

Abstract
Acute lymphoblastic leukemia (ALL) is an aggressive hematological tumor resulting from the malignant transformation of lymphoid progenitors. Despite intensive chemotherapy, 20% of pediatric and over 50% of adult ALL patients fail to achieve a complete remission or relapse after intensified chemotherapy, making disease relapse and resistance to therapy the most significant challenge in the treatment of this disease1,2. Using whole exome sequencing, here we identify mutations in the cytosolic 5'-nucleotidase II gene (NT5C2), which encodes a 5'-nucleotidase enzyme responsible for inactivation of nucleoside analog chemotherapy drugs, in 20/103 (19%) relapse T-ALLs and in 1/35 (3%) relapse B-precursor ALLs analyzed. NT5C2 mutant proteins show increased nucleotidase activity in vitro and conferred resistance to chemotherapy with 6-mercaptopurine and 6-thioguanine when expressed in ALL lymphoblasts. These results support a prominent role for activating mutations in NT5C2 and increased nucleoside analog metabolism in disease progression and chemotherapy resistance in ALL.
Therapy of ALL includes initial treatment with high dose combination chemotherapy, which obtains clinical and hematologic remission in over 90% of cases. This is typically followed by additional rounds of highly intensive therapy aimed to further reduce disease burden; and then by a 2 year long lower intensity maintenance therapy in which treatment with oral 6-mercaptopurine plays a particularly important role3,4. Patients with relapsed ALL generally receive a more intense treatment. However, despite these efforts, their outcome remains unsatisfactory with cure rates of less than 40%5. This is particularly the case in patients with relapsed T-ALL and in cases with primary resistance or early relapse, which is associated with higher risk of failure to achieve a second complete remission, shorter duration of chemotherapy response and poor survival6,7. Much effort has been spent on the study of the molecular basis of relapse and chemotherapy resistance in ALL. However the specific mechanisms mediating escape from therapy, disease progression and leukemia relapse remain largely unknown. To address this question we performed whole exome sequencing of matched diagnosis, remission and relapse DNA samples from 5 pediatric T-ALL patients (Supplementary Table 1). This analysis identified a mean mutation load of 13 somatic mutations per sample (range 5 – 17) (Supplementary Table 2). Out of 60 somatic mutations identified in total, 17 mutations were present at diagnosis and at relapse, 24 genes were selectively mutated in relapsed T-ALL samples and 19 mutations were present only at diagnosis. Moreover, 4 of the 5 relapsed cases analyzed showed the presence of at least one somatic mutation present also at diagnosis, together with secondary mutations specifically acquired at the time of relapse. In addition, 4 out of the 5 cases showed absence of at least one mutation marker present at diagnosis during disease progression leading to relapse. Single nucleotide polymorphism analysis of exome sequencing results ruled out that loss of these markers was due to loss of heterozygosity at relapse (Supplementary Table 3). This result is consistent with previous studies based on copy number alteration analyses8–10 and supports that relapsed ALLs can originate as derivates of ancestral subclones related to, but distinct from the main leukemic population present at diagnosis.
Somatically mutated genes at diagnosis included known T-ALL tumor suppressor genes such as FBXW711, WT18 and DNM212 in addition to numerous new genes not implicated before in the pathogenesis of this disease. Analysis of mutant alleles found at the time of relapse identified mutations in three genes encoding proteins involved in positive regulation of TP53 signaling, including TP53 itself (TP53 R213Q), BANP (BANP H391Y)13 and RPL11 (RPL11 R18P)14. Notably, mutations in TP53 have been reported in about 10% of relapsed ALL cases and are associated with a particularly poor prognosis15. Given the prominent role of TP53 pathway in DNA damage induced apoptosis16, we performed extended mutation analysis of the TP53, BANP and RPL11 genes in 18 additional diagnostic and relapsed T-ALL samples (Supplementary Table 1). This analysis failed to identify additional TP53 or BANP mutations, but showed the presence two additional somatic RPL11 mutant alleles; one present both at diagnosis and relapse (RPL11 X178Q); and the other one (RPL11 G30fs) specifically mutated at relapse. Relapse-associated mutations also included a prototypical activating mutation in the NRAS oncogene (NRAS G13V). Notably, NRAS mutations in ALL have been associated with poor outcome17 and are particularly prevalent in early T-cell precursor ALLs12,18, a group of high risk leukemias with poor prognosis19. Extended mutation analysis of NRAS in relapsed T-ALL cases demonstrated the presence of 2 diagnostic and relapse sample pairs harboring a prototypical NRAS G12S activating allele and a third patient with a heterozygous activating NRAS G12R mutation, which was present at diagnosis and showed loss of heterozygosity at the time of relapse.
