

Abstract
Objectives
Chronic azithromycin therapy has been associated with improved clinical outcomes in patients with cystic fibrosis (CF) who are chronically infected with Pseudomonas aeruginosa. We have previously demonstrated that azithromycin polarizes macrophages towards an alternatively activated phenotype, thereby blunting inflammation associated with infection. Because this phenotype is pro-fibrotic, it is important to evaluate azithromycin's consequential effects upon fibroblast function and extracellular matrix (ECM) protein production.
Methods
We co-cultured macrophages and fibroblasts together and stimulated them by adding P. aeruginosa or lipopolysaccharide to assess the ability of azithromycin to alter the macrophage phenotype, along with the impact exerted upon the production of fibronectin and other effectors that govern tissue remodelling, including transforming growth factor β (TGFβ), matrix metalloproteinase-9 (MMP-9) and arginase. We supported these studies by evaluating the impact of azithromycin treatment on these proteins in a mouse model of P. aeruginosa infection.
Results
Azithromycin increased arginase expression in vitro, as well as the activation of latent TGFβ, consistent with polarization to the alternative macrophage phenotype. While the drug increased fibronectin concentrations after stimulation in vitro, secretion of the ECM-degrading enzyme MMP-9 was also increased. Neutralization of active TGFβ resulted in the ablation of azithromycin's ability to increase fibronectin concentrations, but did not alter its ability to increase MMP-9 expression. In P. aeruginosa-infected mice, azithromycin significantly decreased MMP-9 and fibronectin concentrations in the alveolar space compared with non-treated, infected controls.
Conclusions
Our results suggest that azithromycin's effect on MMP-9 is regulated independently of TGFβ activity. Additionally, the beneficial effects of azithromycin may be partially due to effects on homeostasis in which ECM-degrading mediators like MMP-9 are up-regulated early after infection. This may impact the damaging effects of inflammation that lead to fibrosis in this patient population.
Keywords: P. aeruginosa, bacterial infections, lung inflammation
Introduction
The fibrotic changes in the lungs that characterize the pathophysiology of cystic fibrosis (CF) are often caused by the Gram-negative bacterium Pseudomonas aeruginosa. The inability of patients with CF to clear this organism from the lungs leads to multiple cycles of a dysregulated innate immune response that results in pulmonary fibrosis. While classical activation of macrophages (CAM) produces a cell type that initiates and coordinates inflammation and kills invading pathogens, macrophages can also undergo alternative activation, giving rise to a phenotype with distinctly different functions. Classical activation is induced through exposure to interferon γ (IFNγ) in addition to a microbial trigger [e.g. lipopolysaccharide (LPS)] and has been extensively studied. Conversely, macrophages can be polarized to an alternatively activated macrophage (AAM) phenotype by interleukin (IL)-4 and IL-13, cytokines mainly produced by Th2-type CD4+ T lymphocytes.1 The role of alternative macrophage activation in the setting of extracellular bacterial infection is unknown. The AAM phenotype produces anti-inflammatory cytokines and is responsible for coordinating repair mechanisms in surrounding tissues following an acute infection through the production of the effector molecules arginase-1, transforming growth factor β (TGFβ) and IL-10.1
Arginase controls tissue repair mechanisms through its catalysis of l-arginine to urea and l-ornithine, which serve as critical precursors in the production of collagen, fibronectin and other proteins involved in tissue remodelling and fibrosis accumulation after inflammatory injury.1 Arginase production has been correlated with activation of the type II cytokine TGFβ in several studies of pulmonary repair and fibrosis.2,3 In target cells, such as fibroblasts and epithelial cells, TGFβ activation leads to build-up of extracellular matrix (ECM) components and a decrease in matrix metalloproteinase (MMP) expression.4,5 This imbalance can eventually lead to fibrosis and scar formation in pulmonary tissues. Excess TGFβ activation is associated with the airway remodelling characteristic of multiple lung pathologies, including CF and chronic obstructive pulmonary disease (COPD).6
The azalide antimicrobial agent azithromycin, utilized chronically for its effects on inflammation, has been shown in three randomized, placebo-controlled trials to improve clinical and outcome measures in patients with CF in the short term.7–9 The anti-inflammatory activity of azithromycin has been partially characterized.10–12 Previous work by our group demonstrated that azithromycin induces properties of alternative macrophage activation in vitro and in a mouse model of P. aeruginosa infection, despite the presence of classical activation stimuli.13,14 The drug induces an increase in arginase production, a decrease in the CAM effector protein inducible nitric oxide synthase (iNOS) and a characteristic AAM cytokine production profile.13,14 Oral azithromycin treatment of mice infected with P. aeruginosa led to a reduction in mortality and a change in the influx of immune cells to a more monocytic and less neutrophilic population.14
To date, the impact of macrophage polarization with azithromycin upon fibrosis has not been studied. Because AAMs coordinate increased activity of TGFβ, we tested the hypothesis that macrophages treated with azithromycin would increase the production of the pro-fibrotic protein fibronectin when co-cultured with fibroblasts and stimulated with P. aeruginosa. Here we demonstrate that azithromycin increases the production of fibronectin in vitro, an effect that was dependent upon the drug's ability to increase the activation of TGFβ. Surprisingly, we also found that azithromycin increased the production of MMP-9, a result that is counter to our hypothesized outcome. In vivo data from a mouse model of P. aeruginosa pneumonia support these in vitro findings. A better understanding of the interplay between the azithromycin-treated macrophages and fibroblasts will help define the effects of the drug upon fibrosis development.
