T, Wimmer E, Dasgupta A. Poliovirus proteinase 3C converts an active form of transcription factor IIIC to an inactive form: a mechanism for inhibition of host cell polymerase III transcription by poliovirus. EMBO J. 1991;10:2941–2947. doi: 10.1002/j.1460-2075.1991.tb07844.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Rana T, Misra S, Mittal MK, Farrow AL, Wilson KT, Linton MF, Fazio S, Willis IM, Chaudhuri G. Mechanism of down-regulation of RNA polymerase III-transcribed non-coding RNA genes in macrophages by Leishmania. J Biol Chem. 2011;286:6614–6626. doi: 10.1074/jbc.M110.181735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Conesa C, Swanson RN, Schultz P, Oudet P, Sentenac A. On the subunit composition, stoichiometry, and phosphorylation of the yeast transcription factor TFIIIC/tau. J Biol Chem. 1993;268:18047–18052. [PubMed] [Google Scholar]
- 91.Wang Z, Roeder RG. DNA topoisomerase I and PC4 can interact with human TFIIIC to promote both accurate termination and transcription reinitiation by RNA polymerase III. Mol Cell. 1998;1:749–757. doi: 10.1016/s1097-2765(00)80074-x. [DOI] [PubMed] [Google Scholar]
- 92.Tavenet A, Suleau A, Dubreuil G, Ferrari R, Ducrot C, Michaut M, Aude JC, Dieci G, Lefebvre O, Conesa C, Acker J. Genome-wide location analysis reveals a role for Sub1 in RNA polymerase III transcription. Proc Natl Acad Sci U S A. 2009;106:14265–14270. doi: 10.1073/pnas.0900162106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rosonina E, Willis IM, Manley JL. Sub1 functions in osmoregulation and in transcription by both RNA polymerases II and III. Mol Cell Biol. 2009;29:2308–2321. doi: 10.1128/MCB.01841-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Conesa C, Acker J. Sub1/PC4 a chromatin associated protein with multiple functions in transcription. RNA Biol. 2010;7:287–290. doi: 10.4161/rna.7.3.11491. [DOI] [PubMed] [Google Scholar]
- 95.Stillman DJ. Nhp6: a small but powerful effector of chromatin structure in Saccharomyces cerevisiae. Biochim Biophys Acta. 2010;1799:175–180. doi: 10.1016/j.bbagrm.2009.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kruppa M, Moir RD, Kolodrubetz D, Willis IM. Nhp6, an HMG1 protein, functions in SNR6 transcription by RNA polymerase III in S. cerevisiae. Mol Cell. 2001;7:309–318. doi: 10.1016/s1097-2765(01)00179-4. [DOI] [PubMed] [Google Scholar]
- 97.Martin MP, Gerlach VL, Brow DA. A novel upstream RNA polymerase III promoter element becomes essential when the chromatin structure of the yeast U6 RNA gene is altered. Mol Cell Biol. 2001;21:6429–6439. doi: 10.1128/MCB.21.19.6429-6439.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lopez S, Livingstone-Zatchej M, Jourdain S, Thoma F, Sentenac A, Marsolier MC. High-mobility-group proteins NHP6A and NHP6B participate in activation of the RNA polymerase III SNR6 gene. Mol Cell Biol. 2001;21:3096–3104. doi: 10.1128/MCB.21.9.3096-3104.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Arimbasseri AG, Bhargava P. Chromatin structure and expression of a gene transcribed by RNA polymerase III are independent of H2A.Z deposition. Mol Cell Biol. 2008;28:2598–2607. doi: 10.1128/MCB.01953-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Braglia P, Dugas SL, Donze D, Dieci G. Requirement of Nhp6 proteins for transcription of a subset of tRNA genes and heterochromatin barrier function in Saccharomyces cerevisiae. Mol Cell Biol. 2007;27:1545–1557. doi: 10.1128/MCB.00773-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Eriksson P, Biswas D, Yu Y, Stewart JM, Stillman DJ. TATA-binding protein mutants that are lethal in the absence of the Nhp6 high-mobility-group protein. Mol Cell Biol. 2004;24:6419–6429. doi: 10.1128/MCB.24.14.6419-6429.