

Abstract
Celiac disease (CD) is an immune-mediated, inflammatory disorder of the small intestines with a defined genetic etiological component associated with the expression of HLA-DQ2 and/or HLA-DQ8 haplotypes. The dietary consumption of gluten-rich cereals triggers a gluten-specific immune response in genetically susceptible individuals leading to a spectrum of clinical manifestations ranging from an inapparent subclinical disease, to overt enteropathy that can in some individuals progress to enteropathy-associated T cell lymphoma (EATL). The tissue-destructive pathologic process of CD is driven by activated NK-like intraepithelial CD8+ lymphocytes and the proinflammatory cytokine IL-15 has emerged to be pivotal in orchestrating this perpetual tissue destruction and inflammation. Moreover, transgenic mice that over-express human IL-15 from an enterocyte-specific promoter (T3b-hIL-15 Tg) recapitulate many of the disease-defining T and B cell-mediated pathologic features of CD, further supporting the evolving consensus that IL-15 represents a valuable target in devising therapeutic interventions against the form of the disease that is especially refractory to gluten-free diet. In the present study, we evaluated the potential efficacy of tofacitinib, a pan-JAK inhibitor that abrogates IL-15 signaling, as a therapeutic modality against CD using T3b-hIL-15 Tg mice. We demonstrate that tofacitinib therapy leads to a lasting reversal of pathologic manifestations in the treated mice, thereby highlighting the potential value of tofacitininb as a therapeutic modality against refractory CD for which no effective therapy exists currently. Additionally, the visceral adiposity observed in the tofacitinib-treated mice underscores the importance of continued evaluation of the drug's impact on the lipid metabolism.
Introduction
Celiac disease is an immune-mediated, inflammatory disorder of the small intestine with well-defined genetic and dietary components involved in its pathogenesis. The prevalence of this disorder appears to be highest in the western hemisphere approaching 1.0–1.5% of the general population [1]. The expression of HLA-DQ2 and/or HLA-DQ8 haplotypes is near universal in the afflicted individuals, although an array of other non-HLA genes has also been implicated to be contributory in the susceptibility to CD [reviewed in refs. 2–5]. The dietary consumption of gluten-rich cereals triggers a robust anti-gluten immune response in genetically susceptible individuals resulting in a spectrum of clinical manifestations ranging from inapparent disease to overt malabsorptive enteropathy, with some individuals even progressing to develop intestinal malignancies (EATL) that carry a poor prognosis [6]. Extra-intestinal manifestations such as dermatitis herpetiformis or ataxia are also occasionally seen clinically [2]. Although CD4+ T cell driven anti-gluten immune responses lead to accumulation of inflammatory mediators such as IFN-gamma as well as B cell expansion in situ with the production of antibodies to gluten/gliadin and tissue trans-glutaminase (TTG), the role of these entities in the actual tissue-destructive pathologic process of CD however, remains obscure [4]. On the other hand, one of the disease defining features of CD is the massive influx of CD8+ intraepithelial lymphocytes (IEL) and these infiltrating IEL display an array of NK-like activation markers including NKG2D but are devoid of any demonstrable gluten-specificity. It is these infiltrating IEL that cause extensive tissue damage in the affected intestinal mucosa via T cell receptor (TCR)-independent by-stander mechanisms with the involvement of NKG2D and other co-activating NK cell receptors [7, 8].
In CD patients the over-expression of the proinflammatory cytokine IL-15 is a consistent feature in the affected small intestinal mucosa although the underlying mechanisms that trigger local over-expression of IL-15 remain to be elucidated. Nonetheless, accumulating evidence suggests that the locally expressed IL-15 acts as a central driver that orchestrates and perpetuates CD8+ T cell-mediated tissue destruction in CD. Specifically, not only is IL-15 an essential growth factor for the maintenance and proliferation of IEL, but it also enhances their cytolytic activity including the reprogramming of these cells phenotypically to be LAK or NK-like CD8+ effectors that undergo oligoclonal expansion resisting activation-induced apoptosis, to cause extensive epithelial and submucosal tissue destruction leading to luminal and trans-mural inflammatory lesions [9 and refs cited therein]. IL-15 promotes further perpetuation and potentiation of intestinal tissue destruction by the induction of NKG2D expression in IEL effectors and by the up-regulation of its cognate ligands MICA, MICB, ULBPs and HLA-E on enterocytes [7]. In addition, the aberrant over-expression of IL-15 also disrupts the immune homeostasis in the gut by disabling Smad-dependent TGF-beta signaling that is pivotal in maintaining an anti-inflammatory milieu and also subverts the generation of retinoic acid/TGF-beta dependent Tregs in the intestinal mucosa resulting in the breaching of immune tolerance to dietary antigens [10–12].