Materials and methods
Fibroblast/macrophage co-culture
The mouse cell lines NIH/3T3 and J774A.1 (ATCC, Manassas, VA, USA) were used. The NIH/3T3 line (ATCC CRL-1568) is a fibroblast cell line originally obtained from embryonic cultures of NIH Swiss mice.15 The J774 (J774A.1, ATCC TIB-67) cell line is an immortalized macrophage cell line derived from BALB/cN adult mice.16 Cells were grown in RPMI 1640 medium (Invitrogen, Carlsbad, CA, USA) supplemented with 5% fetal calf serum and 2 × 10–5 M 2-mercaptoethanol and incubated at 37°C and 5% CO2. NIH/3T3 cells were added to culture-treated 6-well plates at a density of 2.5 × 105 cells/mL and allowed to incubate overnight. The medium was then removed and replaced with 5 mL of fresh medium along with 2.5 × 105 J774 cells and again allowed to incubate overnight. The next morning, cells were stimulated by adding IFNγ (R&D systems, Minneapolis, MN, USA) (100 ng/mL) and either 2.5 × 105 cfu of live heat-killed P. aeruginosa [PA39018, a non-biofilm-producing strain obtained from ATCC (Manassas, VA, USA)] or 50 ng/mL LPS, depending on the experiment. Certain wells were additionally treated with 30 μM (22.5 μg/mL) azithromycin (Sigma Aldrich, St Louis, MO, USA). This concentration was chosen based upon optimal macrophage polarization conditions delineated in our previous studies.13 Supernatants were saved for TGFβ, MMP-9 and fibronectin analyses. Cells were washed and counted using trypan blue staining to assess viability, and aliquots were taken and lysed for the arginase activity assay and qRT–PCR. Supernatants and aliquots of cell lysates were frozen at −80°C in protease inhibitor buffer (Roche Mini-tablet Protease Inhibitor Cocktail) or RNAlater (Applied Biosystems, Foster City, CA, USA) for subsequent analysis.
In vivo infection
C57Bl/6 mice were purchased from The Jackson Laboratory (Bar Harbor, ME, USA) and utilized for all experiments. All animal studies were approved by the University of Kentucky Institutional Animal Care and Use Committee and comply with all national standards and regulations governing the use of animals for research. Mice were housed in pathogen-free isolation and transferred to a biosafety level 2 housing unit after infection. Tablets of azithromycin (Pliva Inc., Zagreb, Croatia) were triturated and the powder was suspended in 2% methylcellulose. Beginning 4 days prior to infection, mice were given azithromycin at 0.16 g/kg in 150 μL or vehicle-only control via oral gavage in order to reach a steady-state concentration of drug exposure at the time of infection. We dosed azithromycin in excess to increase the likelihood of producing the phenotypic changes observed within 4 days and because interspecies differences between intracellular azithromycin concentrations are unknown. Administration of drug or vehicle was continued daily until the time of sacrifice. The clinical mucoid strain P. aeruginosa M57-15 was grown in Trypticase soy broth (TSB) to late log phase or early stationary phase. The method for incorporation of the bacteria into agarose beads, essential to induce prolonged infection in these mice, was adapted from previously described methods.17,18 The P. aeruginosa-laden agarose beads were then diluted to achieve the desired inoculum of 1 to 2 × 105 cfu in 100 μL, which represents approximately the 10% lethal dose. Beads were instilled intratracheally using a blunted 24-gauge inoculation needle while the animals were under isofluorane anaesthesia.
Mice were humanely killed on day 0 and on post-infection days 3, 7 and 14, and bronchial alveolar lavage samples were obtained as representative of the airway compartment. Lungs were lavaged with 5 mL of buffered solution containing 0.3 mM EDTA in 1 mL aliquots. Cell lysates were frozen at −80°C for subsequent protein quantification. Lungs were removed and digested in RPMI medium containing 5% heat-inactivated fetal calf serum with 1 mg/mL collagenase A and 50 U/mL DNase for 1 h at 37°C. An aliquot was plated to assess bacterial burden by manual cfu counting on Pseudomonas selection agar.
Arginase activity
The total protein concentration of each sample was analysed using the bicinchoninic acid protein assay (Pierce Biotechnology, Rockford, IL, USA). Arginase activity was quantified by measuring the conversion of l-arginine into urea.19 In brief, 10 mM MnCl2 in 50 mM Tris–HCl was added to the lysed sample and the mixture was then incubated at 55°C to activate the arginase enzyme. l-Arginine was then added and samples were incubated overnight and then terminated by the addition of an acid solution. The colorimetric indicator 9% α-isonitrosopropiophenone in 100% ethanol was then added to each tube, heated to 95°C and the OD of each sample was read using a 490 nm filter. Readings were compared with a standard curve of known urea concentration.
MMP-9 and fibronectin concentrations
Cell culture supernatants and lung lavage samples were analysed for MMP-9 and fibronectin concentrations by indirect ELISA. Samples were diluted in coating buffer and incubated at 4°C overnight for adherence. Wells were blocked with 0.5% BSA in 20 mM Tris plus 150 mM sodium chloride, then incubated with an antibody specific to MMP-9 (Abcam, Cambridge, MA, USA) or fibronectin (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) followed by an anti-rabbit HRP-conjugated secondary antibody (Millipore, CA, USA). Wells were analysed by OD reading at 450 nm, using OptEIA detection reagents (BD, CA, USA). Readings were compared with a standard curve using MMP-9 or fibronectin recombinant protein (R&D Systems) and normalized to viable cell count (MMP-9) or total protein (fibronectin).
TGFβ activity
Cell co-culture supernatants were analysed by ELISA using the TGFβ1 Emax ImmunoAssay System (Promega, Madison, WI, USA). Samples were diluted in PBS according to the manufacturer's instructions. Plates were coated with a monoclonal antibody specific for bioactive TGFβ and samples were added to the wells and a standard curve was prepared using the supplied TGFβ1 standard. Polyclonal anti-TGFβ1 antibodies were applied, followed by washing and incubation with HRP conjugate. Development solution was applied to the samples and the OD readings were obtained at 450 nm and compared with the TGFβ standard curve to find the concentration of the active form of TGFβ in each test sample. Concentrations were normalized to viable cell count values.