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kassavetis GA, Steiner DF. Nhp6 is a transcriptional initiation fidelity factor for RNA polymerase III transcription in vitro and in vivo. J Biol Chem. 2006;281:7445–7451. doi: 10.1074/jbc.M512810200. [DOI] [PubMed] [Google Scholar]
- 103.Szerlong H, Saha A, Cairns BR. The nuclear actin-related proteins Arp7 and Arp9: a dimeric module that cooperates with architectural proteins for chromatin remodeling. EMBO J. 2003;22:3175–3187. doi: 10.1093/emboj/cdg296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Parnell TJ, Huff JT, Cairns BR. RSC regulates nucleosome positioning at Pol II genes and density at Pol III genes. EMBO J. 2008;27:100–110. doi: 10.1038/sj.emboj.7601946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Raha D, Wang Z, Moqtaderi Z, Wu L, Zhong G, Gerstein M, Struhl K, Snyder M. Close association of RNA polymerase II and many transcription factors with Pol III genes. Proc Natl Acad Sci U S A. 2010;107:3639–3644. doi: 10.1073/pnas.0911315106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Venters BJ, Wachi S, Mavrich TN, Andersen BE, Jena P, Sinnamon AJ, Jain P, Rolleri NS, Jiang C, Hemeryck-Walsh C, Pugh BF. A comprehensive genomic binding map of gene and chromatin regulatory proteins in Saccharomyces. Mol Cell. 2011;41:480–492. doi: 10.1016/j.molcel.2011.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Dieci G, Ruotolo R, Braglia P, Carles C, Carpentieri A, Amoresano A, Ottonello S. Positive modulation of RNA polymerase III transcription by ribosomal proteins. Biochem Biophys Res Commun. 2009;379:489–493. doi: 10.1016/j.bbrc.2008.12.097. [DOI] [PubMed] [Google Scholar]
- 108.Ghavi-Helm Y, Michaut M, Acker J, Aude JC, Thuriaux P, Werner M, Soutourina J. Genome-wide location analysis reveals a role of TFIIS in RNA polymerase III transcription. Genes Dev. 2008;22:1934–1947. doi: 10.1101/gad.471908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Busti S, Coccetti P, Alberghina L, Vanoni M. Glucose signaling-mediated coordination of cell growth and cell cycle in Saccharomyces cerevisiae. Sensors (Basel) 2010;10:6195–6240. doi: 10.3390/s100606195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.McCudden C, Hains M, Kimple R, Siderovski D, Willard F. G-protein signaling: back to the future. Cellular and Molecular Life Sciences. 2005;62:551–577. doi: 10.1007/s00018-004-4462-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zaman S, Lippman SI, Zhao X, Broach JR. How Saccharomyces Responds to Nutrients. Annu Rev Genet. 2008 doi: 10.1146/annurev.genet.41.110306.130206. [DOI] [PubMed] [Google Scholar]
- 112.Colombo S, Ronchetti D, Thevelein JM, Winderickx J, Martegani E. Activation state of the Ras2 protein and glucose-induced signaling in Saccharomyces cerevisiae. J Biol Chem. 2004;279:46715–46722. doi: 10.1074/jbc.M405136200. [DOI] [PubMed] [Google Scholar]