Paralleling these IL-15 mediated effects seen in the human disease, the hyper-expression of human IL-15 driven from an enterocyte-specific promoter in a transgenic mouse model (T3b-hIL-15 Tg) recapitulates many of the disease defining pathologic features of CD [9, 13, 14]. Although, as expected these T3b-hIL-15 Tg mice do not show gluten sensitivity, but display inflammatory pathologic lesions with extensive infiltration of NKG2D-expressing CD8+ IEL along with profound blunting and atrophy of intestinal villi that are anatomically confined to the duodeno-jejunal region of the small intestines. In addition, the presence of hypergammaglobulinemia with auto-antibodies including those against tissue-transglutaminase 2, and the accumulation of plasma cells in the lamina propria of the affected intestinal mucosa of these T3b-hIL-15 Tg mice collectively represent both T and B cell mediated effects of CD in this mouse model [9, 13]. The recapitulation of most of the disease-defining pathologic manifestations in T3b-hIL-15 Tg mice therefore further strengthen the notion that the locally over-expressed IL-15 is indeed a central driver of the inflammatory pathology as opposed to being a mere correlate of the disease. Equally important is the fact that these observations collectively make a compelling rationale that targeting IL-15 represents an attractive approach in developing therapies for CD that is especially refractory to dietary gluten withdrawal.
IL-15 signals through a tripartite receptor complex, consisting of IL-15Rα, a private receptor subunit that binds IL-15 with high affinity, along with CD122 (IL-2/IL-15Rβ) and CD132 (the common γc chain) that together form the primary signaling module of the receptor complex that transduces IL-15 mediated effects in responsive cells. The protein tyrosine kinases JAK1 and JAK3 (Janus Kinase-1 and -3) that associate with CD122 and CD132 respectively, are two pivotal scaffolds that integrate signal transduction via this tripartite IL-15 receptor complex [reviewed in refs 15, 16]. In the present study we evaluated the potential efficacy of tofacitinib, an orally active small molecule pan-JAK inhibitor that abrogates IL-15 signaling, with a favorable benefit-risk profile in several late-stage trials [17–19] and was approved by the FDA recently for treating rheumatoid arthritis, as a therapeutic modality against CD using T3b-hIL-15 Tg mice. We demonstrate that a 42-day course of tofacitinib therapy leads to a lasting reversal of the pathologic manifestations in the treated mice, thereby highlighting the potential value of tofacitininb as a therapeutic modality against refractory CD for whom no effective therapy exists currently.
Materials and Methods
Mice, drugs and devices
The generation of T3b-hIL-15 Tg mice have been reported previously [14] and female mice that were 4 months of age were used in most experiments unless indicated otherwise. All animal experiments were approved by the institutional animal care and use committee of Tokyo Metropolitan Institute of Medical Science. Tofacitinib was purchased from LC Laboratories, Woburn, MA, and was dissolved in a sterile solution of 50% DMSO, 10% PEG 300 and 40% water. For in vivo delivery of tofacitinib, subcutaneously placed (dorsally between the scapulae) ALZET mini osmotic pumps (model 2006) from Durect Corp, Cupertino, CA were used to deliver a dose of 30 mg/kg/day for 42 days.
Flow cytometry
For phenotypic characterization of various lymphocyte subsets, lymphocytes were isolated by Ficoll density gradient centrifugation either from blood collected from the retro-orbital plexus of living animals or from spleens harvested from euthanized mice. For certain experiments we also isolated lymphocytes from the intraepithelial (IEL) or laminar proprial (LPL) compartments as previously described [14]. Flow cytometric analysis was performed using a BD FACSCanto II workstation with Flo Jo data analysis software (Tree Star, Ashland, OR).