However, the most remarkable finding in our exome sequence analysis was the presence of a relapse-associated heterozygous mutation in the NT5C2 gene (NT5C2 K359Q). NT5C2 is a ubiquitous enzyme responsible for the final dephosphorylation of 6-hydroxypurine nucleotide monophosphates such as IMP, dIMP, GMP, dGMP and XMP before they can be exported out of the cell20,21. In addition, and most notably, NT5C2 can also dephosphorylate and inactivate 6-thioinositol monophosphate and 6-thioguanosine monophosphate which mediate the cytotoxic effects of 6-mercaptopurine (6-MP) and 6-thioguanine (6-TG)22, two nucleoside analogs commonly used in the treatment of ALL. Mutation analysis of an extended panel of 98 relapse T-ALL (Supplementary Table 1) and 35 relapse B-precursor ALL samples (Supplementary Table 1) identified 22 additional mutations T-ALL and one additional NT5C2 mutation in a B-precursor ALL patient in first relapse (Fig. 1 and Supplementary Table 4). Strikingly, 13 of these samples harbored the same NT5C2 R367Q mutation, 4 cases showed a recurrent NT5C2 R238W mutation and 2 samples harbored a L375F single amino acid substitution (Fig. 1 and Supplementary Table 4). In each of the 9 cases for which original diagnostic DNA was available for analysis, NT5C2 mutations showed to be specifically acquired at the time of relapse. No NT5C2 mutations were identified in 23 T-ALL and 27 B-precursor ALL additional diagnostic samples, further supporting the specific association of NT5C2 mutations with relapsed disease. Analysis of clinical and molecular features associated with NT5C2 mutant relapsed T-ALLs treated in Berlin Frankfurt Münster (BFM) group based clinical trials (ALL-BFM 95, ALL-BFM 2000, COALL 06-97, NHL-BFM 95 and Euro-LB 02) (Supplementary Table 1) showed an association of NT5C2 mutations with early disease recurrence (very early or early relapse vs. late relapse, P <0.05) and relapse under treatment (P = 0.002) independently of treatment protocol (Supplementary Tables 5–10).
Figure 1. NT5C2 mutations in relapsed pediatric T-ALL.

(a) Schematic representation of the structure of the NT5C2 protein. The haloacid dehalogenase (HAD) and the substrate binding domains (SB) are indicated. NT5C2 mutations identified in relapsed pediatric samples are shown. Filled circles represent heterozygous mutations. Multiple circles in the same amino acid position account for multiple patients with the same variant. (b) DNA sequencing chromatograms of paired diagnosis and relapse genomic T-ALL DNA samples showing representative examples of relapse specific heterozygous NT5C2 mutations, with the mutant allele sequence highlighted in red.