Neutralization/inhibition experiments
Certain experiments used neutralizing antibody or small-molecule inhibitors to assess the resulting absence of function for each corresponding protein. Materials included TGFβ-1,2,3-neutralizing antibody (R&D Systems), which was used at 0.25 μg/mL, the arginase-1 inhibitor S-(2-boronoethyl)-l-cysteine (BEC), synthesized as described previously and used at a concentration of 500 μM,20,21 and the MMP-9 inhibitor SB-3CT (Enzo Life Sciences, Farmingdale, NY, USA), used at 600 nM, which specifically blocks MMP-9.22 Inhibitors were added concurrently with cytokines and azithromycin.
qRT–PCR
RNA was isolated using RNeasy Mini Kits (Qiagen, Valencia, CA, USA) and quantified using a Nanodrop 2000 (Thermo Scientific, Wilmington, DE, USA). Reverse transcription was performed on equal amounts of RNA utilizing Taqman Reverse Transcriptase Reagents (Applied Biosystems) according to the manufacturer's protocols. qRT–PCR was initiated utilizing Taqman gene expression arrays for murine TGFβ1, MMP-9 and GAPDH using an ABI Prism 7000 (Applied Biosystems).
Western blot
To assess Smad2 and pSmad2 expression, samples were run with β-mercaptoethanol, in denaturing conditions, on 10% SDS–PAGE gels and then transferred to polyvinylidene fluoride membranes at 100 V for 1 h. Membranes were blocked in 5% dry milk or BSA and, when phospho-antibodies were used, in Tris-buffered saline (TBS), for at least 4 h. Primary antibodies specific for Smad2 or pSmad2 (Cell Signaling Technology, Boston, MA, USA) were diluted 1:1000 in 1% dry milk or BSA in TBS and incubated overnight. Blots were washed and incubated for 2 h at room temperature in secondary HRP conjugate at 1:4000 dilution [goat anti-mouse HRP conjugated (BD Pharmingen, San Diego, CA, USA)] and detected using Pierce ECL Western Blotting Substrate (Thermo Scientific, Rockford, IL, USA).
Statistical analysis
Results are reported as mean ± SD and were analysed using GraphPad Prism (GraphPad Software, La Jolla, CA, USA). Data were compared among groups at each timepoint via one-way ANOVA, followed by Bonferroni's post test for individual comparisons. Differences were deemed statistically significant at a P value <0.05.
Results
Macrophage arginase production
We assessed the ability of azithromycin to polarize macrophages in the presence of P. aeruginosa using co-culture conditions as we previously published for macrophages cultured alone.13 There was a significant increase in arginase concentrations in lysates after 24 h (P < 0.01) and 48 h (P < 0.01) of bacterial stimulation in cells treated with azithromycin (Figure 1a). Arginase concentrations predictably decreased when the organism was introduced, as this shifts macrophages towards a pro-inflammatory state. Furthermore, there was no statistically significant impact upon cell number or viability between azithromycin-treated and control cells (Figure 1b). Azithromycin had no effect on arginase production from NIH/3T3 fibroblast cells that had been exposed to P. aeruginosa in the absence of macrophages (data not shown).
Figure 1.

Arginase production in co-cultured cells. NIH/3T3 and J774 cells were cultured together overnight with IFNγ with or without 30 μM azithromycin (AZM) and stimulated the following day with P. aeruginosa (PA). (a) Cells were harvested at 0, 6, 12, 24 and 48 h, and arginase activity, a measure of arginase concentration, was assessed in cell lysates. (b) Cell viability was determined using trypan blue staining and manual counting. Samples were run in triplicate and data are shown as means ± SD. Results are representative of three replicated experiments and were analysed using one-way ANOVA with Bonferroni's post test. **P < 0.01.
Supernatant TGFβ activation
Next we examined the activation characteristics of TGFβ as a result of drug exposure. When azithromycin was added along with IFNγ and P. aeruginosa, the amount of active TGFβ was increased compared with control conditions (including IL-4/IL-13 treatment) after 4 h of culture (Figure 2a). Subsequently we assessed TGFβ activation temporally when the cells were stimulated with LPS. Azithromycin was found to have the same effect up to 48 h after stimulation (Figure 2b). We additionally assessed tgfβ gene expression via qRT–PCR over time (Figure 2c). Relative to time 0, tgfβ mRNA expression was down-regulated in both groups, with the azithromycin-treated cells maintaining higher levels of mRNA throughout the experiment, although differences did not reach statistical significance. Finally, appreciable TGFβ activation did not occur when the cell lines were cultured individually (Figure 2d and e).
Figure 2.

Disposition of TGFβ in the co-culture system over time. (a) Macrophages were co-cultured with fibroblasts and treated with IFNγ, IL-4/IL-13 or medium alone overnight. At time 0, azithromycin (AZM) and/or P. aeruginosa (PA) were added and the cells were incubated for 4 h. (b–e) Macrophages were co-cultured with fibroblasts and treated with IFNγ overnight then stimulated with LPS in the presence or absence of AZM. Cells and supernatants were collected at 6, 12, 24 and 48 h after the addition of LPS. (b) Activated TGFβ concentrations in the culture supernatant as measured using the TGFβ1 Emax ImmunoAssay System are graphed over time for the AZM treatment and non-treatment groups. (c) qRT–PCR analysis of tgfβ mRNA was normalized to GAPDH expression in each sample and then normalized to expression of tgfβ at time 0. Single-cell controls were treated similarly to measure active TGFβ concentration in (d) J774 macrophages alone and (e) NIH/3T3 fibroblasts alone. Samples were run in triplicate and data are shown as means ± SD with active TGFβ concentrations normalized to viable cell count. Results are representative of four replicated experiments and were analysed using one-way ANOVA with Bonferroni's post test. Significance is indicated for P values <0.05 (*), <0.01 (**) and <0.001 (***).
MMP-9 secretion
We then sought to determine whether azithromycin increases the expression of MMP-9, which cleaves and activates matrix-bound TGFβ, which then leads to downstream fibrogenesis.23 Figure 3(a) shows that MMP-9 secretion was significantly increased at 4 h by the addition of azithromycin (P < 0.05), even in the absence of cytokine treatment. Cells exposed to IFNγ, P. aeruginosa and azithromycin produced concentrations of MMP-9 that were significantly higher than the control conditions. Upon examining this effect over time (Figure 3b), the steady-state level of MMP-9 was higher in the cells treated with azithromycin. Evaluation of gene expression showed that azithromycin treatment prevented the decrease in MMP-9 mRNA expression in the cells treated with IFNγ and P. aeruginosa (Figure 3c). For each cell line cultured individually, the drug dramatically increased the concentration of MMP-9 in the supernatant of the J774 macrophages (Figure 3d) and while production of MMP-9 from the fibroblast cell line was high (Figure 3e) results were inconsistent across experimental timepoints.