Tissue staining and histology
For histologic assessment, tissues from small intestines were first fixed with 4% para-formadehyde, followed by embedding in paraffin. Four micrometer sections were cut and fixed onto slides prior to deparaffinization. Afixed, deparaffinized sections were then stained with hematoxylin and eosin. For immunohistochemical assessment of infiltrating CD3+ T cells, deparaffinized tissue sections were dehydrated through xylene and ethanol followed by incubation with a rabbit polyclonal antibody against CD3 (cat#ab5690) from abcam Inc (Cambridge, MA). Immunoreactive cells were visualized with MACH4 universal HRP-polymer kit with DAB (Biocare Medical, Concord, CA) according to manufacturer's instructions. Sections were counterstained with hematoxylin lightly. For morphological assessment, stained sections were examined by light microscopy.
Statistics
All data are expressed as mean ± SD and Student's t test was used for analysis of data and a P value less than 0.05 was considered significant.
Results
In T3b-hIL-15 Tg mice, the human IL-15 trans-gene expression is strictly confined to the intestinal enterocytes as has been demonstrated previously [14]. However, there is considerable seepage of enterocyte-expressed IL-15 into the circulation that can be readily detected in the sera of these mice [9]. Associated with these elevated levels of circulatory IL-15 in T3b-hIL-15 Tg mice there is a concordant expansion of CD8+ T cells in the peripheral blood of these transgenic mice which provides a reliable biomarker to monitor the impact of administered tofacitinib longitudinally over the course of the treatment. One group of T3b-hIL-15 Tg mice (n=6) that were 4 months of age were treated with tofacitinib while another group of 6 animals was sham-treated with the vehicle control. It should be noted that at 4 months of age these transgenic mice display both microscopic and macroscopic pathologic lesions in the small intestines with 100% penetrance. At this age, T3b-hIL-15 Tg mice also show signs of abdominal discomfort and thus necessitated us to deliver tofacitinib via implanted pumps rather than twice daily by oral gavage to minimize the stress of frequent handling. The implanted pumps delivered a dose of tofacitinib that was equivalent to the dose that had been used in several phase III human trials (30 mg/kg/day) for period of 42 days [17].
The impact of tofacitinib was evident by 10 days post treatment by the assessment of peripheral blood cell subset profiles in the treated animals in comparison to their pre-treatment profiles by flow cytometry. As shown in Fig. 1, there was a substantial reduction in the percentage of circulating NK cells with the CD3−NK1.1+ phenotype at this early time point (5.3% ± 1.6 SD in pre-treatment versus 2.1% ± 1.8 SD at day 10 post-treatment, p=0.004) consistent with their exquisite requirement for IL-15 for survival and maintenance. On the other hand, the impact of tofacitinib on the CD3+ cell subset was modest at this early time point (81.4% ± 4.1 SD before initiation of therapy versus 64.9± 4.5 SD day 10 post-treatment; p=0.08) and this reduction was primarily confined to CD8+ T cells (Supplementary Fig. 1). Unlike its effects on NK cells, the impact of tofacitinib on the CD8+ T cell subset was however, more gradual (see Supplementary Fig. 1). Nonetheless, at the conclusion of tofacitinib therapy, the peripheral blood cell profiles as assessed by flow cytometry revealed a dramatic picture. The tofacitinib treatment regimen profoundly affected the expanded CD8+ T cell subset in the T3b-hIL-15 Tg mice leading to normal CD4+ and CD8+ percentage profiles in the peripheral blood of the treated mice (Fig. 2, panel A). The reemergence of the CD4+ T cell subset in normal frequency in the treated mice is due to the unmasking of this subset with the ablation of grossly expanded CD8+ T cells with tofacitinib treatment. It is note-worthy that the major CD8+ T cell subset that was ablated by tofacitinib therapy was the subset that displayed the highest expression levels of NKG2D (Fig. 2, panel B). Importantly, this CD8+ T cell subset with enhanced NKG2D expression is the very same population that is implicated to be the principal driver of pathologic intestinal tissue destruction in CD. Having demonstrated that this putative pathogenic T cell population is ablated in the peripheral blood of tofacitinib-treated mice, we examined whether these CD8+ T cells with NKG2D expression were also eliminated from the proximal small intestinal mucosa. Consistent with what we observed in the peripheral blood, there was a substantial reduction in CD8+NKG2D+ cells in the intraepithelial lymphocytic fraction of the tofacitinib treated mice, namely, 12.6% in sham-treated versus 7.6% in tofacitinib treated mice as shown in Fig. 2, panel C.