- 113.Peeters T, Versele M, Thevelein JM. Directly from Galpha to protein kinase A: the kelch repeat protein bypass of adenylate cyclase. Trends Biochem Sci. 2007;32:547–554. doi: 10.1016/j.tibs.2007.09.011. [DOI] [PubMed] [Google Scholar]
- 114.Nikawa J, Cameron S, Toda T, Ferguson KM, Wigler M. Rigorous feedback control of cAMP levels in Saccharomyces cerevisiae. Genes Dev. 1987;1:931–937. doi: 10.1101/gad.1.9.931. [DOI] [PubMed] [Google Scholar]
- 115.Rubio-Texeira M, Van Zeebroeck G, Voordeckers K, Thevelein JM. Saccharomyces cerevisiae plasma membrane nutrient sensors and their role in PKA signaling. FEMS Yeast Res. 2010;10:134–149. doi: 10.1111/j.1567-1364.2009.00587.x. [DOI] [PubMed] [Google Scholar]
- 116.Portela P, Moreno S. Glucose-dependent activation of protein kinase A activity in Saccharomyces cerevisiae and phosphorylation of its TPK1 catalytic subunit. Cell Signal. 2006;18:1072–1086. doi: 10.1016/j.cellsig.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 117.Griffioen G, Branduardi P, Ballarini A, Anghileri P, Norbeck J, Baroni MD, Ruis H. Nucleocytoplasmic distribution of budding yeast protein kinase A regulatory subunit Bcy1 requires Zds1 and is regulated by Yak1-dependent phosphorylation of its targeting domain. Mol Cell Biol. 2001;21:511–523. doi: 10.1128/MCB.21.2.511-523.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Griffioen G, Swinnen S, Thevelein JM. Feedback inhibition on cell wall integrity signaling by Zds1 involves Gsk3 phosphorylation of a cAMP-dependent protein kinase regulatory subunit. J Biol Chem. 2003;278:23460–23471. doi: 10.1074/jbc.M210691200. [DOI] [PubMed] [Google Scholar]
- 119.Griffioen G, Thevelein JM. Molecular mechanisms controlling the localisation of protein kinase A. Curr Genet. 2002;41:199–207. doi: 10.1007/s00294-002-0308-9. [DOI] [PubMed] [Google Scholar]
- 120.Schmelzle T, Beck T, Martin DE, Hall MN. Activation of the RAS/cyclic AMP pathway suppresses a TOR deficiency in yeast. Mol Cell Biol. 2004;24:338–351. doi: 10.1128/MCB.24.1.338-351.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Tudisca V, Recouvreux V, Moreno S, Boy-Marcotte E, Jacquet M, Portela P. Differential localization to cytoplasm, nucleus or P-bodies of yeast PKA subunits under different growth conditions. Eur J Cell Biol. 2010;89:339–348. doi: 10.1016/j.ejcb.2009.08.005. [DOI] [PubMed] [Google Scholar]
- 122.Ptacek J, Devgan G, Michaud G, Zhu H, Zhu X, Fasolo J, Guo H, Jona G, Breitkreutz A, Sopko R, McCartney RR, Schmidt MC, Rachidi N, Lee SJ, Mah AS, Meng L, Stark MJ, Stern DF, De Virgilio C, Tyers M, Andrews B, Gerstein M, Schweitzer B, Predki PF, Snyder M. Global analysis of protein phosphorylation in yeast. Nature. 2005;438:679–684. doi: 10.1038/nature04187. [DOI] [PubMed] [Google Scholar]
- 123.Smets B, Ghillebert R, De Snijder P, Binda M, Swinnen E, De Virgilio C, Winderickx J. Life in the midst of scarcity: adaptations to nutrient availability in Saccharomyces cerevisiae. Curr Genet. 2010;56:1–32. doi: 10.1007/s00294-009-0287-1. [DOI] [PubMed] [Google Scholar]
- 124.Budovskaya YV, Stephan JS, Deminoff SJ, Herman PK. An evolutionary proteomics approach identifies substrates of the cAMP-dependent protein kinase. Proc Natl Acad Sci U S A. 2005;102:13933–13938. doi: 10.1073/pnas.0501046102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Cameron S, Levin L, Zoller M, Wigler M. cAMP-independent control of sporulation, glycogen metabolism, and heat shock resistance in S. cerevisiae. Cell. 1988;53:555–566. doi: 10.1016/0092-8674(88)90572-7. [DOI] [PubMed] [Google Scholar]