Figure 3.

Impact of azithromycin (AZM) upon MMP-9. (a) Macrophages were co-cultured with fibroblasts and treated with IFNγ, IL-4/IL-13 or medium alone overnight. At time 0, AZM and/or P. aeruginosa (PA) were added and the cells were incubated for 4 h. Supernatants were collected to measure MMP-9 protein concentrations by indirect ELISA. (b) Co-cultured cells were treated with IFNγ overnight and stimulated with PA in the presence or absence of AZM. Cells and supernatants were collected at 0, 6, 12, 24, 48 and 72 h. Supernatants were analysed for MMP-9 protein concentration via indirect ELISA. (c) Co-cultured cells were treated as in (a), but harvested at earlier timepoints to measure mRNA for MMP-9. Cells were collected at timepoints 0 and 15 min, 30 min, 1 h, 3 h and 6 h after the addition of PA to determine the impact of AZM treatment. Single-cell controls were treated similarly to measure MMP-9 concentration in (d) J774 macrophages alone and (e) NIH/3T3 fibroblasts alone. Samples were run in triplicate and data are shown as means ± SD and normalized to viable cell count. Results are representative of three replicated experiments and were analysed using one-way ANOVA with Bonferroni's post test. Significance is indicated for P values <0.05 (*), <0.01 (**) and <0.001 (***).
Fibronectin concentration
At specified timepoints fibronectin was quantified by indirect ELISA and normalized to total protein concentration from cell lysates. When measured after 4 h of co-culture, treatment with azithromycin alone caused a slight reduction in fibronectin concentration compared with untreated controls (Figure 4a). When IFNγ and P. aeruginosa were added, fibronectin concentrations returned to baseline and were slightly elevated by the addition of azithromycin. Significantly higher fibronectin concentrations were observed at 12 and 24 h after stimulation in cells exposed to IFNγ plus azithromycin compared with IFNγ alone (P < 0.001 and P < 0.05, respectively) (Figure 4b). Further analysis revealed that azithromycin had no effect on fibronectin concentration when either J774 or NIH/3T3 cells were separately cultured and stimulated with LPS (Figure 4c and d).
Figure 4.

Impact of azithromycin (AZM) upon fibronectin. (a) Macrophages were co-cultured with fibroblasts and treated with IFNγ, IL-4/IL-13 or medium alone overnight. At time 0, AZM and/or P. aeruginosa (PA) were added and the cells were incubated for 4 h. Supernatants were collected to measure MMP-9 protein concentrations by indirect ELISA. (b–d) Macrophages and/or fibroblasts were co-cultured and treated with IFNγ and LPS with or without 30 µM AZM at time 0. Cells and supernatants were collected at 0, 6, 12, 24 and 48 h after stimulation. (b) Fibronectin concentration in the cell pellet lysate, as a measure of protein incorporated into the matrix, was determined by indirect ELISA and depicted over time. Macrophages (c) or fibroblasts (d) were cultured alone and treated as in (a) with fibronectin concentration shown over time post-stimulation with LPS. Results are normalized to total protein in each sample. Samples were run in triplicate and data are shown as means ± SD. Results are representative of three replicated experiments and were analysed using one-way ANOVA with Bonferroni's post test. Significance is indicated for P values <0.05 (*) and <0.001 (***).
Arginase inhibition
To assess whether the ability of azithromycin to alter TGFβ activation and fibronectin production was dependent upon its effect upon arginase, macrophages and fibroblasts in co-culture were stimulated with LPS, this time with the addition of the arginase inhibitor BEC. Figure 5(a) shows results similar to those presented above, as azithromycin increased TGFβ activation over time post-stimulation. With the addition of BEC, activation of TGFβ was increased at the 12 h timepoint by azithromycin, but subsequent increases over the control condition were ablated (Figure 5b). Similarly, the statistically significant increase in fibronectin concentration at the 24 h timepoint by azithromycin (Figure 5c) did not occur in the presence of BEC (Figure 5d).
Figure 5.

Impact of arginase inhibition upon ability of azithromycin (AZM) to activate TGFβ and fibronectin. Macrophages and fibroblasts were co-cultured, treated with IFNγ and stimulated with LPS in the presence or absence of 30 μM AZM. (a and b) Activated TGFβ concentrations in the culture supernatant as measured using the TGFβ1 Emax ImmunoAssay System are graphed over time for the AZM treatment and non-treatment groups with (b) or without (a) addition of the arginase inhibitor BEC. (c and d) Fibronectin concentration in the cell pellet lysate, as a measure of protein incorporated into the matrix, was determined by indirect ELISA and depicted over time with (d) or without (c) the addition of the arginase inhibitor BEC. Data are presented as means ± SD, with active TGFβ values normalized to viable cell counts and fibronectin concentrations normalized to amount of total protein. Results are representative of two replicated experiments and were analysed using one-way ANOVA with Bonferroni's post test. Significance is indicated for P values <0.05 (*).
TGFβ neutralization
We conducted neutralization experiments to determine whether blocking TGFβ activation would affect fibronectin production from the co-cultured cells. The anti-TGFβ antibody utilized for this purpose selectively binds to the activated form of the protein only. While fibronectin concentrations were increased by azithromycin (Figure 6a), the addition of anti-TGFβ antibody resulted in ablation of the effect of the drug (Figure 6b). Azithromycin caused the MMP-9 concentrations to stabilize at a higher level than that of the control condition (Figure 6c) and the addition of the TGFβ-blocking antibody did not change the impact of azithromycin treatment on MMP-9 concentration (Figure 6d). To ensure that the neutralizing antibody was indeed effective in blocking TGFβ function, we performed a western blot to detect Smad2 activation. Smad2 phosphorylation was inhibited by the addition of the neutralizing antibody, while the concentration of total Smad2 was unaffected, suggesting successful TGFβ neutralization (Figure 6e).