Given the described role of NT5C2 in the metabolism and inactivation of nucleoside analog drugs22–24; the recurrent finding of the NT5C2 R367Q, NT5C2 R238W and NT5C2 L375F alleles; and the reported association of increased levels of nucleotidase activity with thiopurine resistance and worse clinical outcome25, we hypothesized that relapse-associated NT5C2 mutations may represent gain of function alleles with increased enzymatic activity. Detailed structure-function analysis of the NT5C2 K359Q mutation further supported this hypothesis. Thus, comparison of the wild type NT5C2 structure and models of the mutant NT5C2 K359Q protein show that this mutation could result in increased NT5C2 activity by mimicking the effect of positive allosteric regulators (Fig. 2a). Allosteric activation of NT5C2 is mediated by binding of ATP, dATP, Ap4A and 2,3-BPG to an allosteric pocket proximal to the NT5C2 active site (Fig. 2a). Occupancy of this regulatory site results in increased ordering of an alpha helix formed by residues G355-E364 (Helix A), which in turn displaces F354 from the catalytic center and moves D356 into the active site of the protein (Fig. 2b,c). Similarly, our model predicts that the NT5C2 K359Q mutation could increase the Helix A stability and reduce its solvent accessibility, resulting in an active configuration with displacement of F354 out of the NT5C2 active site and positioning D356 into the catalytic center of the enzyme (Fig. 2b–e). Consistent with this prediction, 5'-nucleotidase assays using NT5C2 K359Q recombinant protein demonstrated a 48-fold increase in enzymatic activity compared wild type NT5C2 (Fig. 3). An additional structurally interesting allele is the NT5C2 Q523* nonsense mutation, which removes an inhibitory region located in the C-terminal segment of the NT5C2 protein26. In addition, and despite the absence of clear structural cues suggesting a role of other mutations in NT5C2 activation, nucleotidase activity analysis of NT5C2 R367Q and NT5C2 D407A mutant proteins revealed an 18 fold and a 16 fold increase in their 5'-IMP nucleotidase activity compared with wild type NT5C2, respectively (Fig. 3).
Figure 2. Structure-function analysis of the NT5C2 K359Q mutant protein.

(a) Molecular surface representation of NT5C2 protein structure. The position of the NT5C2 K359Q mutation found is highlighted in red. The substrate inosine monophosphate (IMP) is depicted in purple; the ATP allosteric activator is shown in yellow. (b) Structure representation of the NT5C2 catalytic center and allosteric regulatory site devoid of substrate or ligands (PDB 2XCX). (c) Structure representation of the NT5C2 catalytic center and allosteric regulatory site bound to IMP and ATP, respectively (PDB 2XCW). (d) Structure representation of the NT5C2 K359Q mutant model corresponding to the catalytic center and allosteric regulatory sites. (e) Overlay of the structures shown in b–d. The white arrow indicates the repositioning of Phe354 from the inactive NT5C2 configuration to the active –ATP-bound NT5C2 and NT5C2 K359Q– structures. Mg2+ ions are depicted as green spheres.
Figure 3. Increased 5'-IMP nucleotidase activity in NT5C2 mutant proteins.

5'-Nucleotidase activity levels of recombinant mutant proteins relative to wild type NT5C2 control are shown. Data are means ± s.d.
Finally, and to formally test the role of NT5C2 mutations in chemotherapy resistance we analyzed the effects of wild type and relapse-associated mutant NT5C2 expression in the response of CCRF-CEM T-ALL cells to 6-mercaptopurine (6-MP) and 6-thyogunanine (6-TG) (Fig. 4). Cell viability analysis in the presence of increased drug concentrations demonstrated increased resistance to 6-MP and 6-TG therapy in cells expressing NT5C2 K359Q, NT5C2 R367Q and NT5C2 D407A compared with empty vector and wild type NT5C2 controls (Fig. 4, Supplementary Fig. 2 and Supplementary Table 11). Similar results were obtained in the CUTLL1 T-ALL cell line (Fig. 4, Supplementary Fig. 2 and Supplementary Table 11). Finally, we tested the effects of relapsed-associated NT5C2 mutations in the response to nelarabine – an AraG precursor highly active in relapsed T-ALL– and AraG 27–30. Strikingly, both nelarabine and AraG showed to be equally active in cells expressing relapse-associated NT5C2 mutations compared to controls (Supplementary Fig. 3).