- 126.Roberts GG, Hudson AP. Transcriptome profiling of Saccharomyces cerevisiae during a transition from fermentative to glycerol-based respiratory growth reveals extensive metabolic and structural remodeling. Mol Genet Genomics. 2006;276:170–186. doi: 10.1007/s00438-006-0133-9. [DOI] [PubMed] [Google Scholar]
- 127.Ciesla M, Towpik J, Graczyk D, Oficjalska-Pham D, Harismendy O, Suleau A, Balicki K, Conesa C, Lefebvre O, Boguta M. Maf1 is involved in coupling carbon metabolism to RNA polymerase III transcription. Mol Cell Biol. 2007;27:7693–7702. doi: 10.1128/MCB.01051-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Toda T, Uno I, Ishikawa T, Powers S, Kataoka T, Broek D, Cameron S, Broach J, Matsumoto K, Wigler M. In yeast, RAS proteins are controlling elements of adenylate cyclase. Cell. 1985;40:27–36. doi: 10.1016/0092-8674(85)90305-8. [DOI] [PubMed] [Google Scholar]
- 129.Zurita-Martinez SA, Cardenas ME. Tor and cyclic AMP-protein kinase A: two parallel pathways regulating expression of genes required for cell growth. Eukaryot Cell. 2005;4:63–71. doi: 10.1128/EC.4.1.63-71.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Zaman S, Lippman SI, Schneper L, Slonim N, Broach JR. Glucose regulates transcription in yeast through a network of signaling pathways. Mol Syst Biol. 2009;5:245. doi: 10.1038/msb.2009.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ramachandran V, Herman PK. Antagonistic interactions between the cAMP-dependent protein kinase and Tor signaling pathways modulate cell growth in Saccharomyces cerevisiae. Genetics. 2011;187:441–454. doi: 10.1534/genetics.110.123372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Loewith R, Hall MN. Target of rapamycin (TOR) in nutrient signaling and growth control. Genetics. 2011;189:1177–1201. doi: 10.1534/genetics.111.133363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.van Dam TJ, Zwartkruis FJ, Bos JL, Snel B. Evolution of the TOR pathway. J Mol Evol. 2011;73:209–220. doi: 10.1007/s00239-011-9469-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Desai BN, Myers BR, Schreiber SL. FKBP12-rapamycin-associated protein associates with mitochondria and senses osmotic stress via mitochondrial dysfunction. Proc Natl Acad Sci U S A. 2002;99:4319–4324. doi: 10.1073/pnas.261702698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Drenan RM, Liu X, Bertram PG, Zheng XF. FKBP12-rapamycin-associated protein or mammalian target of rapamycin (FRAP/mTOR) localization in the endoplasmic reticulum and the Golgi apparatus. J Biol Chem. 2004;279:772–778. doi: 10.1074/jbc.M305912200. [DOI] [PubMed] [Google Scholar]
- 136.Li H, Tsang CK, Watkins M, Bertram PG, Zheng XF. Nutrient regulates Tor1 nuclear localization and association with rDNA promoter. Nature. 2006;442:1058–1061. doi: 10.1038/nature05020. [DOI] [PubMed] [Google Scholar]
- 137.Urban J, Soulard A, Huber A, Lippman S, Mukhopadhyay D, Deloche O, Wanke V, Anrather D, Ammerer G, Riezman H, Broach JR, De Virgilio C, Hall MN, Loewith R. Sch9 is a major target of TORC1 in Saccharomyces cerevisiae. Mol Cell. 2007;26:663–674. doi: 10.1016/j.molcel.2007.04.020. [DOI] [PubMed] [Google Scholar]