Figure 6.

Result of TGFβ neutralization on ability of azithromycin (AZM) to affect fibronectin and MMP-9. Macrophages and fibroblasts were co-cultured, treated with IFNγ and stimulated with LPS in the presence or absence of 30 μM AZM. Cells and supernatants were harvested at successive timepoints to measure fibronectin (by indirect ELISA) without (a) and with (b) the addition of TGFβ-neutralizing antibody (α-TGFβ). Likewise, MMP-9 concentrations were measured in culture over time without (c) and with (d) the addition of α-TGFβ. (e) α-TGFβ's effect upon phosphorylation of Smad2. Smad2 and pSmad2 expression at 0 and 48 h of culture (with β-actin as a loading control) in cells co-cultured with LPS, IFNγ and AZM. Samples were run in triplicate and data are shown as means ± SD. Fibronectin values were normalized to the total amount of protein in each sample and MMP-9 concentrations were normalized to viable cell counts. Results are representative of three replicated experiments and were analysed using one-way ANOVA with Bonferroni's post test. Significance is indicated for P values <0.05 (*), <0.01 (**) and <0.001 (***).
MMP-9 inhibition
In order to determine whether MMP-9 was activating TGFβ directly, an MMP-9 inhibitor, SB-3CT (Enzo Life Sciences, Farmingdale, NY, USA), was added to the co-culture at 600 nM, at which concentration the inhibition is specific for MMP-9 inhibition.22 Consistent with previous experiments, azithromycin increased the concentration of activated TGFβ in the co-culture supernatants when cells were stimulated with IFNγ and LPS (Figure 7a). Here, there was no decrease in TGFβ activation when MMP-9 was inhibited. TGFβ was activated to an even higher degree in cells to which SB-3CT was added at the 48 h timepoint (Figure 7b).
Figure 7.
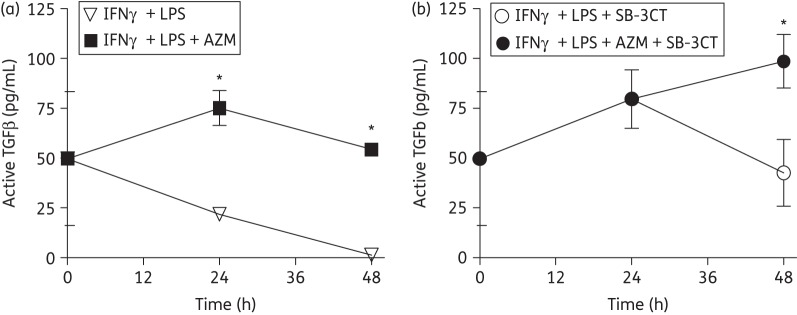
Effect of MMP-9 inhibition upon the activation of TGFβ. Macrophages and fibroblasts were co-cultured, treated with IFNγ and stimulated with LPS in the presence or absence of 30 μM azithromycin (AZM). Cells and supernatants were harvested at successive timepoints to measure active TGFβ without (a) and with (b) addition of the MMP-9 inhibitor SB-3CT. Concentrations of TGFβ were measured by using a TGFβ1 Emax ImmunoAssay System and reported as means ± SD and normalized to viable cell counts. Results are representative of three replicated experiments and were analysed using one-way ANOVA with Bonferroni's post test. Significance is indicated for P values <0.05 (*).
In vivo mouse infection
To support the relevance of these in vitro experiments, the effects of azithromycin upon both MMP-9 and fibronectin concentrations in a mouse model of P. aeruginosa were evaluated. We have previously published data that demonstrate that azithromycin increases arginase concentrations in the lungs of mice infected with P. aeruginosa.14 Normal C57Bl/6 mice receiving daily oral doses of azithromycin had significantly higher concentrations of MMP-9 and fibronectin in the alveolar space on day 7 post-infection compared with infected control animals (Figure 8a and b). Interestingly, both MMP-9 and fibronectin rebounded in the control animals to markedly higher concentrations on day 14 post-infection—evidence that is suggestive of resultant inflammatory injury, which probably led to more severe lung damage with associated fibrosis. As has been the case in all of our investigations, azithromycin treatment did not alter the clearance of the pathogen (data not shown).
Figure 8.

Expression of MMP-9 and fibronectin in a mouse model of P. aeruginosa pneumonia. C57Bl/6 mice were infected intratracheally with agarose beads containing 1.5 × 105 cfu of bacteria. Mice were dosed daily via oral gavage with 0.16 g/kg azithromycin (AZM) starting 4 days before infection. (a) MMP-9 and (b) fibronectin were measured by indirect ELISA from alveolar lavage samples at post-infection timepoints and expressed as concentration per 105 cells. Means ± SD are plotted for experiments utilizing two or three mice per treatment group per timepoint. Significance is indicated for P values <0.05 (*) as analysed using one-way ANOVA with Bonferroni's post-test and the results are representative of two experimental replicates.
Discussion
We report here that azithromycin increases the amount of active TGFβ and the concentration of fibronectin in co-cultured macrophages and fibroblasts stimulated with P. aeruginosa. Our previous findings that evaluated the ability of azithromycin to polarize macrophages to an alternatively activated-like phenotype are again demonstrated here.13 Additionally, we found that azithromycin increased the ratio of active to total TGFβ and resulted in increased concentrations of MMP-9 when compared with non-treated cells. This effect on MMP-9 was also observed in the acute phases of response to P. aeruginosa infection in the lungs of mice. Azithromycin also increased the concentration of fibronectin when macrophages and fibroblasts were stimulated with LPS, as well as in a mouse model of P. aeruginosa pneumonia, results that can be considered a functional indicator of the increase in TGFβ activation. Indeed, our results demonstrate that the ability of azithromycin to induce fibronectin production is dependent on the activation of TGFβ. Likewise, the drug's effect on TGFβ activation is probably through the increase in arginase expression, confirmed by our utilization of arginase inhibition. These results link the generation of alternative macrophage activation by azithromycin (thereby up-regulating arginase expression) with increases in ECM protein production through the key mediator of TGFβ.