Figure 4. Expression of NT5C2 mutations in ALL cells induces resistance to chemotherapy with 6-MP and 6-TG.
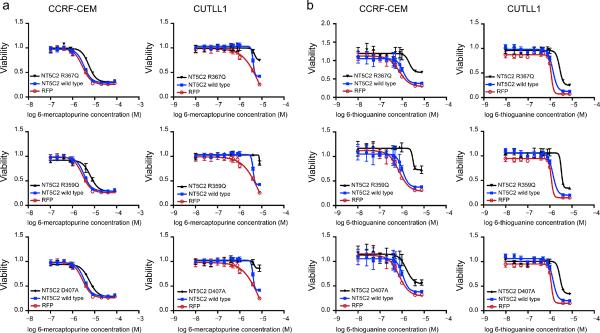
(a) Viability assays in CCRF-CEM and CUTLL1 T-ALL cells expressing wild type NT5C2, relapse-associated mutant NT5C2 alleles or a red fluorescent protein control (RFP), treated with increased concentrations of 6-mercaptopurine (6-MP). (b) 6-Thioguanine (6-TG) dose response cell viability curves. Data is shown as means ± s.d.
Prolonged maintenance treatment with 6-mercaptopurine is essential to obtain durable remissions in the treatment of ALL3,4. Indeed, low-adherence to 6-mercaptopurine treatment, defined as less than 95% compliance, results in increased relapsed rates and may account for as much as 59% of all ALL relapses31. In this context, our results highlight the prominent role of relapse-specific mutations in NT5C2 as a mechanism of resistance to 6-MP and a genetic driver of relapse in ALL. In addition, and most notably, the lack of nelarabine cross resistance in cells expressing activating NT5C2 alleles analyzed here suggests that these mutations may not impair the effectiveness of nelarabine-based salvage therapies in relapsed T-ALL.
Supplementary Material
ACKNOWLEDGEMENTS
We thank A. A. Da Silva for technical support in the production of recombinant NT5C2 proteins and R. Parsons for insightful comments and advice. We also thank P. H. Wiernik as ECOG leukemia committee chair at the time the E2993 study was initiated. This work was supported by the St. Baldrick's Foundation (A.F.), the Partnership for Cure Foundation (R.R), the Innovative Research Award by the Stand Up to Cancer Foundation (A.F.) the Chemotherapy Foundation (A.F), the Leukemia & Lymphoma Society Scholar Award (A.F.), the German Federal Ministry for Education and Research in the National Genome Research Network (grant 01GS0870 to RK-S); the German Foundation for Childhood Cancer (grants no. DKS 2003.08 and 2007.02 to RK-S), the National Institutes of Health (U24 CA114737 to E.P) and the ECOG Leukemia Tissue Bank. A. P. G. is a postdoctoral researcher funded by the Rally Foundation.
Footnotes
AUTHOR CONTRIBUTIONS GT and APG performed validation and recurrence mutation analysis, enzymatic activity and cell drug resistance assays; ZC performed structure function analysis and analyzed Illumina sequence data; HK analyzed Illumina sequence data; VT analyzed genomic data from diagnostic and relapse T-ALLs; MA performed validation analysis of Illumina sequencing results; MP, GB, EP, JR, JH and RK-S contributed clinical samples and clinical correlative information; TP directed and supervised mutation analysis; RR directed and supervised the analysis of Illumina sequencing data; AF designed the study, directed and supervised research and wrote the manuscript.