- 138.Tsang CK, Liu H, Zheng XF. mTOR binds to the promoters of RNA polymerase I- and III-transcribed genes. Cell Cycle. 2010;9:953–957. doi: 10.4161/cc.9.5.10876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Wei Y, Zheng XS. Maf1 regulation: a model of signal transduction inside the nucleus. Nucleus. 2010;1:162–165. doi: 10.4161/nucl.1.2.11179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Toda T, Cameron S, Sass P, Wigler M. SCH9, a gene of Saccharomyces cerevisiae that encodes a protein distinct from, but functionally and structurally related to, cAMP-dependent protein kinase catalytic subunits. Genes Dev. 1988;2:517–527. doi: 10.1101/gad.2.5.517. [DOI] [PubMed] [Google Scholar]
- 141.Huang X, Liu J, Dickson RC. Down-regulating sphingolipid synthesis increases yeast lifespan. PLoS Genet. 2012;8:e1002493. doi: 10.1371/journal.pgen.1002493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Di Como CJ, Arndt KT. Nutrients, via the Tor proteins, stimulate the association of Tap42 with type 2A phosphatases. Genes Dev. 1996;10:1904–1916. doi: 10.1101/gad.10.15.1904. [DOI] [PubMed] [Google Scholar]
- 143.Duvel K, Santhanam A, Garrett S, Schneper L, Broach JR. Multiple roles of Tap42 in mediating rapamycin-induced transcriptional changes in yeast. Mol Cell. 2003;11:1467–1478. doi: 10.1016/s1097-2765(03)00228-4. [DOI] [PubMed] [Google Scholar]
- 144.Yan G, Shen X, Jiang Y. Rapamycin activates Tap42-associated phosphatases by abrogating their association with Tor complex 1. EMBO J. 2006;25:3546–3555. doi: 10.1038/sj.emboj.7601239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Willis IM, Desai NA, Upadhya R. Signaling repression of transcription by RNA polymerase III in yeast. Prog Nucl Acid Res and Mol Bio. 2004;77:323–353. doi: 10.1016/S0079-6603(04)77009-9. [DOI] [PubMed] [Google Scholar]
- 146.Reinke A, Anderson S, McCaffery JM, Yates J, III, Aronova S, Chu S, Fairclough S, Iverson C, Wedaman KP, Powers T. TOR complex 1 includes a novel component, Tco89p (YPL180w), and cooperates with Ssd1p to maintain cellular integrity in Saccharomyces cerevisiae. J Biol Chem. 2004;279:14752–14762. doi: 10.1074/jbc.M313062200. [DOI] [PubMed] [Google Scholar]
- 147.Jansen JM, Wanless AG, Seidel CW, Weiss EL. Cbk1 regulation of the RNA-binding protein Ssd1 integrates cell fate with translational control. Curr Biol. 2009;19:2114–2120. doi: 10.1016/j.cub.2009.10.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Yan G, Lai Y, Jiang Y. The TOR complex 1 is a direct target of Rho1 GTPase. Mol Cell. 2012;45:743–753. doi: 10.1016/j.molcel.2012.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Levin DE. Regulation of cell wall biogenesis in Saccharomyces cerevisiae: the cell wall integrity signaling pathway. Genetics. 2011;189:1145–1175. doi: 10.1534/genetics.111.128264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Mizuta K, Warner JR. Continued functioning of the secretory pathway is essential for ribosome synthesis. Mol Cell Biol. 1994;14:2493–2502. doi: 10.1128/mcb.14.4.2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Li Y, Moir RD, Sethy-Coraci IK, Warner JR, Willis IM. Repression of ribosome and tRNA synthesis in secretion-defective cells is signaled by a novel branch of the cell integrity pathway. Mol Cell Biol. 2000;20:3843–3851. doi: 10.1128/mcb.20.11.3843-3851.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Nierras CR, Warner JR. Protein kinase C enables the regulatory circuit that connects membrane synthesis to ribosome synthesis in Saccharomyces cerevisiae. J Biol Chem. 1999;274:13235–13241. doi: 10.1074/jbc.274.19.13235. [DOI] [PubMed] [Google Scholar]