We designed this investigation to evaluate the impact of azithromycin on a particular effector function of AAMs—to govern ECM production in the tissue remodelling process that results from inflammatory injury. We utilized the co-culture of macrophages and fibroblasts in order to explore the basic functionality of the remodelling process induced by the drug. Previous studies by our group have focused upon the impact of azithromycin on inflammation, but to this point we are unsure of whether the improvement in morbidity, mortality and lung tissue damage observed in mice infected with P. aeruginosa is due only to a decrease in inflammation or whether the remodelling process is also affected. We suspect the latter because of the profound impact that azithromycin has in the face of bacterial challenge upon the production of arginase, the effector protein that governs ECM production.14 We included in this investigation important mediators of this response that are downstream of arginase and attempted to determine interdependence among the changes of each mediator, culminating in the impact observed upon the ECM protein fibronectin. The inclusion of data from the P. aeruginosa mouse model validates the in vitro results and lays the groundwork for future in vivo experiments evaluating the impact of azithromycin on remodelling and fibrosis.
The type II cytokine TGFβ is implicated in multiple models of pulmonary insult as having a pro-fibrotic role. It also imparts anti-inflammatory effects, as the absence of TGFβ leads to widespread inflammation and dysregulation of immune function.24 Conversely, overexpression of TGFβ leads to increased immune cell migration, fibrosis and organ remodelling.25 Expression of TGFβ in the lungs has been shown to be elevated in patients with fibrotic lung diseases, including idiopathic pulmonary fibrosis and CF.26,27 Patients with CF who have TGFβ concentrations at both high and low extremes in the lungs were shown to have the worst outcomes in a phenotypic gene study performed in the Czech Republic,28 while a study performed in the USA correlated high TGFβ expression levels in bronchoalveolar lavage samples to subjects with the lowest degree of pulmonary function.27
While increased concentrations of activated TGFβ were observed when cells were treated with azithromycin, MMP-9 concentrations were also increased. Typically, TGFβ is activated by several molecules, including integrins,29 thrombospondin6 and the MMPs. We are interested in MMP-9 because of its proteolytic function,30 complementary to the pro-fibrotic function of TGFβ.31 While TGFβ primarily results in build-up of the ECM, MMP-9 is important in ECM turnover, directly degrading fibronectin as well as several types of collagen.30,32 Our results suggest that azithromycin may impact this homeostatic balance to induce a mechanism for matrix clearance. In patients with pulmonary infections, by facilitating both of these processes, overall matrix turnover is probably increased, which could help to restore homeostasis in a hyper-inflammatory lung environment.33
We hypothesized that the observed effect of azithromycin on proteins involved in remodelling was through the increase in activated TGFβ (tied to the drug's ability to induce an AAM-like polarization), but this was proved to be only partially true. While increased TGFβ activation appears to be responsible for the increase in fibronectin in the system, MMP-9 was unaffected by loss of TGFβ activity. The increase in MMP-9 induced by azithromycin is counter to what would be predicted by the drug's ability to induce alternative activation in macrophages. Indeed, our data confirm that normal induction of alternative macrophage activation through IL-4 and IL-13 does not increase MMP-9. Although MMP-9 can directly activate TGFβ,34 our data suggest that, in the co-culture model, azithromycin's affects are not reliant on a direct relationship between these two proteins. An increase in MMP-9 activity without a corresponding increase in natural inhibitors could cause more proteolysis than by simply activating TGFβ. The major function of MMP-9 is to degrade collagen IV, present in basement membranes of epithelial cells, forming the bulk of the pulmonary structure.35 Therefore, increasing MMP-9 production may have wider implications than our in vitro model would demonstrate. If azithromycin indeed increases both the production of MMP-9 and activation of TGFβ in patients, this could provide an explanation for its clinical efficacy. The fact that azithromycin improves clinical outcome measures in patients with CF may hinge upon its ability to also increase ECM turnover. We are currently addressing the effects of azithromycin treatment upon other MMPs and other molecules of similar function.
We did not examine any potential direct effects of azithromycin on P. aeruginosa in this study. While the spectrum of activity of azithromycin does not extend to P. aeruginosa, the drug has been shown to affect quorum-sensing gene expression, as well as to alter biofilm production from P. aeruginosa.36 Changes in these functions of the bacterium could alter its virulence.37 Likewise, P. aeruginosa produces elastases and other proteins that may result in the cleavage and activation of latent TGFβ or degradation of TGFβ or MMP-9, or provide other mechanisms of immune evasion.38 We cannot eliminate the possibility that alterations of P. aeruginosa contributed to the results we observed, although there were direct changes to the macrophages induced by azithromycin. Because we have generated similar results using LPS instead of live bacteria, there is a decreased likelihood that direct drug effects on the organism were responsible for our observations.
In addition, the ability of azithromycin to dampen the initial inflammatory response, as we have previously published,14 could be responsible for improved outcomes and limited fibrosis in the long term by controlling the amount of damage done to the lung tissue at the outset of an infectious challenge. Pre-treatment with azithromycin in mice infected with P. aeruginosa decreases the inflammatory response to the infection by reducing neutrophilia, as well as the concentrations of tumour necrosis factor-α and IL-6 in the lungs.14 These data suggest that azithromycin could provide its benefit through a decrease in inflammation controlled by an increase in AAM effector proteins during the early stages of infection. The results presented here in the mouse model of infection suggest that the protective effect of azithromycin leads to an overall decrease in the amount of remodelling needed, as MMP-9 and fibronectin concentrations in the post-acute phase were lower in mice that received the drug. These results support the data we previously published showing decreased pulmonary damage at these late timepoints in mice receiving azithromycin.14 Additionally, in a clinical study published by our group in 2010, characteristics of macrophage activation in sputum and lavage samples obtained from patients with CF were profiled.39 The results showed trends towards increases in arginase and mannose receptor expression in macrophages of subjects receiving azithromycin and colonized with P. aeruginosa, suggesting that alternative macrophage activation is of potential clinical importance.39
The clinical trials that evaluated chronic azithromycin therapy in the treatment of patients with CF followed subjects for 6 months to 1 year.7–9 Additional investigation is warranted to determine the clinical importance of our data, as durations of therapy with azithromycin in clinical practice go well beyond this timeframe. Future work will include human clinical studies to assess this, as well as in vivo experiments to assess the effect of azithromycin on fibrosis development in the lungs during sub-chronic infections with P. aeruginosa. The fact that azithromycin is effective in improving clinical outcomes in patients with chronic inflammatory lung pathology is highly supportive of future evaluation and therapeutic target development involving effector proteins and functional aspects of alternative macrophage activation.