Methods are included in the supplementary material
REFRENCES
- 1.Moricke A, et al. Long-term results of five consecutive trials in childhood acute lymphoblastic leukemia performed by the ALL-BFM study group from 1981 to 2000. Leukemia. 2010;24:265–284. doi: 10.1038/leu.2009.257. [DOI] [PubMed] [Google Scholar]
- 2.Salzer WL, et al. Long-term results of the pediatric oncology group studies for childhood acute lymphoblastic leukemia 1984–2001: a report from the children's oncology group. Leukemia. 2010;24:355–370. doi: 10.1038/leu.2009.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Koren G, et al. Systemic exposure to mercaptopurine as a prognostic factor in acute lymphocytic leukemia in children. N Engl J Med. 1990;323:17–21. doi: 10.1056/NEJM199007053230104. [DOI] [PubMed] [Google Scholar]
- 4.Relling MV, Hancock ML, Boyett JM, Pui CH, Evans WE. Prognostic importance of 6-mercaptopurine dose intensity in acute lymphoblastic leukemia. Blood. 1999;93:2817–2823. [PubMed] [Google Scholar]
- 5.Pui CH, Evans WE. Treatment of acute lymphoblastic leukemia. N Engl J Med. 2006;354:166–178. doi: 10.1056/NEJMra052603. [DOI] [PubMed] [Google Scholar]
- 6.Tallen G, et al. Long-term outcome in children with relapsed acute lymphoblastic leukemia after time-point and site-of-relapse stratification and intensified short-course multidrug chemotherapy: results of trial ALL-REZ BFM 90. J Clin Oncol. 2010;28:2339–2347. doi: 10.1200/JCO.2009.25.1983. [DOI] [PubMed] [Google Scholar]
- 7.Gaynon PS, et al. Survival after relapse in childhood acute lymphoblastic leukemia: impact of site and time to first relapse--the Children's Cancer Group Experience. Cancer. 1998;82:1387–1395. doi: 10.1002/(sici)1097-0142(19980401)82:7<1387::aid-cncr24>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 8.Tosello V, et al. WT1 mutations in T-ALL. Blood. 2009;114:1038–1045. doi: 10.1182/blood-2008-12-192039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mullighan CG, et al. Genomic analysis of the clonal origins of relapsed acute lymphoblastic leukemia. Science. 2008;322:1377–1380. doi: 10.1126/science.1164266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clappier E, et al. Clonal selection in xenografted human T cell acute lymphoblastic leukemia recapitulates gain of malignancy at relapse. J Exp Med. 2011;208:653–661. doi: 10.1084/jem.20110105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thompson BJ, et al. The SCFFBW7 ubiquitin ligase complex as a tumor suppressor in T cell leukemia. J Exp Med. 2007;204:1825–1835. doi: 10.1084/jem.20070872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang J, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012;481:157–163. doi: 10.1038/nature10725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jalota A, et al. Tumor suppressor SMAR1 activates and stabilizes p53 through its arginine-serine-rich motif. J Biol Chem. 2005;280:16019–16029. doi: 10.1074/jbc.M413200200. [DOI] [PubMed] [Google Scholar]
- 14.Zhang Y, et al. Ribosomal protein L11 negatively regulates oncoprotein MDM2 and mediates a p53-dependent ribosomal-stress checkpoint pathway. Mol Cell Biol. 2003;23:8902–8912. doi: 10.1128/MCB.23.23.8902-8912.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hof J, et al. Mutations and deletions of the TP53 gene predict nonresponse to treatment and poor outcome in first relapse of childhood acute lymphoblastic leukemia. J Clin Oncol. 2011;29:3185–3193. doi: 10.1200/JCO.2011.34.8144. [DOI] [PubMed] [Google Scholar]