Figure 1.
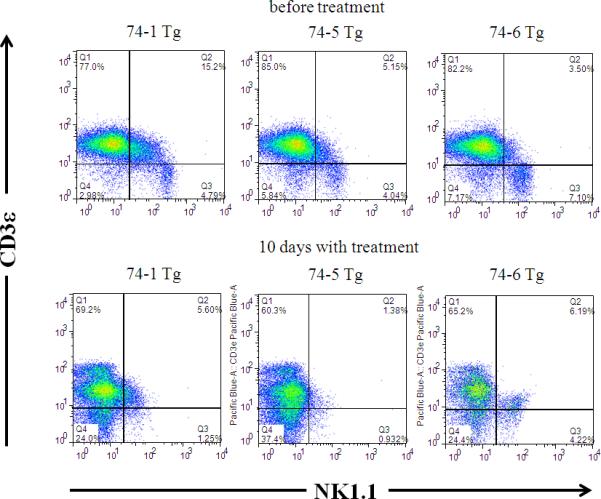
The impact of tofacitinib treatment on NK cells. PBMC were isolated from T3b-hIL-15 Tg mice prior to the initiation tofacitinib therapy and 10 days post-treatment and the impact of the drug on peripheral blood NK cells were assessed by flow cytometry. The modulation of NK cell subsets in three individual mice treated with tofacitinib is shown.
Figure 2.

Ablative effects of Tofacitinib therapy are primarily confined to CD8+ T lymphocytes of T3bhIL-15 Tg mice. After 6 weeks of tofacitinib treatment, the impact of drug therapy on the peripheral blood or intra-epithelial CD3+ T cell subsets in (gated on CD3+ population) was assessed by flow cytometry. For comparison, wild type litter-mates (WT) and sham-treated T3b-hIL-15 Tg mice were also included in the assessment and the data shown are from one representative mouse from each group of 6 mice and the other mice displayed similar profiles. Panel A shows peripheral blood CD4+ and CD8+ T cell subsets; Panel B shows the impact of tofacitinib on peripheral blood CD8+ T cells that express NKG2D; and Panel C shows the impact of tofacitinib on intraepithelial CD8+ T cells that express NKG2D.
From the flow cytometric data shown above it was apparent that tofacitinib therapy was effectively ablating the pathogenic CD8+NKG2D+ T cells that are critically dependent on IL-15 for survival and proliferation. It is important to appreciate that there is a degree of selectivity among different T cell subsets to the inhibitory effects of the pan-JAK inhibitor tofacitinib. As evident in Fig. 2, tofacitinib therapy did not render treated animals lymphopenic, but eliminated only the IL-15 driven activated CD8+ expanded cell population while sparing and retaining normal CD4+ and CD8+ cell numbers in the treated animals thus suggesting perhaps the existence of threshold differences in sensitivity to tofacitinib that is linked to the activation status of a particular cell.
Concordant with the observation that a 42-day regimen of tofacitinib therapy leads to a virtual elimination of CD8+NKG2D+ pathogenic T cells that are incriminated in tissue destruction, on examination of the treated animals following necropsy, the impact of tofacinib treatment on intestinal pathology of these T3b-hIL-15 Tg mice was striking. As has been reported previously [9, 14], in T3b-hIL-15 Tg mice, the expansion of CD8+ T cells in the peripheral blood is evident very early in life, but macroscopic evidence of splenomegaly, enlarged lymph nodes and gross pathologic manifestations of the small intestines become visible when these mice are around 3 months of age and continue progressively thereafter until they succumb to death around 10–12 months of age. At the time of necropsy, our study animals were 6 months of age and all mice in the sham-treated group displayed massive splenomegaly, visible inflammation of the proximal small intestines with swelling and distention of the affected region, hyperemic granular serosal surface with distended blood vessels as well as considerable shortening of the intestinal length (see Fig. 3). In striking contrast, in all mice that were treated with tofacitinib, the spleens were of normal size and the intestines were of normal length and were devoid of any evidence of inflammation. Moreover, consistent with gross pathologic changes that were evident in the sham-treated T3b-hIL-15 Tg mice, tissue sections made from the affected area of the proximal intestines of these sham-treated mice revealed a spectrum microscopic abnormalities that included massive infiltration of IEL, vacuolar degenerative changes in enterocytes, and blunting and disfigurement of intestinal villi. However, all animals in the tofacitinib treated group were free of any aberrant histologic changes and displayed reestablishment of normal villous morphology that resembled those of non-transgenic littermates (Fig. 4, panel A). We confirmed that the infiltrating lymphocytes in the proximal small intestines of T3b-hIL-15 Tg mice were primarily cells bearing CD3 by immunohistochemistry as shown in panel B of Fig. 4. These macroscopic and microscopic observations collectively attest to the efficacy of tofacitinib therapy in reversing the pathologic manifestations associated with these T3b-hIL-15 Tg mice due to the local over-expression of IL-15 in the small intestines.