Funding
This work was supported by the National Center for Research Resources (NCRR; TL1 RR033172), funded by the Office of the Director, National Institutes of Health (NIH) and supported by the NIH Roadmap for Medical Research.
Transparency declarations
None to declare.
Disclaimer
The content is solely the responsibility of the authors and does not necessarily represent the official views of NCRR and NIH.
Acknowledgements
We thank Melissa Hollifield, Heather Hoy, Jennifer Hagadone, Jennifer Strange and Greg Bauman for their technical assistance with this work.
References
- 1.Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- 2.Kitowska K, Zakrzewicz D, Konigshoff M, et al. Functional role and species-specific contribution of arginases in pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2008;294:L34–45. doi: 10.1152/ajplung.00007.2007. [DOI] [PubMed] [Google Scholar]
- 3.Liu Y, Van Ginderachter JA, Brys L, et al. Nitric oxide-independent CTL suppression during tumor progression: association with arginase-producing (M2) myeloid cells. J Immunol. 2003;170:5064–74. doi: 10.4049/jimmunol.170.10.5064. [DOI] [PubMed] [Google Scholar]
- 4.Heine UI, Munoz EF, Flanders KC, et al. Colocalization of TGF-beta 1 and collagen I and III, fibronectin and glycosaminoglycans during lung branching morphogenesis. Development. 1990;109:29–36. doi: 10.1242/dev.109.1.29. [DOI] [PubMed] [Google Scholar]
- 5.Vaday GG, Schor H, Rahat MA, et al. Transforming growth factor-β suppresses tumor necrosis factor α-induced matrix metalloproteinase-9 expression in monocytes. J Leukoc Biol. 2001;69:613–21. [PubMed] [Google Scholar]
- 6.Sheppard D. Transforming growth factor β: a central modulator of pulmonary and airway inflammation and fibrosis. Proc Am Thorac Soc. 2006;3:413–7. doi: 10.1513/pats.200601-008AW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Saiman L, Marshall BC, Mayer-Hamblett N, et al. Azithromycin in patients with cystic fibrosis chronically infected with Pseudomonas aeruginosa: a randomized controlled trial. JAMA. 2003;290:1749–56. doi: 10.1001/jama.290.13.1749. [DOI] [PubMed] [Google Scholar]
- 8.Wolter J, Seeney S, Bell S, et al. Effect of long term treatment with azithromycin on disease parameters in cystic fibrosis: a randomised trial. Thorax. 2002;57:212–6. doi: 10.1136/thorax.57.3.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Equi A, Balfour-Lynn IA, Bush A, et al. Long term azithromycin in children with cystic fibrosis: a randomised, placebo-controlled crossover trial. Lancet. 2002;360:978–84. doi: 10.1016/s0140-6736(02)11081-6. [DOI] [PubMed] [Google Scholar]
- 10.Scaglione F, Rossoni G. Comparative anti-inflammatory effects of roxithromycin, azithromycin and clarithromycin. J Antimicrob Chemother. 1998;41(Suppl B):47–50. doi: 10.1093/jac/41.suppl_2.47. [DOI] [PubMed] [Google Scholar]
- 11.Verleden GM, Vanaudenaerde BM, Dupont LJ, et al. Azithromycin reduces airway neutrophilia and interleukin-8 in patients with bronchiolitis obliterans syndrome. Am J Respir Crit Care Med. 2006;174:566–70. doi: 10.1164/rccm.200601-071OC. [DOI] [PubMed] [Google Scholar]
- 12.Meyer M, Huaux F, Gavilanes X, et al. Azithromycin reduces exaggerated cytokine production by M1 alveolar macrophages in cystic fibrosis. Am J Respir Cell Mol Biol. 2009;41:590–602. doi: 10.1165/rcmb.2008-0155OC. [DOI] [PubMed] [Google Scholar]
- 13.Murphy BS, Sundareshan V, Cory TJ, et al. Azithromycin alters macrophage phenotype. J Antimicrob Chemother. 2008;61:554–60. doi: 10.1093/jac/dkn007. [DOI] [PubMed] [Google Scholar]
- 14.Feola DJ, Garvy BA, Cory TJ, et al. Azithromycin alters macrophage phenotype and pulmonary compartmentalization during lung infection with Pseudomonas. Antimicrob Agents Chemother. 2010;54:2437–47. doi: 10.1128/AAC.01424-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jainchill JL, Aaronson SA, Todaro GJ. Murine sarcoma and leukemia viruses: assay using clonal lines of contact-inhibited mouse cells. J Virol. 1969;4:549–53. doi: 10.1128/jvi.4.5.549-553.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ralph P, Nakoinz I. Phagocytosis and cytolysis by a macrophage tumour and its cloned cell line. Nature. 1975;257:393–4. doi: 10.1038/257393a0. [DOI] [PubMed] [Google Scholar]
- 17.van Heeckeren AM, Schluchter MD. Murine models of chronic Pseudomonas aeruginosa lung infection. Lab Anim. 2002;36:291–312. doi: 10.1258/002367702320162405. [DOI] [PubMed] [Google Scholar]
- 18.Nacucchio MC, Cerquetti MC, Meiss RP, et al. Short communication. Role of agar beads in the pathogenicity of Pseudomonas aeruginosa in the rat respiratory tract. Pediatr Res. 1984;18:295–6. doi: 10.1203/00006450-198403000-00018. [DOI] [PubMed] [Google Scholar]
- 19.Corraliza IM, Campo ML, Soler G, et al. Determination of arginase activity in macrophages: a micromethod. J Immunol Methods. 1994;174:231–5. doi: 10.1016/0022-1759(94)90027-2. [DOI] [PubMed] [Google Scholar]