- 153.Levin DE. Cell wall integrity signaling in Saccharomyces cerevisiae. Microbiol Mol Biol Rev. 2005;69:262–291. doi: 10.1128/MMBR.69.2.262-291.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Denis V, Cyert MS. Molecular analysis reveals localization of Saccharomyces cerevisiae protein kinase C to sites of polarized growth and Pkc1p targeting to the nucleus and mitotic spindle. Eukaryot Cell. 2005;4:36–45. doi: 10.1128/EC.4.1.36-45.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Anastasia SD, Nguyen DL, Thai V, Meloy M, MacDonough T, Kellogg DR. A link between mitotic entry and membrane growth suggests a novel model for cell size control. J Cell Biol. 2012;197:89–104. doi: 10.1083/jcb.201108108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Torres J, Di Como CJ, Herrero E, De La Torre-Ruiz MA. Regulation of the cell integrity pathway by rapamycin-sensitive TOR function in budding yeast. J Biol Chem. 2002;277:43495–43504. doi: 10.1074/jbc.M205408200. [DOI] [PubMed] [Google Scholar]
- 157.St Denis NA, Litchfield DW. Protein kinase CK2 in health and disease: From birth to death: the role of protein kinase CK2 in the regulation of cell proliferation and survival. Cell Mol Life Sci. 2009;66:1817–1829. doi: 10.1007/s00018-009-9150-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Salvi M, Sarno S, Cesaro L, Nakamura H, Pinna LA. Extraordinary pleiotropy of protein kinase CK2 revealed by weblogo phosphoproteome analysis. Biochim Biophys Acta. 2009;1793:847–859. doi: 10.1016/j.bbamcr.2009.01.013. [DOI] [PubMed] [Google Scholar]
- 159.Filhol O, Cochet C. Protein kinase CK2 in health and disease: Cellular functions of protein kinase CK2: a dynamic affair. Cell Mol Life Sci. 2009;66:1830–1839. doi: 10.1007/s00018-009-9151-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Tripodi F, Cirulli C, Reghellin V, Brambilla L, Marin O, Coccetti P. Nutritional modulation of CK2 in Saccharomyces cerevisiae: regulating the activity of a constitutive enzyme. Mol Cell Biochem. 2011;356:269–275. doi: 10.1007/s11010-011-0958-3. [DOI] [PubMed] [Google Scholar]
- 161.Kubinski K, Domanska K, Sajnaga E, Mazur E, Zielinski R, Szyszka R. Yeast holoenzyme of protein kinase CK2 requires both beta and beta’ regulatory subunits for its activity. Mol Cell Biochem. 2007;295:229–236. doi: 10.1007/s11010-006-9292-6. [DOI] [PubMed] [Google Scholar]
- 162.Meggio F, Pinna LA. One-thousand-and-one substrates of protein kinase CK2? FASEB J. 2003;17:349–368. doi: 10.1096/fj.02-0473rev. [DOI] [PubMed] [Google Scholar]
- 163.Hockman DJ, Schultz MC. Casein kinase II is required for efficient transcription by RNA polymerase III. Mol Cell Biol. 1996;16:892–898. doi: 10.1128/mcb.16.3.892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Maldonado E, Allende JE. Phosphorylation of yeast TBP by protein kinase CK2 reduces its specific binding to DNA. FEBS Lett. 1999;443:256–260. doi: 10.1016/s0014-5793(98)01734-7. [DOI] [PubMed] [Google Scholar]
- 165.Lee K, Du C, Horn M, Rabinow L. Activity and autophosphorylation of LAMMER protein kinases. J Biol Chem. 1996;271:27299–27303. doi: 10.1074/jbc.271.44.27299. [DOI] [PubMed] [Google Scholar]