Figure 3.
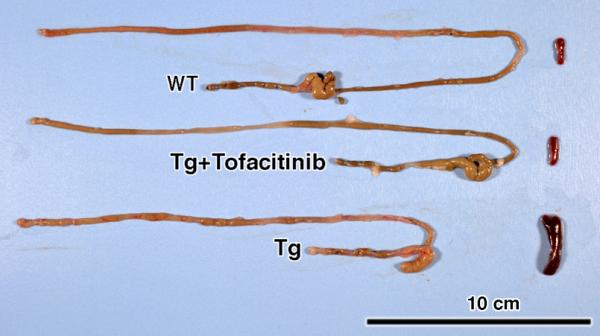
A 42-day course of tofacitinib therapy reverses the macroscopic inflammatory pathologic manifestations in T3b-hIL-15 Tg mice. Following tofacitinib therapy, the intestines and spleens were harvested from T3b-hIL-15 Tg mice and a gross examination of organs for length and size, swelling/distention with serosal surface hyperemia with distended blood vessels was performed. For comparison, wild type litter-mates (WT) and sham-treated T3b-hIL-15 Tg mice were also included in the assessment. Affected organs from one representative mouse from each group of 6 mice are shown and the other mice displayed similar profiles.
Figure 4.

A 42-day course of tofacitinib therapy reverses the microscopic inflammatory pathologic manifestations in T3b-hIL-15 Tg mice. To evaluate the impact of tofacitinib therapy on villous architecture and IEL infiltration, the small intestines were harvested and tissue sections were made from the proximal small intestines. Immunohistochemistry was performed to assess the degree of CD3+ T cell infiltration into the proximal small intestinal tissues and to evaluate the impact of tofacitinib treatment on this CD3+T cell infiltration. Immunostained sections of proximal small intestines from wild type litter mates, sham-treated, and tofacitinib-treated T3b-hIL-15 Tg mice with magnification at 10×. Panel A shows staining with normal rabbit IgG control antibody and panel B shows staining with rabbit polyclonal anti CD3 antibody. Affected small intestines from one representative mouse from each group of 6 mice are shown and the other mice displayed similar profiles.
Clinical management of CD involves a life-long endeavor even in patients who are responsive to a gluten-free diet [2, 3, 5]. Devising a long term therapeutic strategy for refractory CD is even more challenging and understandably would require multiple cycles of therapy even if a single cycle of therapy confers an extended therapeutic benefit. Therefore, in order to have an assessment on the durability of tofacitinib efficacy, we monitored the peripheral blood cell subset profiles by flow cytometry in a group of T3b-hIL-15 Tg mice that had been treated with a 42 day course of tofacitinib, 10 weeks after the conclusion of the treatment. As shown in Fig. 5, there was clear evidence that the re-expansion of CD8+ T cell subset has already begun (87.5% and 73.9% in two treated animals versus 23.8% and 30.8% in wild-type littermates) in these mice within 2 months after completion of tofacitinib therapy although the levels were still lower than those seen in the untreated T3b-hIL-15 Tg mice, reflecting the possibility that multi-cycle, long-term tofacitinib therapy will likely be necessary in managing patients with refractory CD when treated with this drug.
Figure 5.

Assessment of the sustainability of Tofacitinib effects on CD8+ T cells in T3b-hIL-15 Tg mice. Ten weeks after the completion of a 42-day course of tofacitinib therapy, blood was collected and PBMC were isolated from 2 animals from groups consisting of tofacitinib treated; wild-type littermates, and sham treated groups of mice. The modulation of CD8+ T cell subset was examined by flow cytometry after gating on CD3+ lymphocytes.