- 20.Matteson DS, Soloway AH, Tomlinson DW, et al. Synthesis and biological evaluation of water-soluble 2-boronoethylthio compounds. J Med Chem. 1964;7:640–3. doi: 10.1021/jm00335a016. [DOI] [PubMed] [Google Scholar]
- 21.Busnel O, Carreaux F, Carboni B, et al. Synthesis and evaluation of new ω-borono-α-amino acids as rat liver arginase inhibitors. Bioorg Med Chem. 2005;13:2373–9. doi: 10.1016/j.bmc.2005.01.053. [DOI] [PubMed] [Google Scholar]
- 22.Lee M, Villegas-Estrada A, Celenza G, et al. Metabolism of a highly selective gelatinase inhibitor generates active metabolite. Chem Biol Drug Des. 2007;70:371–82. doi: 10.1111/j.1747-0285.2007.00577.x. [DOI] [PubMed] [Google Scholar]
- 23.Corbel M, Belleguic C, Boichot E, et al. Involvement of gelatinases (MMP-2 and MMP-9) in the development of airway inflammation and pulmonary fibrosis. Cell Biol Toxicol. 2002;18:51–61. doi: 10.1023/a:1014471213371. [DOI] [PubMed] [Google Scholar]
- 24.Christ M, McCartney-Francis NL, Kulkarni AB, et al. Immune dysregulation in TGF-beta 1-deficient mice. J Immunol. 1994;153:1936–46. [PubMed] [Google Scholar]
- 25.Pulichino AM, Wang IM, Caron A, et al. Identification of transforming growth factor β1-driven genetic programs of acute lung fibrosis. Am J Respir Cell Mol Biol. 2008;39:324–36. doi: 10.1165/rcmb.2007-0186OC. [DOI] [PubMed] [Google Scholar]
- 26.Kaminski N. Microarray analysis of idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol. 2003;29(Suppl):S32–6. [PubMed] [Google Scholar]
- 27.Harris WT, Muhlebach MS, Oster RA, et al. Plasma TGF-β1 in pediatric cystic fibrosis: potential biomarker of lung disease and response to therapy. Pediatr Pulmonol. 2011;46:688–95. doi: 10.1002/ppul.21430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brazova J, Sismova K, Vavrova V, et al. Polymorphisms of TGF-beta1 in cystic fibrosis patients. Clin Immunol. 2006;121:350–7. doi: 10.1016/j.clim.2006.08.015. [DOI] [PubMed] [Google Scholar]
- 29.Koth LL, Alex B, Hawgood S, et al. Integrin β6 mediates phospholipid and collectin homeostasis by activation of latent TGF-β1. Am J Respir Cell Mol Biol. 2007;37:651–9. doi: 10.1165/rcmb.2006-0428OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Parks WC, Wilson CL, Lopez-Boado YS. Matrix metalloproteinases as modulators of inflammation and innate immunity. Nat Rev Immunol. 2004;4:617–29. doi: 10.1038/nri1418. [DOI] [PubMed] [Google Scholar]
- 31.Lee CG, Homer RJ, Zhu Z, et al. Interleukin-13 induces tissue fibrosis by selectively stimulating and activating transforming growth factor β1. J Exp Med. 2001;194:809–21. doi: 10.1084/jem.194.6.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marom B, Rahat MA, Lahat N, et al. Native and fragmented fibronectin oppositely modulate monocyte secretion of MMP-9. J Leukoc Biol. 2007;81:1466–76. doi: 10.1189/jlb.0506328. [DOI] [PubMed] [Google Scholar]
- 33.Nichols D, Chmiel J, Berger M. Chronic inflammation in the cystic fibrosis lung: alterations in inter- and intracellular signaling. Clin Rev Allergy Immunol. 2008;34:146–62. doi: 10.1007/s12016-007-8039-9. [DOI] [PubMed] [Google Scholar]
- 34.Yu Q, Stamenkovic I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-β and promotes tumor invasion and angiogenesis. Genes Dev. 2000;14:163–76. [PMC free article] [PubMed] [Google Scholar]
- 35.Suki B, Bates JH. Extracellular matrix mechanics in lung parenchymal diseases. Respir Physiol Neurobiol. 2008;163:33–43. doi: 10.1016/j.resp.2008.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hoffmann N, Lee B, Hentzer M, et al. Azithromycin blocks quorum sensing and alginate polymer formation and increases the sensitivity to serum and stationary-growth-phase killing of Pseudomonas aeruginosa and attenuates chronic P. aeruginosa lung infection in Cftr−/− mice. Antimicrob Agents Chemother. 2007;51:3677–87. doi: 10.1128/AAC.01011-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kohler T, Perron GG, Buckling A, et al. Quorum sensing inhibition selects for virulence and cooperation in Pseudomonas aeruginosa. PLoS Pathog. 2010;6:e1000883. doi: 10.1371/journal.ppat.1000883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cosgrove S, Chotirmall SH, Greene CM, et al. Pulmonary proteases in the cystic fibrosis lung induce interleukin 8 expression from bronchial epithelial cells via a heme/meprin/epidermal growth factor receptor/Toll-like receptor pathway. J Biol Chem. 2011;286:7692–704. doi: 10.1074/jbc.M110.183863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Murphy BS, Bush HM, Sundareshan V, et al. Characterization of macrophage activation states in patients with cystic fibrosis. J Cyst Fibros. 2010;9:314–22. doi: 10.1016/j.jcf.2010.04.006. [DOI] [PubMed] [Google Scholar]