- 166.Bullock AN, Das S, Debreczeni JE, Rellos P, Fedorov O, Niesen FH, Guo K, Papagrigoriou E, Amos AL, Cho S, Turk BE, Ghosh G, Knapp S. Kinase domain insertions define distinct roles of CLK kinases in SR protein phosphorylation. Structure. 2009;17:352–362. doi: 10.1016/j.str.2008.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Prasad J, Manley JL. Regulation and Substrate Specificity of the SR Protein Kinase Clk/Sty. Mol Cell Biol. 2003;23:4139–4149. doi: 10.1128/MCB.23.12.4139-4149.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Nikolakaki E, Du C, Lai J, Giannakouros T, Cantley L, Rabinow L. Phosphorylation by LAMMER protein kinases: determination of a consensus site, identification of in vitro substrates, and implications for substrate preferences. Biochemistry. 2002;41:2055–2066. doi: 10.1021/bi011521h. [DOI] [PubMed] [Google Scholar]
- 169.Moeslein FM, Myers MP, Landreth GE. The CLK family kinases, CLK1 and CLK2, phosphorylate and activate the tyrosine phosphatase, PTP-1B. J Biol Chem. 1999;274:26697–26704. doi: 10.1074/jbc.274.38.26697. [DOI] [PubMed] [Google Scholar]
- 170.Kang WH, Park YH, Park HM. The LAMMER kinase homolog, Lkh1, regulates Tup transcriptional repressors through phosphorylation in Schizosaccharomyces pombe. J Biol Chem. 2010;285:13797–13806. doi: 10.1074/jbc.M110.113555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Rodgers JT, Haas W, Gygi SP, Puigserver P. Cdc2-like Kinase 2 Is an Insulin-Regulated Suppressor of Hepatic Gluconeogenesis. Cell Metabolism. 2010;11:23–34. doi: 10.1016/j.cmet.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Rodgers JT, Vogel RO, Puigserver P. Clk2 and B56[beta] Mediate Insulin-Regulated Assembly of the PP2A Phosphatase Holoenzyme Complex on Akt. Molecular Cell. 2011;41:471–479. doi: 10.1016/j.molcel.2011.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Kaidanovich-Beilin O, Woodgett JR. GSK-3: Functional Insights from Cell Biology and Animal Models. Front Mol Neurosci. 2011;4:40. doi: 10.3389/fnmol.2011.00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Cohen P, Frame S. The renaissance of GSK3. Nat Rev Mol Cell Biol. 2001;2:769–776. doi: 10.1038/35096075. [DOI] [PubMed] [Google Scholar]
- 175.Xu C, Kim NG, Gumbiner BM. Regulation of protein stability by GSK3 mediated phosphorylation. Cell Cycle. 2009;8:4032–4039. doi: 10.4161/cc.8.24.10111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Hirata Y, Andoh T, Asahara T, Kikuchi A. Yeast glycogen synthase kinase-3 activates Msn2p-dependent transcription of stress responsive genes. Mol Biol Cell. 2003;14:302–312. doi: 10.1091/mbc.E02-05-0247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Kassir Y, Rubin-Bejerano I, Mandel-Gutfreund Y. The Saccharomyces cerevisiae GSK-3 beta homologs. Curr Drug Targets. 2006;7:1455–1465. doi: 10.2174/1389450110607011455. [DOI] [PubMed] [Google Scholar]
- 178.Hwang CS, Varshavsky A. Regulation of peptide import through phosphorylation of Ubr1, the ubiquitin ligase of the N-end rule pathway. Proc Natl Acad Sci U S A. 2008;105:19188–19193. doi: 10.1073/pnas.0808891105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Sents W, Ivanova E, Lambrecht C, Haesen D, Janssens V. The biogenesis of active protein phosphatase 2A holoenzymes: a tightly regulated process creating phosphatase specificity. FEBS J. 2012 doi: 10.1111/j.1742-4658.2012.08579.x. [DOI] [PubMed] [Google Scholar]