- 16.Bunz F, et al. Disruption of p53 in human cancer cells alters the responses to therapeutic agents. J Clin Invest. 1999;104:263–269. doi: 10.1172/JCI6863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lubbert M, et al. N-ras gene point mutations in childhood acute lymphocytic leukemia correlate with a poor prognosis. Blood. 1990;75:1163–1169. [PubMed] [Google Scholar]
- 18.Van Vlierberghe P, et al. ETV6 mutations in early immature human T cell leukemias. J Exp Med. 2011;208:2571–2579. doi: 10.1084/jem.20112239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Coustan-Smith E, et al. Early T-cell precursor leukaemia: a subtype of very high-risk acute lymphoblastic leukaemia. Lancet Oncol. 2009;10:147–156. doi: 10.1016/S1470-2045(08)70314-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Oka J, Matsumoto A, Hosokawa Y, Inoue S. Molecular cloning of human cytosolic purine 5'-nucleotidase. Biochem Biophys Res Commun. 1994;205:917–922. doi: 10.1006/bbrc.1994.2752. [DOI] [PubMed] [Google Scholar]
- 21.Spychala J, Madrid-Marina V, Fox IH. High Km soluble 5'-nucleotidase from human placenta. Properties and allosteric regulation by IMP and ATP. J Biol Chem. 1988;263:18759–18765. [PubMed] [Google Scholar]
- 22.Brouwer C, et al. Role of 5'-nucleotidase in thiopurine metabolism: enzyme kinetic profile and association with thio-GMP levels in patients with acute lymphoblastic leukemia during 6-mercaptopurine treatment. Clin Chim Acta. 2005;361:95–103. doi: 10.1016/j.cccn.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 23.Hunsucker SA, Mitchell BS, Spychala J. The 5'-nucleotidases as regulators of nucleotide and drug metabolism. Pharmacol Ther. 2005;107:1–30. doi: 10.1016/j.pharmthera.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 24.Galmarini CM, Jordheim L, Dumontet C. Role of IMP-selective 5'-nucleotidase (cNII) in hematological malignancies. Leuk Lymphoma. 2003;44:1105–1111. doi: 10.1080/1042819031000077142. [DOI] [PubMed] [Google Scholar]
- 25.Pieters R, et al. Relation of 5'-nucleotidase and phosphatase activities with immunophenotype, drug resistance and clinical prognosis in childhood leukemia. Leuk Res. 1992;16:873–880. doi: 10.1016/0145-2126(92)90033-4. [DOI] [PubMed] [Google Scholar]
- 26.Allegrini S, et al. Mechanistic studies on bovine cytosolic 5'-nucleotidase II, an enzyme belonging to the HAD superfamily. Eur J Biochem. 2004;271:4881–4891. doi: 10.1111/j.1432-1033.2004.04457.x. [DOI] [PubMed] [Google Scholar]
- 27.Sanford M, Lyseng-Williamson KA. Nelarabine. Drugs. 2008;68:439–447. doi: 10.2165/00003495-200868040-00004. [DOI] [PubMed] [Google Scholar]
- 28.Berg SL, et al. Phase II study of nelarabine (compound 506U78) in children and young adults with refractory T-cell malignancies: a report from the Children's Oncology Group. J Clin Oncol. 2005;23:3376–3382. doi: 10.1200/JCO.2005.03.426. [DOI] [PubMed] [Google Scholar]
- 29.Gokbuget N, et al. High single-drug activity of nelarabine in relapsed T-lymphoblastic leukemia/lymphoma offers curative option with subsequent stem cell transplantation. Blood. 2011;118:3504–3511. doi: 10.1182/blood-2011-01-329441. [DOI] [PubMed] [Google Scholar]
- 30.DeAngelo DJ, et al. Nelarabine induces complete remissions in adults with relapsed or refractory T-lineage acute lymphoblastic leukemia or lymphoblastic lymphoma: Cancer and Leukemia Group B study 19801. Blood. 2007;109:5136–5142. doi: 10.1182/blood-2006-11-056754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bhatia S, et al. Nonadherence to oral mercaptopurine and risk of relapse in Hispanic and non-Hispanic white children with acute lymphoblastic leukemia: a report from the children's oncology group. J Clin Oncol. 2012;30:2094–2101. doi: 10.1200/JCO.2011.38.9924. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.