Although the most pronounced inhibitory activity of tofacitinib is directed against JAK3, it has demonstrable, yet 20-fold less inhibitory activity against JAK2 and 100-fold less activity against JAK1 as well [18]. Ideally, an oral drug that is uniquely inhibitory against JAK3 is the preferred choice to abrogate IL-15 activity in treating CD patients, such agents are currently unavailable to use clinically in patients. Nonetheless, such mono-JAK specific inhibitors are likely to be available for clinical use in the not so distant future as some of these drugs are already in early Phase1/Phase2 trials of rheumatoid arthritis, for example VX-509, a JAK3 inhibitor from Vertex Pharmaceutical is being evaluated in a Phase 2 trial and GLPG0634 a JAK1inhibitor (Galapagos, NV) has completed a Phase 1 trial successfully [17, 19]. However, the adverse effects seen with tofacitinib has not been overly serious from reports of several trials, although anemia and neutropenia related to JAK2 inhibition along with increased low density lipoprotein levels that may be associated with IL-6 signaling inhibition [reviewed in ref 17]. Intriguingly, in our study as shown in Fig. 6, we observed increased visceral adiposity with the accumulation of omental fat in all of the T3b-hIL-15 Tg mice that were treated with a 42-course of tofacitinib, which we believe should be looked at carefully in ongoing trials because the potential for tofacitinib-induced alterations in lipid metabolism and associated dyslipidemia may not have been adequately addressed especially in the context of long term usage.
Figure 6.
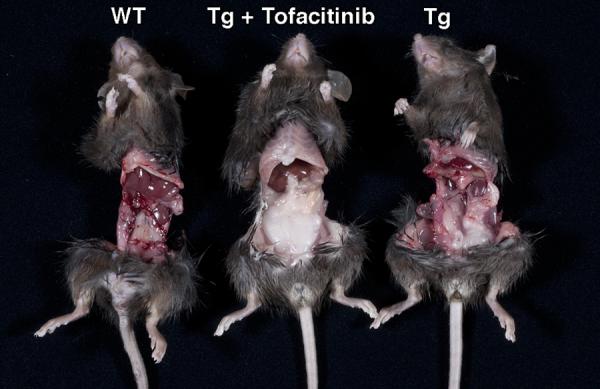
Tofacitinib therapy induces visceral adiposity with the accumulation of omental fat in T3b-hIL-15 Tg mice. Following a 42-day course of tofacitinib, mice were euthanized and eviscerated. For comparison, wild type litter-mates (WT) and sham-treated T3b-hIL-15 Tg mice were also included in the assessment and the data shown are from one representative mouse from each group of 6 mice and the other mice displayed similar manifestations.
Discussion
With the increasing recognition of the central role that locally over-expressed IL-15 plays in orchestrating pathologic tissue destruction in CD, there is growing enthusiasm for exploring IL-15 directed therapeutic strategies especially for refractory CD that does not respond to dietary gluten withdrawal and for which no effective therapy exists currently [6, 20]. Unmitigated refractory CD often progresses to enteropathy associated T cell lymphoma (EATL) which itself is refractory to standard lymphoma regimens and carries a grim prognosis, thus underscoring the urgent need for effective treatment modalities for this disease. Blockade of IL-15 signaling with antibodies either directed against the CD122 receptor subunit that prevents IL-15 transpresentation and binding to its cognate receptor complex or neutralizing antibodies that bind directly to IL-15 have been shown to be potently effective in reversing the intestinal pathologic manifestations in animal models that reasonably reproduce pathologic lesions seen in CD [9, 11, 21]. Based on these very encouraging pre-clinical observations, a Phase II trial is to begin in the very near future with a cohort of refractory CD patients, to evaluate the potential efficacy of humanized Mik-Beta-1 monoclonal antibody directed against CD122 that blocks IL-15 action, in improving the clinical disease. However, the availability of humanized Mik-Beta-1 monoclonal antibody or the IL-15 neutralizing antibody AMG714 for widespread clinical use remains uncertain as their clinical development plans are still at early stages. As an alternative, in the present study, we have provided evidence that tofacitinib is equally effective as anti-IL-15 biologics in reversing IL-15 mediated intestinal pathologic manifestations in the T3b-hIL-15 Tg mouse model of celiac disease pathology [9, 11, 21] and we are cautiously optimistic that tofacitinib with the convenience of its oral administration would prove to be an efficacious therapeutic modality in the long-term management of gluten-refractory CD for which currently no effective therapies exist.