- 180.Gentry MS, Hallberg RL. Localization of Saccharomyces cerevisiae protein phosphatase 2A subunits throughout mitotic cell cycle. Mol Biol Cell. 2002;13:3477–3492. doi: 10.1091/mbc.02-05-0065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.van Zyl W, Huang W, Sneddon AA, Stark M, Camier S, Werner M, Marck C, Sentenac A, Broach JR. Inactivation of the protein phosphatase 2A regulatory subunit A results in morphological and transcriptional defects in Saccharomyces cerevisiae. Mol Cell Biol. 1992;12:4946–4959. doi: 10.1128/mcb.12.11.4946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Hombauer H, Weismann D, Mudrak I, Stanzel C, Fellner T, Lackner DH, Ogris E. Generation of active protein phosphatase 2A is coupled to holoenzyme assembly. PLoS Biol. 2007;5:e155. doi: 10.1371/journal.pbio.0050155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Ronne H, Carlberg M, Hu GZ, Nehlin JO. Protein phosphatase 2A in Saccharomyces cerevisiae: effects on cell growth and bud morphogenesis. Mol Cell Biol. 1991;11:4876–4884. doi: 10.1128/mcb.11.10.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Gingras AC, Caballero M, Zarske M, Sanchez A, Hazbun TR, Fields S, Sonenberg N, Hafen E, Raught B, Aebersold R. A novel, evolutionarily conserved protein phosphatase complex involved in cisplatin sensitivity. Mol Cell Proteomics. 2005;4:1725–1740. doi: 10.1074/mcp.M500231-MCP200. [DOI] [PubMed] [Google Scholar]
- 185.Pedruzzi I, Dubouloz F, Cameroni E, Wanke V, Roosen J, Winderickx J, De Virgilio C. TOR and PKA signaling pathways converge on the protein kinase Rim15 to control entry into G0. Mol Cell. 2003;12:1607–1613. doi: 10.1016/s1097-2765(03)00485-4. [DOI] [PubMed] [Google Scholar]
- 186.Lippman SI, Broach JR. Protein kinase A and TORC1 activate genes for ribosomal biogenesis by inactivating repressors encoded by Dot6 and its homolog Tod6. Proc Natl Acad Sci U S A. 2009;106:19928–19933. doi: 10.1073/pnas.0907027106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Budhwar R, Lu A, Hirsch JP. Nutrient control of yeast PKA activity involves opposing effects on phosphorylation of the Bcy1 regulatory subunit. Mol Biol Cell. 2010;21:3749–3758. doi: 10.1091/mbc.E10-05-0388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Searle JS, Schollaert KL, Wilkins BJ, Sanchez Y. The DNA damage checkpoint and PKA pathways converge on APC substrates and Cdc20 to regulate mitotic progression. Nat Cell Biol. 2004;6:138–145. doi: 10.1038/ncb1092. [DOI] [PubMed] [Google Scholar]
- 189.Searle JS, Wood MD, Kaur M, Tobin DV, Sanchez Y. Proteins in the nutrient-sensing and DNA damage checkpoint pathways cooperate to restrain mitotic progression following DNA damage. PLoS Genet. 2011;7:e1002176. doi: 10.1371/journal.pgen.1002176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Voordeckers K, Kimpe M, Haesendonckx S, Louwet W, Versele M, Thevelein JM. Yeast 3-phosphoinositide-dependent protein kinase-1 (PDK1) orthologs Pkh1-3 differentially regulate phosphorylation of protein kinase A (PKA) and the protein kinase B (PKB)/S6K ortholog Sch9. J Biol Chem. 2011;286:22017–22027. doi: 10.1074/jbc.M110.200071. [DOI] [PMC free article] [PubMed] [Google Scholar]