Supplementary Material
Acknowledgements
This research was supported in part by the intramural programs of the National Cancer Institute and a Grant-in-Aid for 2009 Multidisciplinary Research Project from MEXT in Japan from the Ministry of Education, Science, Sports, and Culture of Japan (T. Hiroi). L.P. Perera gratefully acknowledges the receipt of an invitational fellowship from the Japan Society for the Promotion of Science.
Footnotes
Authorship Contributions
S.Y., P-Y.P., T.A.W., T.H., and L.P.P. designed experiments and interpreted the data S.Y., and L.P.P. performed experiments; L.P.P supervised the study and wrote the paper.
Disclosure of Conflicts of Interest
The authors declare no competing financial interests.
References
- 1.Dubé C, Rostom A, Sy R, Cranney A, Saloojee N, Garritty C, Sampson M, Zhang L, Yazdi F, Mamaladze V, Pan I, Macneil J, Mack D, Patel D, Moher D. The prevalence of celiac disease in average-risk and at-risk Western European populations: a systematic review. Gastroenterology. 2005;128(4 Suppl 1):S57–67. doi: 10.1053/j.gastro.2005.02.014. [DOI] [PubMed] [Google Scholar]
- 2.Armstrong MJ, Hegade VS, Robins G. Advances in coeliac disease. Curr Opin Gastroenterol. 2012;28(2):104–12. doi: 10.1097/MOG.0b013e32834d0844. [DOI] [PubMed] [Google Scholar]
- 3.Scanlon SA, Murray JA. Update on celiac disease - etiology, differential diagnosis, drug targets, and management advances. Clin Exp Gastroenterol. 2011;4:297–311. doi: 10.2147/CEG.S8315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abadie V, Sollid LM, Barreiro LB, Jabri B. Integration of genetic and immunological insights into a model of celiac disease pathogenesis. Annu Rev Immunol. 2011;29:493–525. doi: 10.1146/annurev-immunol-040210-092915. [DOI] [PubMed] [Google Scholar]
- 5.Green PH, Cellier C. Celiac disease. N Engl J Med. 2007;357(17):1731–43. doi: 10.1056/NEJMra071600. [DOI] [PubMed] [Google Scholar]
- 6.Di Sabatino A, Biagi F, Gobbi PG, Corazza GR. How I treat enteropathy-associated T-cell lymphoma. Blood. 2012;119(11):2458–68. doi: 10.1182/blood-2011-10-385559. [DOI] [PubMed] [Google Scholar]
- 7.Hüe S, Mention JJ, Monteiro RC, Zhang S, Cellier C, Schmitz J, Verkarre V, Fodil N, Bahram S, Cerf-Bensussan N, Caillat-Zucman S. A direct role for NKG2D/MICA interaction in villous atrophy during celiac disease. Immunity. 2004;21(3):367–77. doi: 10.1016/j.immuni.2004.06.018. [DOI] [PubMed] [Google Scholar]
- 8.Meresse B, Chen Z, Ciszewski C, Tretiakova M, Bhagat G, Krausz TN, Raulet DH, Lanier LL, Groh V, Spies T, Ebert EC, Green PH, Jabri B. Coordinated induction by IL15 of a TCR-independent NKG2D signaling pathway converts CTL into lymphokine-activated killer cells in celiac disease. Immunity. 2004;21(3):357–66. doi: 10.1016/j.immuni.2004.06.020. [DOI] [PubMed] [Google Scholar]
- 9.Yokoyama S, Watanabe N, Sato N, Perera PY, Filkoski L, Tanaka T, Miyasaka M, Waldmann TA, Hiroi T, Perera LP. Antibody-mediated blockade of IL-15 reverses the autoimmune intestinal damage in transgenic mice that overexpress IL-15 in enterocytes. Proc Natl Acad Sci U S A. 2009;106(37):15849–54. doi: 10.1073/pnas.0908834106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Benahmed M, Meresse B, Arnulf B, Barbe U, Mention JJ, Verkarre V, Allez M, Cellier C, Hermine O, Cerf-Bensussan N. Inhibition of TGF-beta signaling by IL-15: a new role for IL-15 in the loss of immune homeostasis in celiac disease. Gastroenterology. 2007;132(3):994–1008. doi: 10.1053/j.gastro.2006.12.025. [DOI] [PubMed] [Google Scholar]
- 11.DePaolo RW, Abadie V, Tang F, Fehlner-Peach H, Hall JA, Wang W, Marietta EV, Kasarda DD, Waldmann TA, Murray JA, Semrad C, Kupfer SS, Belkaid Y, Guandalini S, Jabri B. Co-adjuvant effects of retinoic acid and IL-15 induce inflammatory immunity to dietary antigens. Nature. 2011;471(7337):220–4. doi: 10.1038/nature09849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zanzi D, Stefanile R, Santagata S, Iaffaldano L, Iaquinto G, Giardullo N, Lania G, Vigliano I, Vera AR, Ferrara K, Auricchio S, Troncone R, Mazzarella G. IL-15 interferes with suppressive activity of intestinal regulatory T cells expanded in Celiac disease. Am J Gastroenterol. 2011;106(7):1308–17. doi: 10.1038/ajg.2011.80. [DOI] [PubMed] [Google Scholar]
- 13.Yokoyama S, Takada K, Hirasawa M, Perera LP, Hiroi T. Transgenic mice that overexpress human IL-15 in enterocytes recapitulate both B and T cell-mediated pathologic manifestations of celiac disease. J Clin Immunol. 2011;31(6):1038–44. doi: 10.1007/s10875-011-9586-7. [DOI] [PubMed] [Google Scholar]
- 14.Ohta N, Hiroi T, Kweon MN, Kinoshita N, Jang MH, Mashimo T, Miyazaki J, Kiyono H. IL-15-dependent activation-induced cell death-resistant Th1 type CD8 alpha beta+NK1.1+ T cells for the development of small intestinal inflammation. J Immunol. 2002;169(1):460–8. doi: 10.4049/jimmunol.169.1.460. [DOI] [PubMed] [Google Scholar]
- 15.Perera PY, Lichy JH, Waldmann TA, Perera LP. The role of interleukin-15 in inflammation and immune responses to infection: implications for its therapeutic use. Microbes Infect. 2012;14(3):247–61. doi: 10.1016/j.micinf.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Waldmann TA. The biology of interleukin-2 and interleukin-15: implications for cancer therapy and vaccine design. Nat Rev Immunol. 2006;6(8):595–601. doi: 10.1038/nri1901. [DOI] [PubMed] [Google Scholar]
- 17.Fleischmann R. Novel small-molecular therapeutics for rheumatoid arthritis. Curr Opin Rheumatol. 2012;24(3):335–41. doi: 10.1097/BOR.0b013e32835190ef. [DOI] [PubMed] [Google Scholar]
- 18.Kontzias A, Laurence A, Gadina M, O'Shea JJ. Kinase inhibitors in the treatment of immune-mediated disease. F1000 Med Rep. 2012;4:5. doi: 10.3410/M4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garber K. Pfizer's JAK inhibitor sails through phase 3 in rheumatoid arthritis. Nature Biotech. 2011;29(6):467–68. doi: 10.1038/nbt0611-467. [DOI] [PubMed] [Google Scholar]
- 20.Chandesris MO, Malamut G, Verkarre V, Meresse B, Macintyre E, Delarue R, Rubio MT, Suarez F, Deau-Fischer B, Cerf-Bensussan N, Brousse N, Cellier C, Hermine O. Enteropathy-associated T-cell lymphoma: a review on clinical presentation, diagnosis, therapeutic strategies and perspectives. Gastroenterol Clin Biol. 2010;34(11):590–605. doi: 10.1016/j.gcb.2010.09.008. [DOI] [PubMed] [Google Scholar]
- 21.Malamut G, El Machhour R, Montcuquet N, Martin-Lannerée S, Dusanter-Fourt I, Verkarre V, Mention JJ, Rahmi G, Kiyono H, Butz EA, Brousse N, Cellier C, Cerf-Bensussan N, Meresse B. IL-15 triggers an antiapoptotic pathway in human intraepithelial lymphocytes that is a potential new target in celiac disease-associated inflammation and lymphomagenesis. J Clin Invest. 2010;120(6):2131–43. doi: 10.1172/JCI41344. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.